
Abstract
The authors examined the involvement of platelet-activating factor (PAF) in mediating leukocyte adherence to brain postcapillary pial venules and altering blood-brain barrier (BBB) permeability during basal conditions and during reoxygenation after asphyxia in newborn piglets. Intravital epifluorescence videomicroscopy, closed cranial windows, and labeling of leukocytes with rhodamine 6G allowed us to obtain serial measurements of adherent leukocytes within postcapillary venules. Blood-brain barrier breakdown was determined by optical measures of cortical extravascular fluorescence intensity after intravenous sodium fluorescein. Superfusion of PAF over the cortex induced a dose-dependent increase in leukocyte adherence to cerebral venules and leakage of fluorescein; with 1 μmol/L PAF, the magnitude of adherence and BBB breakdown was similar to that seen during reoxygenation after 9 minutes of asphyxia. Both adherence and loss of BBB integrity resulting from either exogenous PAF or asphyxia-reoxygenation could be significantly attenuated by intravenous administration of WEB 2086, a PAF receptor antagonist. Window superfusion of superoxide dismutase with PAF attenuated PAF-induced increases in adherence and associated fluorescein leakage. These findings indicate that PAF exhibits proinflammatory effects in piglet brain and that PAF contributes to leukocyte adherence and BBB breakdown after cerebral ischemia. These PAF effects are mediated by increases in superoxide radical generation.
In brain and other organs, the endothelium is a major target of postischemic reperfusion injury, manifested as alterations in vascular reactivity, increases in vascular permeability, and vasogenic edema (Granger et al., 1989; Dirnagl, 1993). Studies of cardiac and skeletal muscle ischemia provided the first compelling evidence that ischemia-induced microvascular injury is caused, in part, by leukocytes that become adherent to microvascular endothelium (Engler et al., 1987; Jerome et al., 1994). Similar studies supported the concept that leukocytes are also important contributors to reperfusion injury in brain (Dirnagl, 1993; del Zoppo, 1994; Härtl et al., 1996). The strongest evidence is provided by reports documenting reductions in leukocyte adherence and accumulation during postischemic reperfusion concomitant with significant attenuation of ischemic brain injury after administration of monoclonal antibodies to leukocyte or endothelial adhesion molecules (Matsuo et al., 1994b) and reductions in ischemic brain injury in adhesion molecule-deficient and other relevant gene-targeted mice (Soriano et al., 1996).
It is presumed that leukocytes must first adhere to the endothelium before they can cause injury to endothelial cells; additional injury may occur during firm adherence and extravasation to the extracellular space (Granger and Kubes, 1994). Leukocyte adherence is a complex process influenced by a variety of factors, e.g., shear stress, oxygen free radicals, nitric oxide, inflammatory mediators, and adhesion receptor molecule expression on both leukocytes and endothelial cells (Granger and Kubes, 1994). Previous studies of leukocyte-mediated ischemic brain injury assayed brain tissue for levels of myeloperoxidase, an enzyme found exclusively in neutrophils, to quantify leukocyte infiltration (Chopp et al., 1994; Matsuo et al., 1994a) or histologically verified the presence of capillary-occluding and extravasated leukocytes (del Zoppo et al., 1991; Caceres et al., 1995). However, these methods do not allow for repeated visualization and quantification of intravascular leukocyte behavior in vivo. We recently reported on our development of an intravital epifluorescence videomicroscopy technique that permits multiple measures of leukocyte dynamics in the postcapillary venules of living animals so that one can elucidate the time course of leukocyte adherence and its functional consequences in response to proinflammatory stimuli like cerebral ischemia (Gidday et al., 1997). In piglets, we observed a progressive increase in leukocytes adherent to cortical venules during the initial 2 hours of reoxygenation after global cerebral ischemia induced either by asphyxia or aortic cross-clamp; 2 hours after the insult, vascular permeability to sodium fluorescein was significantly elevated. Pretreating asphyxic piglets with a monoclonal antibody against the leukocyte adhesion glycoprotein complex CD11/CD18 significantly attenuated both the increase in leukocyte adherence and the increase in vascular permeability, indicative of a causal role for adherent leukocytes in endothelial dysfunction during the early reoxygenation period (Gidday et al., 1997). The present study begins to address the precise mechanisms responsible for leukocyte adherence and leukocyte-mediated cerebral microvascular injury by examining the participation of platelet-activating factor (PAF) in these events.
Studies in peripheral vascular beds reveal that among inflammatory mediators that influence intravascular leukocyte-endothelial interactions, PAF exhibits potent effects in this regard (Braquet et al., 1987). This lipid autacoid is released from many cell types, including endothelial cells, platelets, neutrophils, basophils, neurons, glia, and mast cells. It participates in diverse inflammatory conditions such as allergic reactions, shock, and ischemia-reperfusion (Braquet et al., 1987). In brain, PAF binding sites have been detailed (Domingo et al., 1988). Platelet-activating factor exhibits potent vasoconstrictive effects after topical application to cerebral arterioles (Armstead et al., 1988). With respect to ischemia, PAF production increases during early postischemic reperfusion (Domingo et al., 1994; Nishida and Markey, 1996), and PAF antagonists have shown efficacy in reducing brain injury in both adult and neonatal CNS ischemia models (Kochanek et al., 1987; Liu et al., 1996). Although these studies indicate a deleterious action of PAF in ischemic brain, studies of PAF's role in initiating leukocyte adherence and its effects on leukocyte-dependent and -independent vascular leakage after cerebral ischemia are still lacking. In addition, the down-stream mediators of PAF-induced leukocyte adherence and leakage have not yet been identified. We undertook the present study to address these questions.
METHODS
Animal preparation
Newborn piglets (less than 5 days old) of either sex were used in this study. Institutional approval was obtained for all protocols. Animals were premedicated with ketamine (20 mg/kg intramuscularly), mechanically ventilated with a mix of room air and oxygen after a tracheotomy was performed, and anesthetized with 1.0% to 1.5% isoflurane. Both femoral arteries were cannulated for continuous recording of arterial blood pressure and intermittent withdrawal of arterial blood samples for analyses of blood gases, glucose, and hematocrit. End-tidal CO2 and transcutaneous oxygen saturation were continuously monitored by a capnometer and forepaw sensor, respectively. A femoral vein was cannulated for infusion of 5% dextrose in 0.45% normal saline (6 mL·kg−1·h−1) and paralysis (0.25 mg·kg−1·h−1 pancuronium). Core body temperature was maintained at 38° to 39°C by a thermoregulated heating pad.
A closed cranial window was placed over the right parietal cortex just posterior to the coronal suture after an 18-mm craniotomy and removal of dura, as described previously (Gidday et al., 1997). The window was made completely of Plexiglas, with several circumferentially placed ports for infusion of artificial cerebral spinal fluid buffer with or without PAF and other drugs, and for the placement of various instrumentation, including a cannula for intracranial pressure measurement. The top surface of the cranial window was fashioned to create a 2-mm-deep chamber for holding distilled water, into which a water immersion lens was lowered for observation of the pial microcirculation by epifluorescence videomicroscopy.
Epifluorescence videomicroscopy
We used an epifluorescence microscope with a 100-W mercury arc light source and a 3.3× photoeyepiece (Gidday et al., 1997). Two filter cubes were used: For imaging of rhodaminelabeled leukocytes, the excitation filter was 535/35 nm, the dichroic filter was 565 nm, and the emission filter was 610/75 nm. A standard fluorescein isothiocyanate cube, with excitation, dichroic, and emission filters of 470/40 nm, 505 nm, and 510 nm, respectively, was used for imaging of vascular leakage of sodium fluorescein. The coupling of a 10× immersion lens (Olympus, Lake Success, NY, U.S.A.) featuring a 0.4 numerical aperture and a 3.1-mm working distance with a Newvicon tube camera (Hamamatsu C2400, Bridgewater, NJ, U.S.A.) with contrast and brightness controls provided real-time, high-resolution images of individual fluorescently labeled leukocytes moving through the pial microcirculation on the surface of the brain. Final image magnification was 290×.
Leukocyte imaging
Circulating leukocytes were labeled in situ by intravenous rhodamine 6G as described earlier (Gidday et al., 1997). A loading dose of rhodamine 6G (2 mL/kg of a 0.06 mg/mL solution; Sigma Chemical, St. Louis, MO, U.S.A.) was administered 20 minutes before the first baseline imaging session to identify an optimal venular network. Before each imaging session, the rhodamine infusion was reinitiated at a rate of 800 μL·min−1·kg−1 for 0.5 to 1.0 minute. Immediately thereafter, leukocyte dynamics in the pial microcirculation were videotaped for 30 to 45 seconds (Gidday et al., 1997). Between imaging sessions, the light path was blocked to avoid potential phototoxic effects.
Protocols
Baseline leukocyte dynamics were imaged at two 30-minute intervals. Within 5 minutes thereafter, animals were either rendered asphyxic or drug superfusion through the cranial window was initiated. To induce asphyxia, the ventilator was turned off for 9 minutes and the respiratory tubing was clamped. Blood gases were examined during the last minute of asphyxia, after which mechanical ventilation was resumed. In asphyxiated animals, we obtained videorecordings of cortical leukocyte dynamics at 60 and 120 minutes of reoxygenation. Videorecordings were obtained at equivalent times in nonasphyxiated control animals or nonasphyxiated control animals superfused with PAF. All window superfusions of drug were initiated at 1 mL/min for 1 minute followed by a continuous superfusion at a rate of 50 μL/min; displaced cerebral spinal fluid was vented through a port 180 degrees opposite to the inflow cannula. In all animals, vascular permeability was measured after the 120-minute leukocyte imaging time point as detailed below.
Animals were randomly divided into seven groups. Group 1 included nonasphyxic controls (n = 14). Group 2 animals were rendered asphyxic without pretreatment (n = 14). Group 3 animals were rendered asphyxic and treated with a single dose of the PAF receptor antagonist WEB 2086 (10 mg/kg intravenously) at the onset of reoxygenation (n = 6). Groups 4 and 5 included nonasphyxic animals in which the cranial window was superfused with PAF 10−6 mol/L (n = 9) and PAF 10−5 mol/L (n = 4), respectively, for 120 minutes. Group 6 included non-asphyxic animals pretreated 10 minutes before window superfusion of PAF (10−6 mol/L) with WEB 2086 (10 mg/kg intravenously; n = 6); Group 7 animals were nonasphyxic animals in which we simultaneously superfused PAF (10−6 mol/L) and the superoxide radical scavenger superoxide dismutase (SOD; 60 U/mL; n = 8) to determine whether superoxide or other downstream radicals were important mediators of PAF-induced leukocyte adherence and fluorescein leakage. After leukocyte imaging at 120 minutes, all animals were given intravenous sodium fluorescein for determination of blood-brain barrier (BBB) integrity.
Quantification of leukocyte-endothelial adherence
Individual leukocytes adherent to the venular endothelium were identified during off-line videotape playback on a high-resolution monitor as described previously (Gidday et al., 1997). This involved identifying a network of postcapillary venules and determining the three-dimensional endothelial surface area of this venular network using image analysis software (OPTIMAS, Version 3.01, BioScan, Inc., Edmonds, WA, U.S.A.). The number of leukocytes adherent to the venular endothelium within this defined network was determined at each time point by manually counting all leukocytes that remained stationary for longer than 30 seconds during repeated videotape playback, and converting these values to the corresponding values for the number of leukocytes per square millimeter of endothelial surface. The diameters of the venules in which leukocyte adherence was measured in this study ranged from 26 ± 5 to 92 ± 10 μm.
Vascular permeability
Vascular permeability to low-molecular-weight sodium fluorescein (MW = 376; 5.5 Å radius) was determined in the majority of animals in each group, as described previously (Gidday et al., 1997). In brief, at the end of the 120-minute period of observation, 1 mL/kg of a 1.0% solution of sodium fluorescein was administered intravenously for 1 minute. Videorecords were obtained just before the injection to obtain appropriate background values for intravascular and extravascular optical density, during the initial minute of administration at the peak in fluorescence to obtain a maximum optical density value and to identify at least five distinct and pure extravascular locations that were not confounded by small branches or capillaries, and at 20 minutes after the injection, at which time extravascular optical density values were measured to determine the magnitude of solute leak.
Statistical analyses
Differences in leukocyte adherence and vascular permeability within and between groups were determined by Kruskal-Wallis one-way analysis of variance with comparison of groups by Dunn's method. All values are reported as means ± SD, and statistical significance was accepted at P values less than 0.05.
RESULTS
Hemodynamic parameters
Mean arterial blood pressure, blood glucose, arterial pH, PaCO2, and PaO2 did not differ significantly among the animal groups during baseline conditions or at 60 and 120 minutes of postasphyxic reoxygenation. None of these parameters changed significantly with time in the control group. The short-term changes in systemic physiologic parameters induced by 9 minutes of asphyxia followed by 120 minutes of reoxygenation are shown in Table 1. During the last minute of asphyxia, animals were severely hypoxic, hypotensive, bradycardic, acidotic, and hypercapnic. Some asphyxic animals underwent cardiac arrest for 1 to 2 minutes during asphyxia, but spontaneously resumed normal sinus rhythms thereafter. The monitored physiologic variables typically returned to preasphyxic levels by 30 to 45 minutes of reoxygenation except for arterial pH, which tended to remain slightly acidotic.
Physiologic parameters in asphyxic piglets and asphyxic piglets treated with WEB
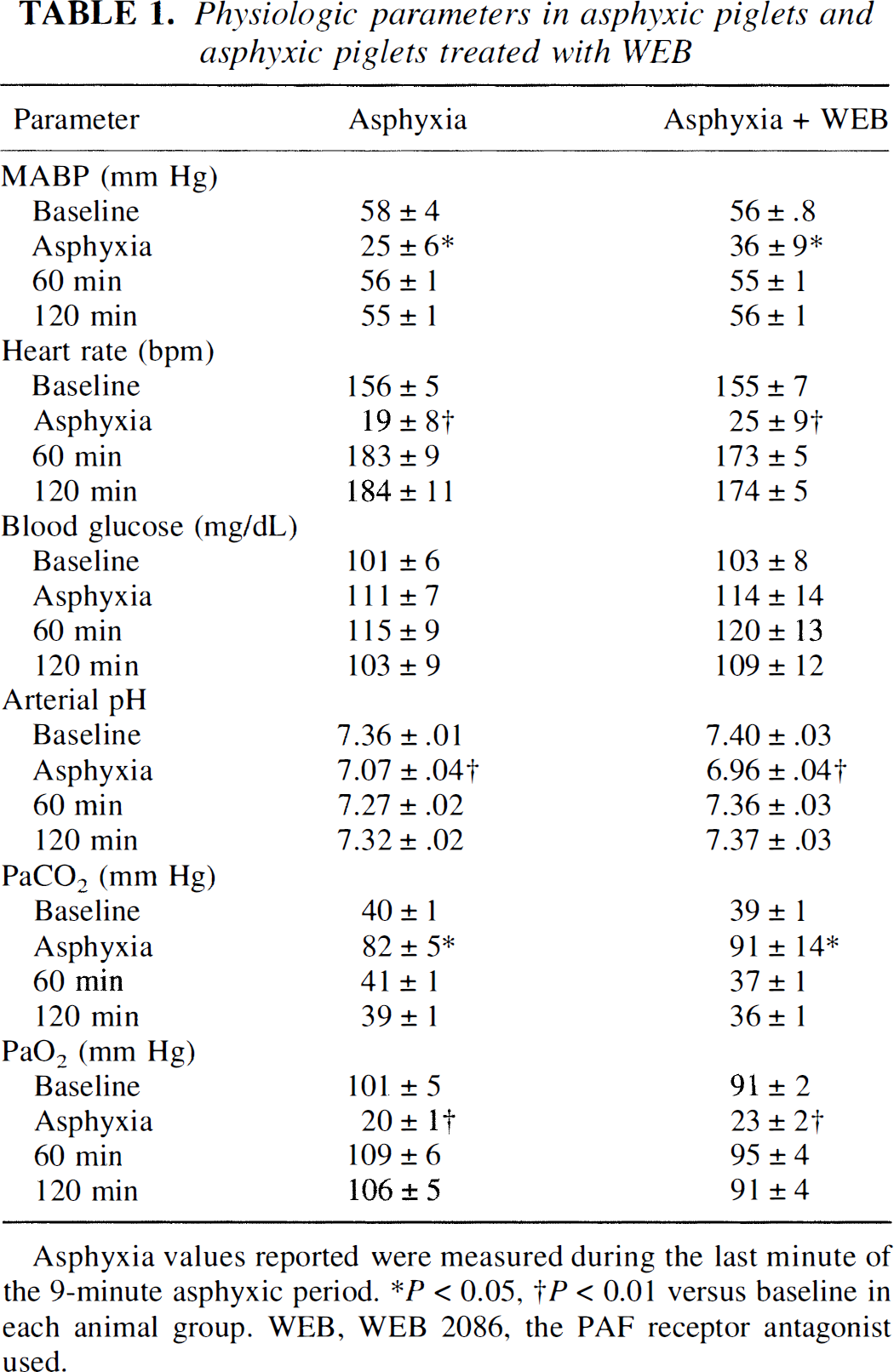
Asphyxia values reported were measured during the last minute of the 9-minute asphyxic period.
P< 0.05,
P < 0.01 versus baseline each animal group. WEB, WEB 2086, the PAF receptor antagonist used.
Asphyxia-reoxygenation and leukocyte-endothelial adherence
The number of adherent leukocytes did not differ among the seven animal groups during baseline conditions. In group 1 control animals, the number of leukocytes adherent to postcapillary venules during baseline was 61 ± 25 leukocytes/mm2; during the ensuing 120 minutes of observation, a gradual increase in the number of adherent leukocytes was noted, which was significant at 120 minutes. In contrast, asphyxia in group 2 animals resulted in a marked increase in the number of adherent leukocytes during 120 minutes of reoxygenation relative to controls (Fig. 1). In addition to increases in the number of adherent leukocytes, there were many more rolling leukocytes during reperfusion after asphyxia relative to nonasphyxic controls, but these data were not analyzed quantitatively. In group 3 animals, in which the role of PAF in asphyxia-induced leukocyte adherence was examined, administration of the PAF receptor antagonist WEB 2086 immediately on reoxygenation abolished the asphyxia-induced increase in leukocyte adherence to cerebral venules (Fig. 1). Representative videoprints of the extent of leukocyte adherence at 2 hours of reoxygenation after asphyxia relative to that observed at the same time in nonasphyxic controls and asphyxic animals treated with WEB 2086 are shown in Fig. 2.
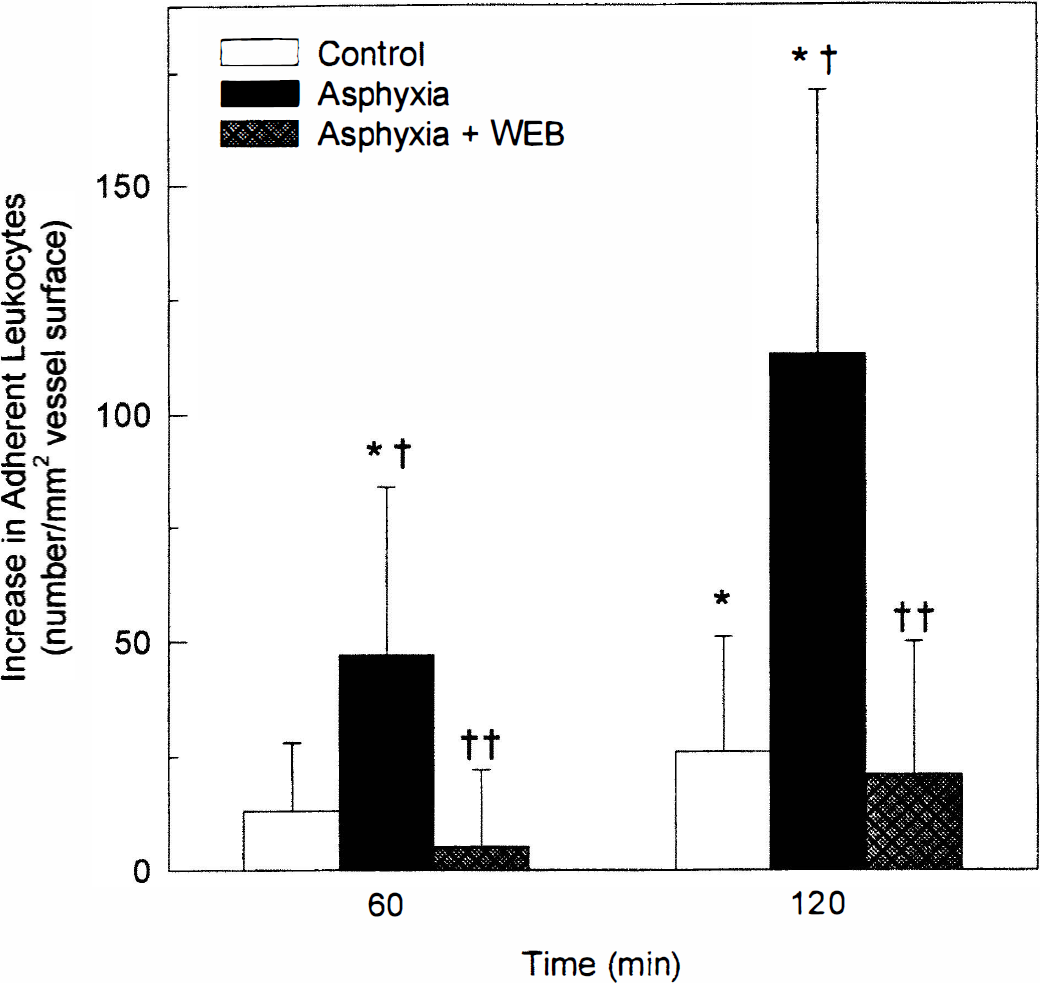
Increases in leukocyte adherence during reoxygenation after asphyxia are blocked by the PAF receptor antagonist WEB 2086. Shown are the increases in the number of leukocytes adherent to cerebral venules at 60 and 120 minutes of reoxygenation after 9 minutes of asphyxia in untreated animals, in animals treated with WEB 2086 (10 mg/kg intravenously) immediately on reoxygenation, and in nonasphyxic controls at equivalent times. (*P < 0.05 versus baseline; † P < 0.05 versus control group at the same time; †† P < 0.05 versus untreated asphyxia group at the same time.)
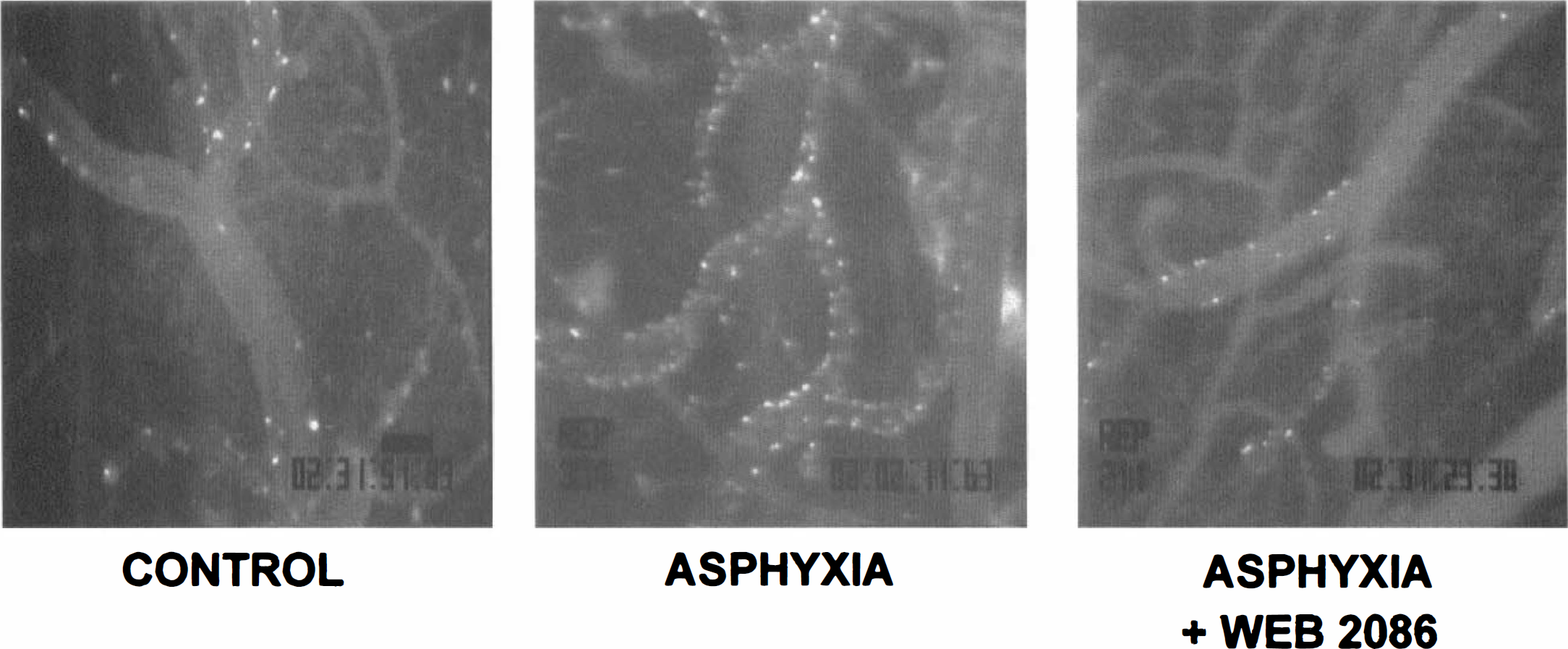
Representative videoprints from a nonasphyxic control animal (left panel), an asphyxic animal (center panel), and an asphyxic animal treated with the PAF antagonist WEB 2086 (right panel). Rhodamine 6G-labeled leukocytes can be seen within cerebral pial venules. Videoimages were obtained at 120 minutes of reoxygenation in the asphyxic groups and after an equivalent period in the control group, as shown by the videotimer in the lower right of each frame. Scale bar above videotimer in left panel = 100 μm.
PAF and leukocyte-endothelial adherence
The proinflammatory action of PAF was directly examined in nonasphyxic animals (groups 4 and 5) by window superfusion. Pilot studies revealed no increases in leukocyte adherence to cerebral venules as a result of superfusion with PAF at concentrations of 10−7 mol/L and less (data not shown). However, window superfusion with PAF at 10−6 mol/L and 10−5 mol/L induced dose-dependent increases in leukocyte-endothelial adherence, which increased in a progressive fashion during 120 minutes of continuous drug presentation by window superfusion (Fig. 3). Qualitatively, we noted that PAF also increased in a dose-dependent fashion the number of rolling leukocytes relative to untreated controls. The magnitude of adherent leukocytes resulting from 120 minutes of window superfusion of 10−6 mol/L PAF was similar to that measured during 120 minutes of reoxygenation after asphyxia. This exogenous PAF-induced leukocyte adherence was completely blocked in animals receiving the PAF receptor antagonist WEB 2086 (Fig. 3, group 6).
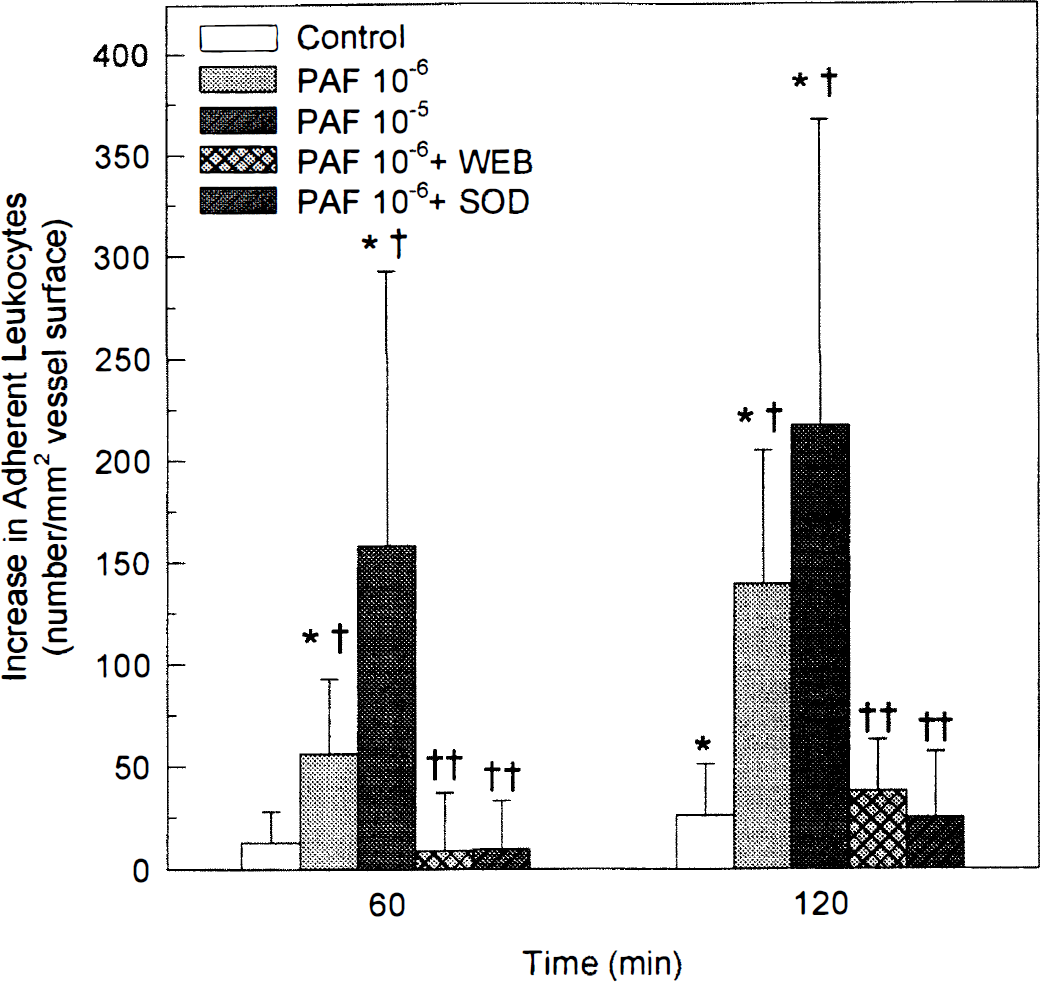
Increases in leukocyte adherence during 120 minutes of continuous window superfusion with PAF (10−6 mol/L) are blocked by the PAF receptor antagonist WEB 2086 (10 mg/kg intravenously 10 minutes before PAF) or cosuperfusion of superoxide dismutase (SOD; 60 U/mL, cosuperfused with PAF). (* P < 0.05 versus baseline; † P < 0.05 versus control group at the same time; †† P < 0.05 versus PAF 10−6 group at the same time.)
Asphyxia-reoxygenation, PAF, and vascular permeability
As reported previously (Gidday et al., 1997), leakage of sodium fluorescein at 120 minutes of reoxygenation after asphyxia was not discretely localized along venules of any particular size, but appeared widespread along all vessels in the venular network. In contrast, no fluorescein leakage was evident in the immediate periarteriolar space. The venular permeability to sodium fluorescein in the different animal groups is shown in Fig. 4. In parallel with the observed increases in leukocyte adherence, asphyxia (group 2) or window superfusion with PAF (groups 4 and 5) was associated with significant increases in vessel permeability to fluorescein. Treating asphyxic animals with the PAF receptor antagonist WEB 2086 on reoxygenation (group 3) resulted in complete blockade of asphyxia-induced increases in vascular permeability; in fact, fluorescein permeability in these WEB 2086-treated animals was significantly less than that measured in untreated, nonasphyxic controls (group 1). The PAF-induced increase in vascular fluorescein permeability was also significantly attenuated by WEB 2086 (group 6).
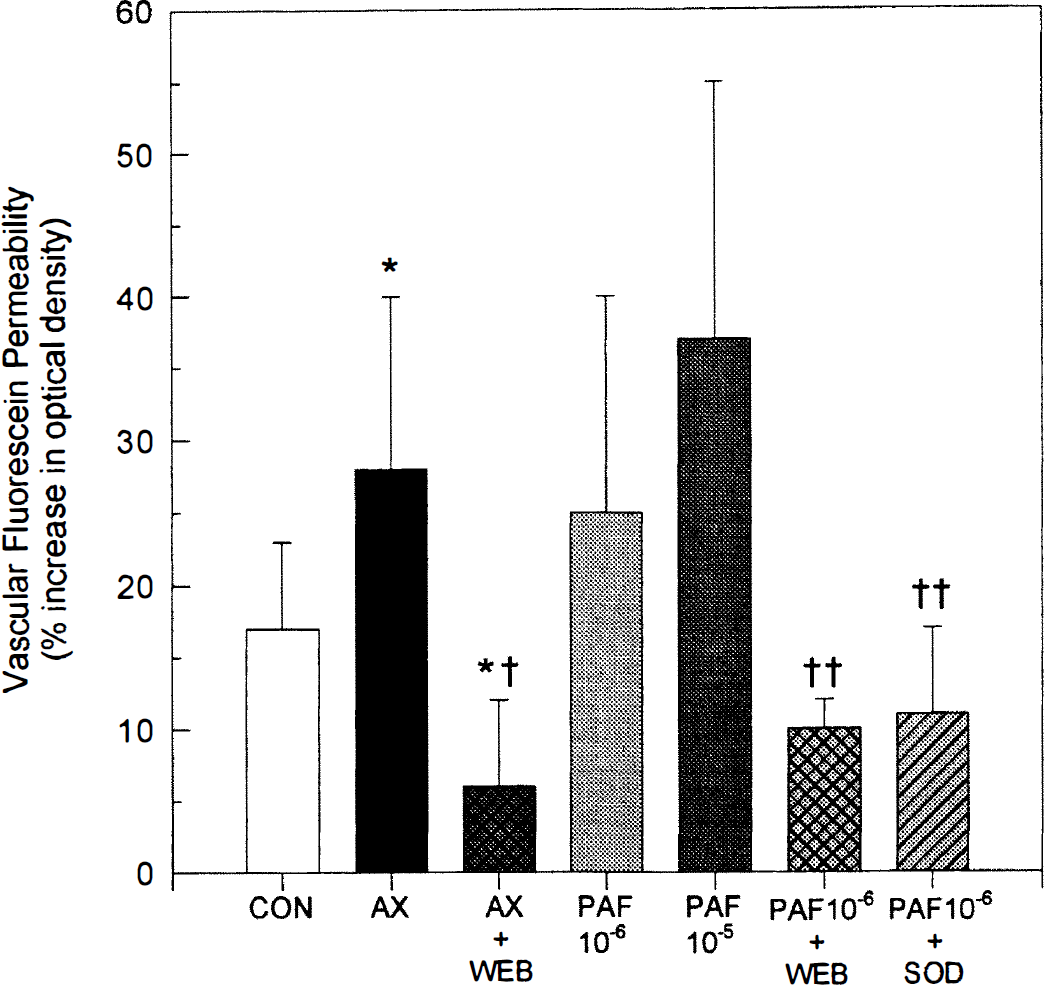
Increases in sodium fluorescein permeability after 120 minutes of postasphyxic reoxygenation or 120 minutes of window superfusion with PAF are blocked by the PAF receptor antagonist WEB 2086 or superoxide dismutase (SOD; 60 U/mL). Shown is the percent increase in optical density of the cortical perivenular extracellular space normalized to background measured 20 minutes after intravenous sodium fluorescein (1 mL/kg of a 1% solution for 1 minute). The seven experimental groups include: non-asphyxic controls (CON; n = 12); asphyxic animals (AX; n = 12); asphyxic animals treated with WEB 2086 at reoxygenation (AX + WEB; n = 5); animals superfused with 10−6 mol/L PAF (n = 8); animals superfused with 10−5 mol/L PAF (n = 4); animals superfused with 10−6 mol/L PAF 10 minutes after WEB 2086 (n = 6); and animals cosuperfused with 10−6 mol/L PAF and 60 U/mL SOD (n = 8). (*P < 0.05 versus control group; † P < 0.05 versus asphyxia group; †† P < 0.05 versus PAF 10−6 group.)
Superoxide radical involvement in PAF-induced leukocyte adherence and vascular permeability
In tests of our hypothesis that oxygen free radicals were important mediators of PAF-induced leukocyte adherence, we found that cosuperfusion of SOD with 10−6 mol/L PAF completely blocked the PAF-induced increase in leukocyte adherence (Fig. 3; group 7). Cosuperfusion with SOD also attenuated PAF-induced increases in vascular permeability (Fig. 4; group 7). In separate studies, we found that window superfusion with the same concentration of SOD significantly attenuated the elevations in leukocyte adherence and fluorescein leakage that occur during postasphyxic reoxygenation (unpublished observations).
DISCUSSION
In the present study, window superfusion of PAF induced marked, dose-dependent increases in both leukocyte adherence to cerebral pial venules and microvascular permeability to sodium fluorescein. The magnitude of these effects after exposure to 10−6 mol/L PAF was similar to that measured during early reoxygenation after asphyxia. The same dose of the PAF receptor antagonist WEB 2086 that completely blocked PAF-induced increases in leukocyte-endothelial adherence and BBB breakdown also completely blocked asphyxia-induced increases in leukocyte adherence and fluorescein leakage, indicating that PAF is an important mediator of these proinflammatory events in the cerebral vasculature of the ischemic brain. Because local administration of SOD blocked PAF- and asphyxia-induced increases in leukocyte adherence and fluorescein leakage, we conclude that postischemic leukocyte adherence and loss of endothelial integrity are ultimately mediated by superoxide radical secondary to the production of PAF.
Platelet-activating factor is synthesized in brain by acetylation of lyso-PAF or by a de novo pathway involving the transfer of phosphocholine to alkylacetyl-sn-glycerol (Braquet et al., 1987). Depending on appropriate stimuli, PAF can be produced by, and act on, endothelial cells, neutrophils, platelets, glia, neurons, and mast cells (Braquet et al., 1987). Platelet-activating factor binding sites are present throughout cortical, hippocampal, and striatal gray matter (Domingo et al., 1988). In response to cerebral ischemia, PAF levels increase promptly (Domingo et al., 1994; Nishida and Markey, 1996). In gerbil brain subjected to transient (10 minutes) forebrain ischemia, PAF levels measured by radioimmunoassay increased during ischemia and gradually returned to baseline levels by 30 minutes of reperfusion (Domingo et al., 1994), whereas in another report, PAF levels measured by gas chromatography/mass spectrometry increased significantly at 30 minutes of reperfusion and reached maximal levels at 1 hour of reperfusion (Nishida and Markey, 1996). Both studies reflect the rapidity with which PAF synthesis occurs in response to cerebral ischemia.
Rapid, ischemia-triggered PAF production and the acute increase in adherent leukocytes we witnessed in the initial hours of postasphyxic reoxygenation suggest a causal relation between PAF and postischemic leukocyte-endothelial adherence in the cerebral circulation. Indeed, PAF is a well-established chemoattractant for leukocytes in peripheral tissues such as mesentery (Kubes et al., 1990b) and hamster cheek pouch (Björk et al., 1983). In a preliminary report, intra-arterial infusion of PAF induced leukocyte adherence to the rat cerebral venular endothelium during resting conditions (Uhl et al., 1993). In our model, the presentation of exogenous extracellular PAF by window superfusion resulted in a dose-dependent increase in leukocyte adherence that was equivalent to that observed during reoxygenation after asphyxia and that was completely blocked by the PAF receptor antagonist WEB 2086. This antagonist showed similar efficacy against PAF-induced leukocyte adherence in the mesenteric circulation (Kubes et al., 1990a).
No in vivo studies have directly examined PAF's ability to damage the BBB, although intra-arterial infusion of PAF increased BBB permeability in an isolated perfused brain model (Kumar et al., 1988) and PAF-induced increases in microvascular permeability have long been recognized in nonneural organs (Björk et al., 1983; Kubes et al., 1990b, 1991; Filep and Földes-Filep, 1993; Kurose et al., 1996). Our results showing a concentration-dependent, WEB 2086-inhibitable increase in fluorescein leakage after superfusion of PAF through the cranial window are consistent with the aforementioned findings in peripheral vascular beds and provide the first evidence that PAF can alter BBB permeability during nonischemic conditions. The small leakage of sodium fluorescein under baseline conditions in the present study, which may have resulted from mast cell activation or histamine and cytokine release secondary to the surgical preparation of the cranial window, appears to be partly PAF-dependent, as fluorescein leak in asphyxic animals treated with WEB 2086 was slightly, but significantly, less than that measured in nonasphyxic, untreated control animals.
The concentration of PAF superfused through the cranial window that was required to elicit an inflammatory response in the cortical microcirculation deserves comment. Measures of postischemic PAF levels in gerbil brain tissue suggest extracellular levels in the 10−8 to 10−7 mol/L range (Nishida and Markey, 1996; Domingo et al., 1994), and in cheek pouch preparations, superfusion of PAF at concentrations as low as 5 × 10−9 mol/L caused leukocyte adherence to venular endothelium and leakage of fluorescently labeled dextran from postcapillary venules (Björk et al., 1983). In contrast, in our study and in a study of adult rats (Uhl et al., 1993), cranial window superfusion of PAF at concentrations less than 10−6 mol/L had no observable effect on leukocyte-endothelial interactions or BBB integrity. The reason that proinflammatory effects are not witnessed at lower PAF concentrations in the cerebral microcirculation relative to peripheral vascular beds may arise from differences in the relative concentration at which the superfused drug reaches its receptors in each preparation; the extent of dilution into the contiguous pool of cerebral spinal fluid for PAF or other solutes infused at low rates through cranial windows will likely be greater than that occurring with the typically high-volume rates of superfusion across spatially well-defined exteriorized tissue preparations. Moreover, the relevant receptors effecting PAF's proinflammatory actions likely reside in the intravascular compartment on endothelial cells and leukocytes, and diffusion of extravascularly administered PAF across cerebral vessels may be more limited than that occurring across peripheral vessels. These uncertainties notwithstanding, by our demonstration that intravascular administration of a PAF receptor antagonist blocked asphyxia-induced and PAF-induced leukocyte-endothelial adherence and fluorescein leakage, we fulfill an essential criterion required to support the hypothesis that PAF plays a key role in the acute inflammatory response to cerebral ischemia.
Endothelial injury, BBB breakdown, and edema are observed in numerous models of cerebral ischemia, including piglet asphyxia models (Caceres et al., 1995). Although leukocyte-dependent endothelial disruption and vascular leakage have been clearly documented in noncerebral tissues (Jerome et al., 1994), and considerable evidence has accumulated attesting to the neuroprotective effects of leukocyte-directed therapies (del Zoppo, 1994; Härtl et al., 1996; Soriano et al., 1996), the possibility that leukocytes participate as mediators of vasogenic edema after stroke has received little experimental attention. In the present study, postischemic treatment with the PAF receptor antagonist WEB 2086 reduced both leukocyte adherence and cerebrovascular fluorescein leakage. We cannot rule out the possibility that distinct leukocyte and endothelial PAF receptors independently regulate leukocyte adherence and microvascular permeability, respectively, and that the PAF receptor antagonist blocked both mechanisms simultaneously. However, we contend that our results are suggestive of a causal relationship between adherent leukocytes and BBB disruption during postischemic reoxygenation. The dependence of endothelial integrity on adherent leukocytes was independently verified in an earlier asphyxia study from our laboratory using an adherence-blocking monoclonal antibody to the leukocyte integrin CD18 (Gidday et al., 1997). Our contention that adherent leukocytes promote endothelial damage is also consistent with studies demonstrating that neutropenia (Matsuo et al., 1994a) or antibodies against leukocyte or endothelial adhesion molecules (Matsuo et al., 1994b) reduced edema formation after transient focal ischemia in rats, prevented the rise of intracranial pressure after thromboembolic stroke in rabbits (Bednar et al., 1991), and abolished the acute development of vasogenic edema in a rabbit meningitis model (Tuomansen et al., 1989). We only measured microvascular permeability at 120 minutes after asphyxia; it is likely that other leukocyte-independent mechanisms also contribute to postischemic vasogenic edema at later times.
That SOD blocked leukocyte adherence and BBB permeability after exposure of the brain to PAF suggests that the proinflammatory properties of exogenous PAF are mediated in large part by enhanced generation of superoxide radical. Results obtained from a number of studies suggest that the cellular source for enhanced superoxide generation after PAF exposure is probably the adherent leukocyte. Leukocytes generate large quantities of superoxide radical by NADPH oxidase as part of their respiratory burst (Shatwell and Segal, 1996). Incubation of isolated neutrophils with PAF results in increased superoxide radical production, degranulation, aggregation, and adherence to cultured endothelium (Ingraham et al., 1982; Kubes et al., 1991). Platelet-activating factor also “primes” neutrophils for oxygen radical generation; exposure of neutrophils to PAF led to enhanced superoxide generation after contact with interleukin-1β-stimulated endothelium (Hill et al., 1994). In the mesenteric vascular bed, PAF-induced leukocyte adherence to venular endothelium occurs concurrently with enhanced oxygen radical production (Kurose et al., 1996). Blocking leukocyte adherence by pretreatment with monoclonal antibodies against CD11b/CD18 on leukocytes or intercellular adhesion molecule-1 on endothelial cells attenuates PAF-induced oxidative stress (Kurose et al., 1996). In a rat model of focal cerebral ischemia, oxygen free radical production in brain was reduced by neutropenia (Matsuo et al., 1995). Similar conclusions have been advanced in studies of the intestinal microcirculation (Kurose et al., 1996) and in experiments with isolated neutrophils (Filep and Földes-Filep, 1993). These studies, in conjunction with our results with SOD, strongly suggest that the increase in barrier permeability we observed after both PAF- and asphyxia-induced elevations in leukocyte adherence was mediated by superoxide radical released from adherent leukocytes.
Several reports have documented that pretreatment of animals with PAF antagonists results in reduced CNS ischemic injury or improved neurologic recovery from ischemia in different species of mature and immature animals (Kochanek et al., 1987; Gilboe et al., 1991; Bielenberg et al., 1992; Liu et al., 1996). The mechanisms by which PAF antagonists exert such protective effects against ischemia have not been clarified, but the common assumption advanced in these papers was that protection was causally related to a reduced inflammatory response or a reduction in PAF's potentially deleterious effects on neurons. Platelet-activating factor antagonists can reduce excitotoxic damage in cultured neuron preparations as well (Prehn and Krieglstein, 1993). In contrast, evidence directly linking the well-recognized proinflammatory action of PAF to cerebral ischemic injury in a mechanistic way is lacking. Our results obtained from asphyxic animals pretreated with WEB 2086, showing a reduction in both leukocyte adherence and BBB disruption, indicate that the protective effects of PAF antagonists in these CNS ischemia models may also result from a blockade of the detrimental effects of neutrophil-endothelial adherence, including microvascular obstruction and loss of BBB integrity.
In conclusion, the present study provides evidence that exogenous extracellular PAF can promote leukocyte adherence to pial venular endothelium and adherence-dependent increases in BBB permeability to small molecular weight tracers. We also show that PAF is an important mediator of the acute elevations in leukocyte adherence and the loss of endothelial barrier integrity during early reoxygenation after asphyxia and that superoxide radical acts downstream from PAF to elicit these effects. Given the accumulating evidence implicating oxygen free radical-mediated vascular and parenchymal injury in stroke, further studies designed to localize and define the mechanisms of PAF-induced free radical formation in ischemic brain are warranted.
Footnotes
Acknowledgments
The authors thank Aarti R. Shah, Joel W. Beetsch, and Ron Perez for excellent technical assistance in these studies.