
Abstract
The mitochondrial permeability transition pore is an inducer of cell death. During the reperfusion phase after cerebral ischemia, calcium accumulates in mitochondria, and a burst of free radical formation occurs, conditions that favor the activation of the mitochondrial permeability transition pore. Here the authors demonstrate that a blocker of the mitochondrial permeability transition pore, the nonimmunosuppressive cyclosporin A analogue N-methyl-Val-4-cyclosporin A (10 mg/kg intraperitoneally), administered during reperfusion and at 24 hours of reperfusion, diminishes infarct size in a rat model of transient focal ischemia of 2 hours' duration. The mitochondrial permeability transition pore may be an important target for drugs against stroke.
Keywords
The importance of mitochondrial function and dysfunction in health and disease is under intense investigations (Brenner et al., 1998; Halestrap et al., 1998). Not only do mitochondria generate ATP needed to maintain the function of ion pumps and secure cell homeostasis, they also play an active role in physiologic cellular signal transduction and in pathophysiologic events (Cai et al., 1998; Ichas and Mazat, 1998). The focus of this interest in mitochondrial pathophysiology is the formation of a tightly regulated pore in the mitochondrial inner membrane, the mitochondrial permeability transition (MPT) pore, under circumstances of oxidative stress and high calcium content (Hunter and Haworth, 1979b). The activation of the MPT pore disrupts the permeability barrier of the inner mitochondrial membrane, causing uncoupling of oxidative phosphorylation, osmotic swelling, and rupture of the outer membrane. The concomitant release of mitochondrial proteins (Liu et al., 1996) may then activate the caspase cascade, in turn activating endonucleases with subsequent irreversible DNA damage and cell death (Thornberry and Lazebnik, 1998). It has been speculated that the extent of open MPT pores in the mitochondrial population of a cell will decide not only whether the cell will live or die, but whether the cell will die acutely from necrosis (–ATP) or suffer a delayed apoptotic death (+ATP) (Qian et al., 1997).
If the MPT pore is a critical factor in the development of ischemia-reperfusion damage, blockade of the MPT pore would be expected to protect from damage. Accordingly, the immunosuppressive compound cyclosporin A, a potent inhibitor of the MPT pore (Crompton et al., 1988), protects from excitotoxic cell death in vitro (Schinder et al., 1996) and from ischemia-reperfusion damage in the isolated heart (Griffiths and Halestrap, 1993) and liver (Travis et al., 1998). Furthermore, its nonimmunosuppressive analogue and MPT pore blocker N-methyl-Val-4-cyclosporin A (MeValCsA) prevents apoptosis (Zamzami et al., 1996) and protects cells from metabolic inhibition (Seaton et al., 1998). In the brain, cyclosporin A (CsA) and its analogues poorly penetrate the blood-brain barrier, but CsA has been reported to reduce damage caused by transient occlusion of the middle cerebral artery (MCAO) in at least two studies preceding the present one (Butcher et al., 1997; Shiga et al., 1992). The mechanism whereby CsA is protective in ischemia-reperfusion damage is however equivocal. Cyclosporin A may act by blocking the MPT, but may also inhibit calcineurin and therefore act as an immunosuppressant and an inhibitor of nitric oxide synthase (Dawson et al., 1993; Liu et al., 1991). In contrast, the CsA analogue MeValCsA has an MPT pore blocking property but does not inhibit calcineurin (Liu et al., 1992), making it a tool to assess the importance of MPT in pathogenic processes.
In this study, we present novel data on the neuroprotective effect of CsA, but also and more importantly, on the neuroprotective effect of its nonimmunosuppressive analogue MeValCsA in a filament model of transient focal ischemia, supporting the hypothesis of MPT pore involvement in brain ischemia-reperfusion damage.
MATERIALS AND METHODS
All animal experiments were approved by the ethical committee at the University of Lund.
Transient MCAO of the rats were performed as described previously (Memezawa et al., 1992) and is summarized below. Male Wistar rats (Mollegaard's Breeding Center, Copenhagen), weighing 310 to 350 g, were fasted overnight with free access to water. Anesthesia was induced by inhalation of 3% halothane in N2O:O2 (70%:30%) and maintained at 1.5% during surgery. The tail artery was cannulated for blood sampling and blood pressure monitoring. Blood pressure, Pa
Drug administration and experimental groups
Three experimental groups were studied: vehicle-treated group (n = 10), cyclosporin A-treated group (n = 6), and MeValCsA treated group (n = 5). Cyclosporin A (10 mg/kg) or MeValCsA (10 mg/kg) were administered intraperitoneally immediately after recirculation, and after 24 hours of recirculation. Stock solutions of CsA or MeValCsA were made up by dissolving 2.5 g of the substances in 32.5 g Cremophore (Sigma, St Louis, MO, U.S.A.), and adding ethanol to a volume of 50 mL. Of this stock solution, 5 mL was then added to 20 mL saline, generating an injectable solution with a final concentration of 10 mg/mL. The same solution was used as vehicle, omitting active substances.
Evaluation of infarct volume with TTC staining
At 48 hours of recovery following MCAO, the rats were anesthetized by inhalation of 3.0% halothane and killed by decapitation. The brain was quickly removed and chilled in ice-cold saline for 10 minutes. Twelve coronal slices were cut (1 mm thick), beginning 1 mm posterior to the anterior pole, and the slices were immersed in a saline solution containing 1.0% 2,3,5-triphenyltetrazolium chloride (TTC)(Sigma) at 37°C for 30 minutes (Bederson et al., 1986a), and fixed by immersion in 4.0% phosphate-buffered formalin solution. Each brain slice was photographed with black and white film, and the unstained area in each photograph was quantified from the developed film, using a CCD video camera and a video image analyzing system (NIH image, version 1.55). The infarct area in the cortex and the striatum was assessed as the percentage of the total area of the contralateral hemisphere (Bederson et al., 1986a). The total infarct volume was determined by summing up the infarct areas of the 12 slices.
Measurements of mitochondrial pore opening
Brain mitochondria from striatum and cortex were prepared as described previously (Friberg et al., 1998; Sims, 1990). The final pellet was resuspended in modified isolation buffer (0.32 mol/L sucrose, 10 mmol/L Tris, pH 7.4) and kept on ice. Mitochondria (≈ 25 µg/mL) were energized with malate/glutamate (2.5 mmol/L) in a buffer containing 250 mmol/L sucrose, 20 mmol/L Mops, 10 mmol/L Tris, 0.5 mmol/L Mg2+, 0.1 mmol/L Pi (K), and 50 µmol/L EGTA, pH 7.0. Experiments were performed in a Perkin-Elmer (Emeryville, CA, U.S.A.) fluorometer by measuring the decrease in light scattering at 520 nm. After an initial 3 minutes, allowing time for energization, swelling was induced by adding 50 µmol/L calcium chloride. When present, CsA or MeValCsA were added at the initiation of each swelling experiment at 1 µmol/L final concentration.
Statistical analysis
One factor analysis of variance followed by Scheffe's test was used to compare physiologic parameters among groups. The infarct size in treated and untreated groups were compared using Kruskal-Wallis test followed by Mann-Whitney U-test.
RESULTS
Blood gases (mm Hg ± SD) at 3 minutes after occlusion were Pa
Figure 1 shows the calculated infarct size. The mean cortical infarct size was 25% in the vehicle-treated animals and 11% in both the CsA- and the MeValCsA-treated groups. The decrease in cortical infarct size in both treated groups was highly significant. In the striatum, the infarct in vehicle-treated animals was 9%, whereas in the CsA- and MeValCsA-treated animals, it decreased to 2% and 3%, respectively. The decrease in striatal damage in the treated animals was also highly significant. The mean volume (mm3 ± SD) of the contralateral hemisphere was 953 ±37 (n = 21), and there were no statistically significant differences among groups (data not shown).
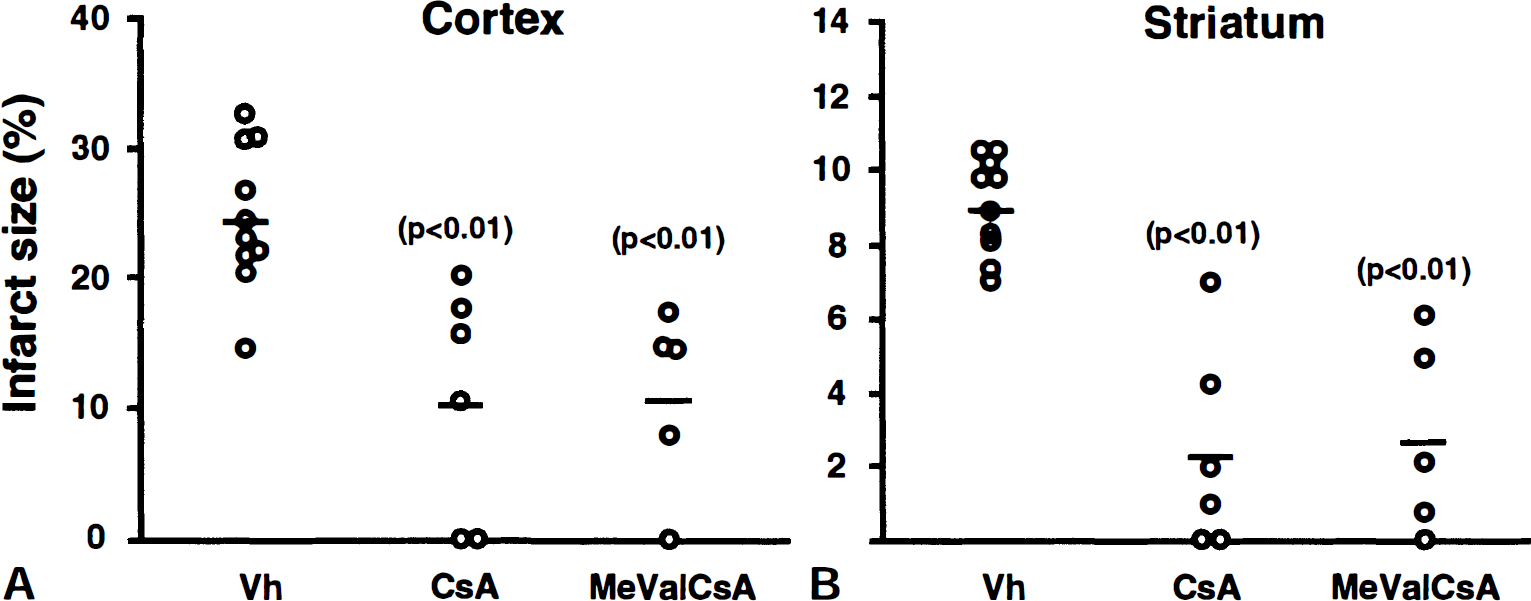
Figure 2 shows representative swelling traces of energized mitochondria prepared from striatum (A) and cortex (B). The addition of calcium chloride initiated swelling of mitochondria from both regions. In the presence of either 1 µmol/L MeValCsA (a) or 1 µmol/L CsA (b), there was a distinct but incomplete inhibition of calcium chloride induced mitochondrial swelling as compared to control (c).
Swelling traces of energized mitochondria prepared from 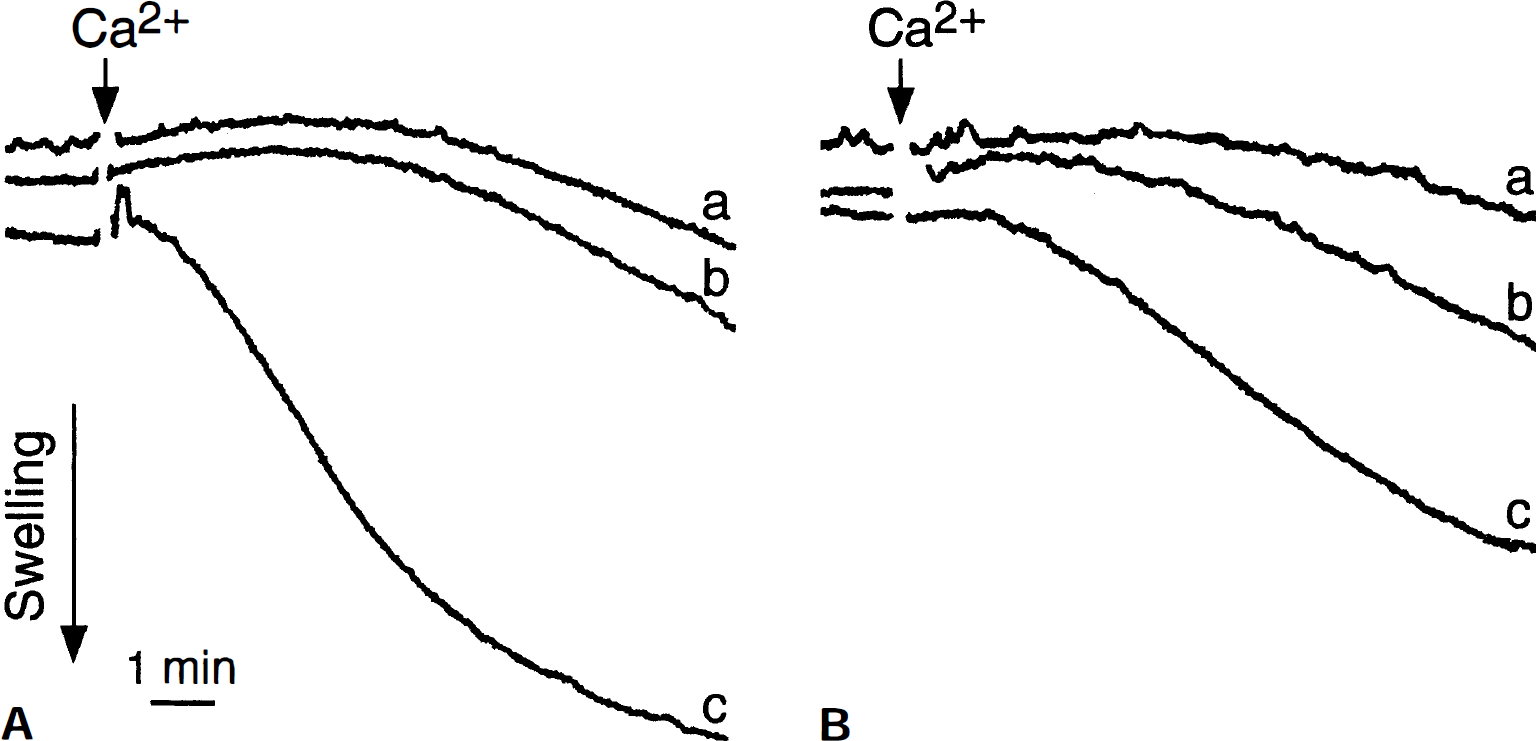
DISCUSSION
The results in the present study clearly show that post-ischemic treatment with either CsA or with its nonimmunosuppressive analogue MeValCsA ameliorates neuronal damage after transient focal ischemia in the rat. Because the immunosuppressive action of CsA does not seem to add further protection as compared to that by its nonimmunosuppressive analogue MeValCsA, and because both compounds are powerful and equipotent inhibitors of the MPT, we argue that their protective effect is caused by inhibition of the MPT.
Mitochondrial permeability transition
Mitochondria are the main source of ATP in cells and play a central role in cellular calcium homeostasis (Gunter and Gunter, 1994). If cytosolic calcium concentrations become too high, as following excessive neuronal firing or severe energy shortage, the subsequent mitochondrial calcium overload will initiate the opening of a nonspecific pore in the inner membrane, the MPT pore (Zoratti and Szabo, 1995). This calcium-dependent formation of the MPT pore represents an abrupt increase of permeability to solutes normally impermeable to the inner membrane (MW <1500 Da), causing osmotic swelling and rupture of the outer membrane, with concomitant loss of mitochondrial proteins (Liu et al., 1996; Susin et al., 1999). First characterized in isolated mitochondria (Hunter and Haworth, 1979a), the phenomenon has now been described in vitro (Kristal and Dubinsky, 1997) and recent studies show that MPT may be of crucial importance in the development of cell damage in vivo (Friberg et al., 1998). The MPT pore acts as a tightly regulated inner membrane channel, but its composition is yet not defined. Widely accepted components are the adenine nucleotide translocase of the inner membrane and matrix specific cyclophilin D (Halestrap et al., 1998), whereas porin of the outer mitochondrial membrane, creatine kinase and Bax may modulate its activity (Beutner et al., 1998; Marzo et al., 1998). Cyclosporin A inhibits opening of the MPT pore through binding to cyclophilin D, thereby preventing its translocation to the inner mitochondrial membrane (Connern and Halestrap, 1994) and MeValCsA prevents translocation of cyclophilin D equally well in brain mitochondria (Friberg et al., 1999). This makes the MPT pore less sensitive to induction by calcium ions.
Ischemia-reperfusion damage and the mitochondrial permeability transition
When blood supply to the brain is blocked because of occlusion of the proximal MCA as in stroke, an infarction develops in the central core (Garcia, 1992), encompassing caudoputamen and the overlaying cortex. The perifocal area surrounding the infarct, the penumbra, is potentially salvageable but will eventually lose viability unless successful intervention is initiated. Recently, caspase inhibitors have been shown to ameliorate stroke damage by 40% to 50% in a mouse model of transient focal ischemia (Namura et al., 1998), supporting the involvement of caspase-mediated cell death, in stroke-damaged neurons. Further support for caspase-mediated cell death in stroke comes from the recent demonstration that cytochrome c is released in the ischemic brain after transient MCAO in the rat (Fujimura et al., 1998). Mitochondrial cytochrome c release precedes caspase activation and cell death in many cell types and may be effectuated through several pathways (Cai et al., 1998), but, because cytochrome c resides in the intermembrane space, an increased permeability of the outer membrane is necessary. Osmotic swelling caused by opening of the MPT pore leads to rupture of the outer membrane and cytochrome c release, but release may also be seen without a preceding loss of mitochondrial potential and swelling in some in vitro systems (Kluck et al., 1997; Yang et al., 1997). This has been explained by the formation of Bax-regulated channels in the outer membrane (Eskes et al., 1998), which also, by unknown mechanisms, may be regulated by CsA (Jürgensmeier et al., 1998). In the rat brain, CsA sensitive mitochondrial swelling has been shown to occur in vivo (Friberg et al., 1998) and in this study we show that treatment with blockers of MPT significantly ameliorates neuronal damage in stroke.
Cyclosporin A and MeValCsA
An anti-ischemic effect of CsA in a model of transient MCAO has been demonstrated (Shiga et al., 1992), in which the animals were administered an oral dose for a week before the insult to suppress the immune system and to reduce edema formation. In a more recent study (Butcher et al., 1997), CsA was protective when administered after endothelin induced focal ischemia. The effect was similar to that of FK 506, an immunosuppressant but not a blocker of MPT (Butcher et al., 1997). Furthermore, CsA has been shown to protect neurons in a model of global ischemia (Uchino et al., 1998) and in a model of insulin-induced hypoglycemic coma (Friberg et al., 1998). The novel part of this study is that not only CsA protects from ischemic damage, but that the nonimmunosuppressive analogue MeValCsA protects equally well. MeValCsA has been shown to prevent loss of mitochondrial potential and induction of apoptosis in a cell free system (Zamzami et al., 1996) and more recently to protect PC 12 cells from metabolic inhibition (Seaton et al., 1998). In this study we show for the first time that MeValCsA is neuroprotective in vivo. Because MeValCsA exerts its main effect through inhibition of MPT in a concentration-dependent manner, similar to CsA (Liu et al., 1992) and not through inhibition of calcineurin-mediated events and immunosuppression, we conclude that the preservation of mitochondrial integrity and function through blockade of the MPT is of crucial importance in the reperfusion phase after ischemia.
Hence, there is increasing evidence that the mitochondrion is a point of convergence for death stimuli and may initiate cell death pathways through the activation of MPT (Halestrap et al., 1998; Kroemer et al., 1998; Lemasters et al., 1998). This can be envisaged to take place through the activation of processes typically occurring in apoptosis as well as during processes leading to necrosis. In the former case, transient activation of MPT leads to the release of mitochondrial proteins and a subsequent activation of caspases followed by DNA fragmentation and cell death. Alternatively, after a period of severe cellular energy shortage, large-scale irreversible MPT may be activated at reperfusion. The subsequent uncoupling of the electron transport chain, profound ATP decrease, and loss of free radical scavengers may lead to a surge of calcium-activated cellular degradation and free radical damage causing cell death. Both these cell death processes are blocked if MPT is inhibited by CsA and MeValCsA.