
Abstract
It was previously reported that prosaposin possesses neurotrophic activity that is ascribed to an 18-mer peptide comprising the hydrophilic sequence of the rat saposin C domain. To evaluate the effect of the 18-mer peptide on ischemic neuronal damage, the peptide was infused in the left lateral ventricle immediately after occlusion of the left middle cerebral artery (MCA) in stroke-prone spontaneously hypertensive (SP-SH) rats. The treatment ameliorated the ischemia-induced space navigation disability and cortical infarction and prevented secondary thalamic degeneration in a dose-dependent manner. In culture experiments, treatment with the 18-mer peptide attenuated free radical-induced neuronal injury at low concentrations (0.002 to 2 pg/mL), and the peptide at higher concentrations (0.2 to 20 ng/mL) protected neurons against hypoxic insult. Furthermore, a saposin C fragment comprising the 18-mer peptide bound to synaptosomal fractions of the cerebral cortex, and this binding decreased at the 1st day after MCA occlusion and recovered to the preischemic level at the 7th day after ischemia. These findings suggest that the 18-mer peptide ameliorates neuronal damage in vivo and in vitro through binding to the functional receptor, although the cDNA encoding prosaposin receptor has not been determined yet.
Keywords
Prosaposin, the precursor of saposins A, B, C, and D, which activate sphingolipid hydrolases in lysosomes, is a 517-amino acid glycoprotein (O'Brien and Kishimoto, 1991; Kishimoto et al., 1992). Besides its role as the precursor of saposins in lysosome, prosaposin itself exists as a secretory protein in human milk, cerebrospinal fluid, and seminal plasma (Hineno et al., 1991; Kondoh et al., 1991; Hiraiwa et al., 1993). Prosaposin is abundant in brain and muscle, whereas processed saposins are predominantly found in spleen, liver, and kidney (O'Brien et al., 1988; Sano et al., 1989). In the brain, prosaposin is localized exclusively in certain neurons and nerve fibers. Furthermore, prosaposin-containing neural elements are abundant in the CA1 field of the hippocampus (Kondoh et al., 1993), which is known to be vulnerable to ischemic insult. With the use of a gerbil ischemia model, we have recently demonstrated that prosaposin has a potent ability to rescue hippocampal neurons from lethal ischemic damage in vivo (Sano et al., 1994), raising the possibility of prosaposin as a candidate for neurotrophic factor. The pivotal role of prosaposin in neurogenesis was also indicated by Harzer et al. (1989) and Schnabel et al. (1992), who reported that prosaposin deficiency causes fetal neurological deficits. O'Brien et al. (1994, 1995) demonstrated that prosaposin stimulates neuritogenesis and increases choline acetyltransferase activity in neuroblastoma cells and that the neurotrophic activity resides, at least in part, in an amino-terminal 12-residue peptide encompassing the hydrophilic region of the human saposin C domain. We further showed that an 18-mer peptide comprising the hydrophilic sequence of the rat saposin C domain protects hippocampal CA1 neurons against lethal forebrain ischemia in gerbils (Kotani et al., 1996). However, the mechanisms by which the 18-mer peptide rescues ischemic CA1 neurons have not been elucidated yet.
The so-called delayed neuronal death in the hippocampal CA1 region of gerbils after transient forebrain ischemia has been a subject of investigation over decades, but other types of ischemic models have also been developed mainly by using rats of several species (Tamura et al., 1981; Coyle, 1982; Coyle and Jokelainen, 1983; Brint et al., 1988; Okuyama et al., 1991). Among them, stroke-prone spontaneously hypertensive (SP-SH) rats with permanent occlusion of the unilateral middle cerebral artery (MCA) above the rhinal fissure and distal to the striate branches exhibit invariably a certain cortical infarct, place navigation disability, and secondary thalamic degeneration focused on the ventroposterior (VP) nucleus (Kumon et al., 1996; Zhang et al., 1998). Therefore, they appear to be an animal model suitable for the study of the effects of peptide growth factors and drugs on the primary ischemic lesion and on the secondary degeneration in brain areas connected synaptically with the primary lesion (Okuyama et al., 1991). There are several advantages of the MCA-occluded SP-SH rat model over the gerbil ischemia model. First, SP-SH rats subjected to permanent occlusion of the MCA mimic human patients suffering from cerebral infarction as compared with gerbils with transient forebrain ischemia. Second, the observation of ischemia-induced behavioral abnormalities is easier in rats than in gerbils. Therefore, it is of interest to see the action of the 18-mer peptide on the brain of MCA-occluded SP-SH rats with a severer ischemic damage than gerbils subjected to transient forebrain ischemia.
The present study was designed first to see whether or not continuous infusion of the 18-mer peptide into the cerebral ventricles of MCA-occluded SP-SH rats, like ciliary neurotrophic factor (CNTF) (Kumon et al., 1996), ameliorated place navigation disability, cortical infarction, and secondary thalamic degeneration. Second, we investigated the effects of the 18-mer peptide on cultured cortical neurons exposed to a free radical-producing agent, FeSO4, or to hypoxic environment. Finally, using the BIAcore system (Karlsson et al., 1991), we studied whether or not subcellular fractions of the cerebral cortex and thalamus in the MCA-occluded rats had binding elements for the 18-mer peptide.
MATERIALS AND METHODS
Synthetic 18-mer peptide
An 18-mer peptide (LSELIINNATEELLIKGL) comprising the hydrophilic sequence of rat saposin C was chemically synthesized by Sawady Technology, Tokyo, Japan (Kotani et al., 1996).
In vivo study
SP-SH rats at the age of 12 to 13 weeks, weighing 250 to 300 g, were housed in an air-conditioned room at constant temperature (22 ± 1°C) with a 12:12 light/dark cycle, and food and water were given ad libitum throughout the experiments. The animals were handled once a week for cage cleaning. The experiments were conducted in accordance with the Guide for Animal Experimentation at Ehime University School of Medicine. Tail systolic blood pressure in each conscious animal was measured before MCA occlusion and at 2 hours, 1 to 7 days after MCA occlusion, with the use of a rat tail manometer tachometer system (KN-210; Natsume, Tokyo, Japan). The mean blood pressure of SP-SH rats before MCA occlusion was 201 ± 22 mm Hg, and it was not affected by MCA occlusion or 18-mer peptide infusion.
SP-SH rats were anesthetized with 1.5% halothane in a 4:3 mixture of nitrous oxide and oxygen, while body temperature was kept at 37 ± 0.2°C. The left MCA was exposed by cutting the temporal muscle and then by making a hole in the temporal bone with a dental drill. Just after MCA occlusion, an osmotic minipump (Alza Corp., Palo Alto, CA, U.S.A.) filled with either the 18-mer peptide or vehicle [0.01 mol/L phosphate-buffered saline (PBS) containing 0.1% bovine serum albumin (BSA)] was implanted subcutaneously into the back of each animal, and a needle from the minipump was placed in the left lateral ventricle. The 18-mer peptide in a dose of 4, 20, or 60 ng/day was continuously infused through the minipump for 28 days (n = 8/group). Sham-operated animals and control rats with MCA occlusion received vehicle infusion (n = 8/group).
Brain and body temperatures in rats infused with vehicle or 60 ng/day of the 18-mer peptide were monitored at the 1st to 7th hours (n = 4/group). Body temperature was measured with a rectal probe connected to a digital thermometer (Yokogawa, Tokyo, Japan) just after diethylether anesthesia, and brain temperature was monitored by combining a temperature-sensitive probe (Mini Mitter Co., Sunriver, OR, U.S.A.) inserted into the right frontal lobe (1.0 mm anterior to bregma, 1.5 mm lateral to the midline, and 2.5 mm in depth) with the telemetry system receiving signals from the probe (Data Sciences, Minneapolis, MN, U.S.A.).
At the 2nd and 4th weeks after MCA occlusion, the rats were subjected to repeated Morris water maze tests three times per day for 4 consecutive days. Each rat was allowed to swim until reaching a submerged platform and to stay there for at least 10 seconds. The escape latency, i.e., time until each rat reached the invisible submerged platform, was measured (Morris, 1981). In the cases where the rats could not escape onto the platform within 90 seconds, they were placed by hand onto the platform for 15 seconds and their escape latency was recorded as 90 seconds. The mean latency of finding the invisible platform was measured for individual animals on each day.
After the water maze tests, the animals were anesthetized by an intraperitoneal injection of chloral hydrate (300 mg/kg), and the osmotic minipump was disconnected from the needle placed in the left lateral ventricle. Bromophenol blue was injected through the needle to test the peptide or vehicle infusion into the cerebral ventricles. Then the rats were perfused transcardially with 4% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.4) and the brain was removed. The animals not showing dye diffusion into the cerebral ventricles were excluded from the experimental groups. The brain with dye diffusion was embedded in paraffin. Three serial coronal sections 5 μm thick were cut at 1.2, 2.3, and 3.6 mm posterior to bregma and stained with 0.1% cresyl violet. The size of cortical infarction was assessed by measuring the area of the cerebral cortex in individual sections with a planimeter, and the ratio of the cerebrocortical area on the MCA-occluded side to that on the contralateral side was calculated in the three sections. This was termed left-to-right cerebrocortical ratio.
To assess secondary thalamic atrophy, the ratio of thalamic area on the MCA-occluded side to that on the contralateral side was calculated in the section 3.6 mm posterior to bregma. The thalamus at this level is known to constitute reciprocal fiber connections with the temporoparietal cortex, and thus it is likely to exhibit secondary degeneration after MCA occlusion that causes an infarct in the temporoparietal cortex. The degeneration of neurons in the VP thalamic nucleus was evaluated microscopically by measuring the size of each cell with a computerized image-processing system (Nexus 6400 System; Kashiwagi, Tokyo, Japan). A square of 0.33 ± 0.30 mm (510 ± 478 pixels) was designated as the region of interest on the video image, and the size of each cell within the square was measured and expressed by its pixel size. Cells larger than 200 pixels were counted as viable neurons (Kumon et al., 1996; Watanabe et al., 1998).
All experiments were done blindly with respect to the experimental group.
Cortical neuron cultures
The cerebral cortices of 17-day rat embryos were aseptically dissected out, and cortical neurons were dissociated from the tissues as described by Kira et al. (1995). The dissociated cells were seeded on 24-well plastic plates (Corning, Corning, NY, U.S.A.) coated with poly-L-lysine (Sigma, St. Louis, MO, U.S.A.) at a density of 1 × 105 cells/cm2. The cells were cultured at 37°C in Dulbecco's modified Eagle's medium (DMEM; 0.1% glucose; Nipro, Osaka, Japan) supplemented with 10% fetal calf serum (Life Technologies, Rockville, MD, U.S.A.) under a humidified atmosphere containing 5% CO2 for 24 hours. Then the medium was replaced with a serum-free DMEM containing graded concentrations (0.2 fg/mL to 200 ng/mL) of the 18-mer peptide, 20 mmol/L N-(2-hydroxyethyl)piperazine-N′,2-ethanesulfonic acid (HEPES), 4.5 mg/mL glucose, 1 mg/mL BSA, 5 mg/mL insulin, 5 nmol/L sodium selenite, and 5 mg/mL transferrin (Boehringer-Mannheim, Mannheim, Germany). Forty-eight hours later, the neurons were subjected to free radical-induced injury (Lim et al., 1997). Briefly, 30 μmol/L FeSO4 was added to each well, and 1.5 hours later the neurons were solubilized with Laemmli's sample solution containing sodium dodecyl sulfate (Laemmli, 1970) and electrophoresed on a 6% polyacrylamide gel. The electrophoretic bands were transferred to nitrocellulose sheets and immunoblotted with a monoclonal antibody (Sternberger Monoclonals, Baltimore, MD, U.S.A.) against microtubule-associated protein 2 (MAP2), which is known to be a specific marker for neuronal cells. Anti-mouse IgG coupled with alkaline phosphatase (Promega, Madison, WI, U.S.A.) was used for the second immunoreaction. The density of each MAP2-positive band was measured by NIH Image (National Institutes of Health, Bethesda, MD, U.S.A.).
To confirm the antioxidant action of the 18-mer peptide, we measured membrane lipid peroxidation of neurons treated with the 18-mer peptide and FeSO4. Cortical neurons were seeded on 35-mm-diameter dishes (Sumitomo, Tokyo, Japan) at a density of 1 × 105 cells/cm2, pretreated with 2 fg/mL to 200 ng/mL of the 18-mer peptide, and exposed to 30 μmol/L FeSO4 as mentioned above. One and one-half hours later, the neurons were lysed in a solution containing 15% trichloroacetic acid, 0.375% thiobarbituric acid (TBA; Wako, Osaka, Japan), 0.25 mol/L HCl, and 0.015% butylated hydroxytoluene (Wako). The lysate was heated for 15 minutes in boiling water. After centrifugation to remove flocculent precipitates, light absorbance at 535 nm was measured. The amount of TBA-reactive substances (TBARS) was calculated by using an extinction coefficient, 1.56 × 105 (mol/L)−1 cm−1 (Buege and Aust, 1987).
In the study of hypoxic injury, cortical neurons were cultured with DMEM supplemented with 10% fetal calf serum for 24 hours as described above, and the culture medium was replaced with an 18-mer peptide-containing serum-free medium that was bubbled with nitrogen gas to remove dissolved oxygen gas beforehand. The neuronal cultures were incubated under an atmosphere of 95% N2 and 5% CO2 for 6 hours, followed by incubation for 1 hour under the conventional atmosphere containing 5% CO2. Then 50 μL of the medium in each well was subjected to measurement of the activity of lactate dehydrogenase (LDH) leaked from damaged neurons by using a kit (Wako). Cytotoxicity was expressed by the ratio of LDH activity in each well to that in the well of neurons completely lysed with Tween 20 (Decker and Lohmann-Matthes, 1988).
Binding site of 18-mer peptide
The 18-mer peptide binding sites in the subcellular fractions of brain tissue were characterized with the BIAcore 2000TM system (Pharmacia Biotech AB, Uppsala, Sweden), which monitors real-time molecular interaction as detected by surface plasmon resonance phenomenon.
A 44-mer peptide sequence comprising the 18-mer peptide was selected from the rat saposin C domain as a ligand fixed to a sensor chip of the BIAcore system, as the 18-mer peptide was too small in molecular size to bind to the sensor chip. The 44-mer peptide (60 μg/mL) in 10 mmol/L acetate buffer (pH 4.5) was immobilized to the sensor chip CM5 by the amine-coupling method (Ward et al., 1992). Another sensor chip devoid of the immobilized ligand was used as a blank in control experiments.
The SP-SH rats with sham operation, those at the 1st and 7th days after MCA occlusion, were anesthetized with diethylether and transcardially perfused with saline (n = 4/group). The brain was removed and divided into the cerebral cortex and thalamus on ice. Both the membrane fraction and the synaptosomal fraction were prepared according to the method of Gray and Whittaker (1962) with slight modifications. In brief, the brain was homogenized with a Teflon homogenizer in 0.32 mol/L sucrose buffer. The homogenate was centrifuged at 1,000g for 10 minutes to obtain the crude nuclear fraction in the pellet. To remove the crude mitochondrial fraction, the supernatant was centrifuged at 12,500g for 20 minutes, and the resulting supernatant was ultracentrifuged at 100,000g for 60 minutes. The pellet was suspended in a modified BIAcore running buffer, which consisted of 10 mmol/L HEPES buffer (pH 7.4) containing 0.15 mol/L NaCl, 3.4 mmol/L ethylenediamine-tetraacetate, and 0.1% Tween-20, and ultracentrifuged at 100,000 g for 30 minutes. This final pellet suspended in the modified BIAcore running buffer was used as the membrane fraction. On the other hand, the crude nuclear fraction suspended in 0.32 mol/L sucrose was overlaid on two layers of 0.8 and 1.2 mol/L sucrose and ultracentrifuged at 51,500g for 90 minutes in a swing rotor. The fraction collected from the interface between 0.8 and 1.2 mol/L sucrose layers was suspended in the modified BIAcore running buffer and ultracentrifuged at 100,000g for 30 minutes. The resultant pellet suspended in the modified BIAcore running buffer was used as the synaptosomal fraction.
Protein concentration of each fraction was adjusted to 0.15 mg/mL by employing the BCA Protein Assay Kit (Pierce, Rockford, IL, U.S.A.) to determine the protein concentration with BSA as a standard. Each sample was injected both to the peptide-immobilized chip and to the blank chip at a flow rate of 5 mL/min for 900 seconds. The next sample was injected after washing both chips with 200 mmol/L glycine-HCl buffer (pH 2.7). The BIAcore sensorgram shows changes in response expressed as resonance units along the time after sample injection, and the maximal response was observed at 850 seconds. Therefore, we measured responses at 850 seconds, and binding quantity to the peptide was measured by subtracting response bound to the blank chip from that bound to the peptide-immobilized chip.
Statistics
Experimental data were evaluated by the two-tailed Mann-Whitney U test or by analysis of variance, followed by post hoc test (Scheffé's F). All data are presented as means ± SD.
RESULTS
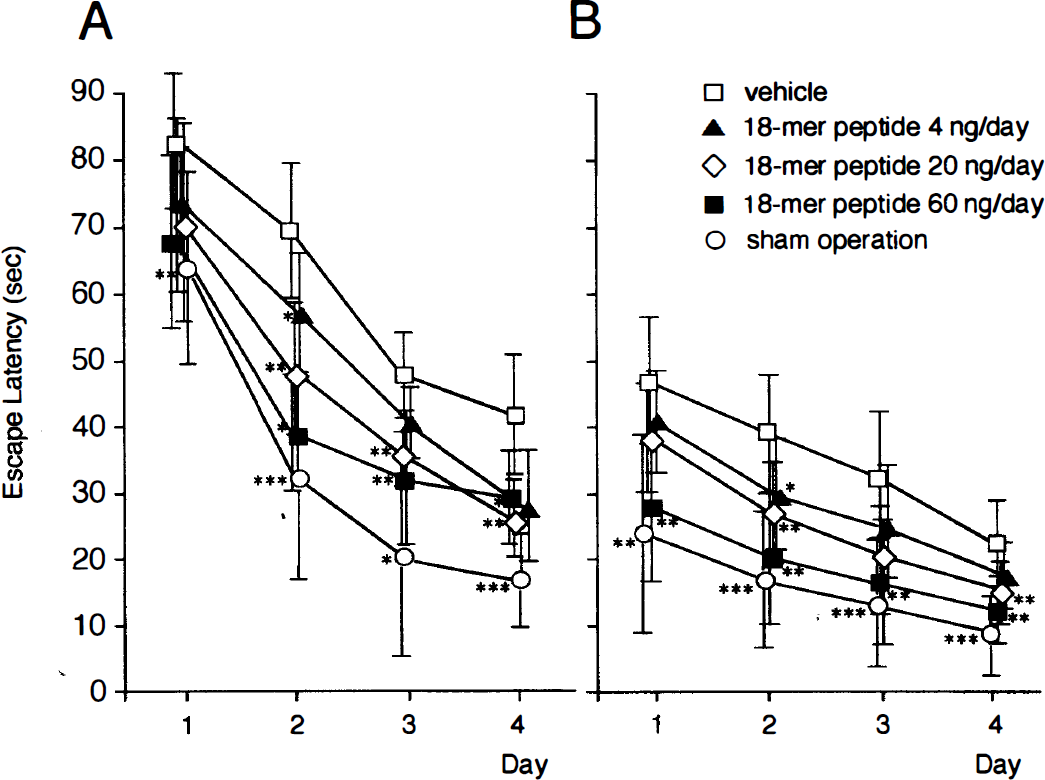
Morris water maze tests at the 2nd week after MCA occlusion revealed that the mean escape latency of vehicle-infused ischemic rats was significantly longer than that of sham-operated rats throughout the trial days. However, the mean escape latency of ischemic rats treated with 20 or 60 ng/day of the 18-mer peptide was shorter than that of the vehicle-infused ischemic rats on the 2nd to 4th trial days. The 18-mer peptide at a dose of 4 ng/day was less effective in shortening the escape latency (Fig. 1A). At the 4th week after MCA occlusion, there was also a significant difference in the escape latency between the vehicle-treated ischemic rats and the sham-operated rats. At this period, the ischemic rats treated with the 18-mer peptide at a dose of 60 ng/day exhibited a shorter escape latency than the vehicle-infused ischemic rats throughout the 4 trial days (Fig. 1B). The 18-mer peptide at a dose of 20 ng/day also reduced the escape latency significantly on the 2nd and 4th trial days. The MCA-occluded rats infused with 4 ng/day of the 18-mer peptide showed a significant improvement in the escape latency only on the 2nd trial day (Fig. 1B). There were no significant differences in swimming speed among all the experimental groups (data not shown). The infusion of 60 ng/day of the 18-mer peptide did not significantly affect brain or body temperature during 7 hours after MCA occlusion (Table 1).
Changes in body and brain temperatures in rats with the infusion of vehicle or the 18-mer peptide (60 ng/d)
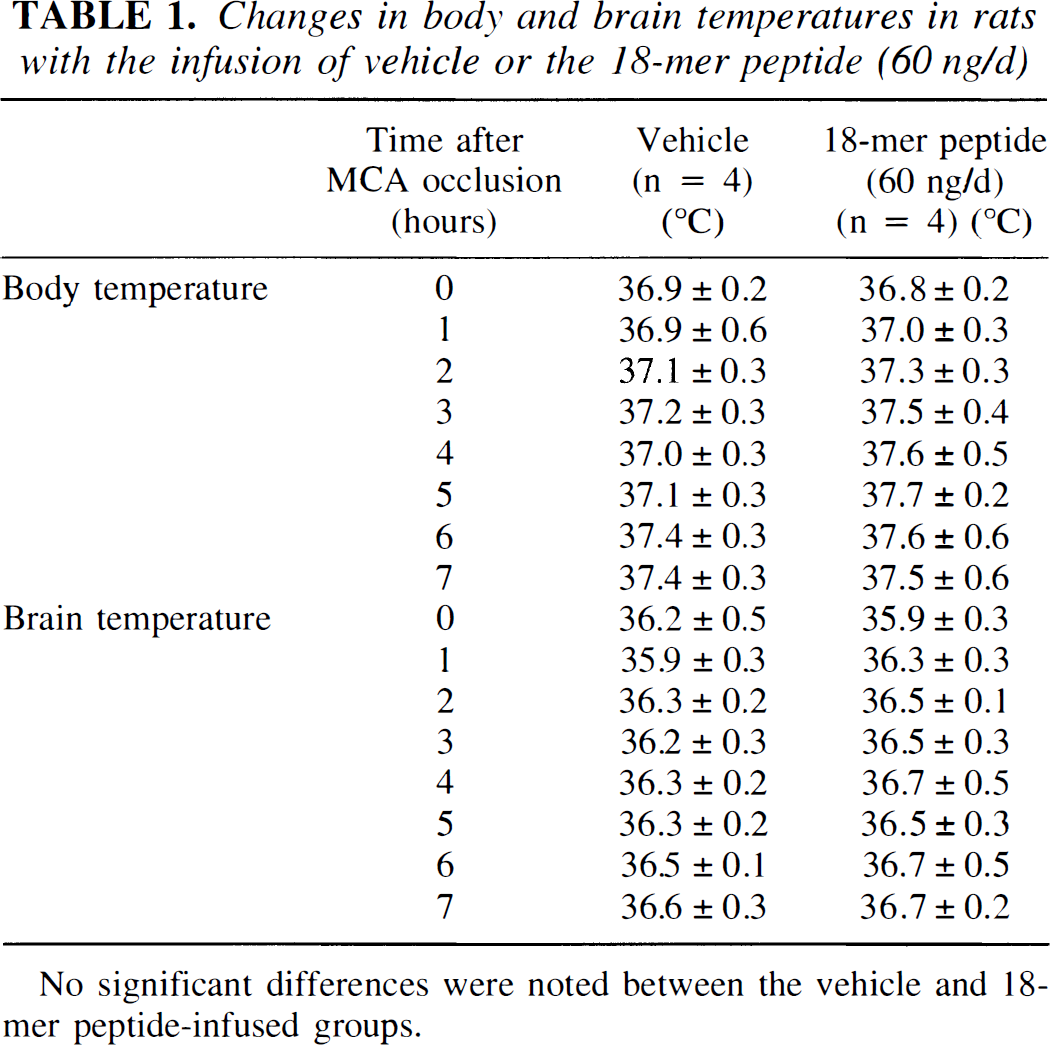
No significant differences were noted between the vehicle and 18-mer peptide-infused groups.
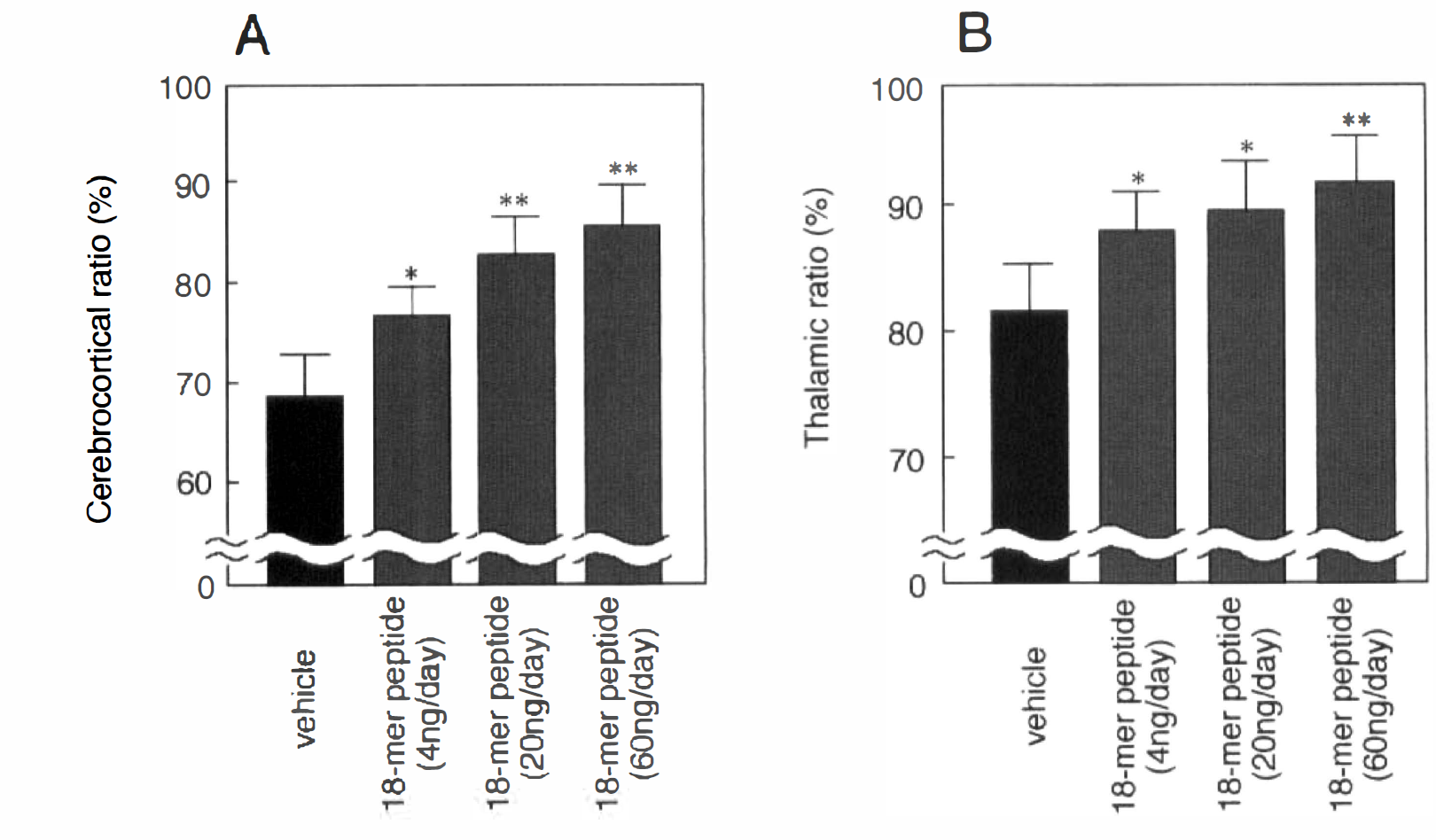
The left-to-right ratio of cerebrocortical area in vehicle-infused ischemic rats was 68.8 ± 4.4%, whereas the ratios in MCA-occluded rats with 18-mer peptide treatment at doses of 4, 20, and 60 ng/day were 76.8 ± 2.9, 82.4 ± 3.5, and 84.8 ± 3.9%, respectively. The ratio of the ischemic rats treated with 4, 20, or 60 ng/day of the 18-mer peptide was significantly larger than that of the vehicle-infused rats with MCA occlusion (Fig. 2A). The left-to-right ratio of thalamic area in the vehicle-infused ischemic rats was 78.2 ± 3.0%, whereas the ratios in MCA-occluded rats with the 18-mer peptide treatment at doses of 4, 20, and 60 ng/day were 87.7 ± 2.2, 91.8 ± 3.2, and 92.5 ± 2.5%, respectively. The 18-mer peptide at doses of 4 to 60 ng/day significantly prevented the secondary thalamic degeneration in MCA-occluded rats (Fig. 2B).
In Nissl-stained sections, neurons in the VP thalamic nucleus of vehicle-infused ischemic rats were much fewer than those of sham-operated rats, and the vehicle-treated thalamic neurons showed shrinkage of cell body at the 1st month after MCA occlusion (Fig. 3A and B). Treatment with the 18-mer peptide increased the number of VP thalamic neurons with normal morphological appearance (Fig. 3C). Neuron counts using a computerized image-processing system revealed that significant numbers of VP thalamic neurons were rescued from secondary degeneration by treatment with 4 to 60 ng/day of the 18-mer peptide (Fig. 4).
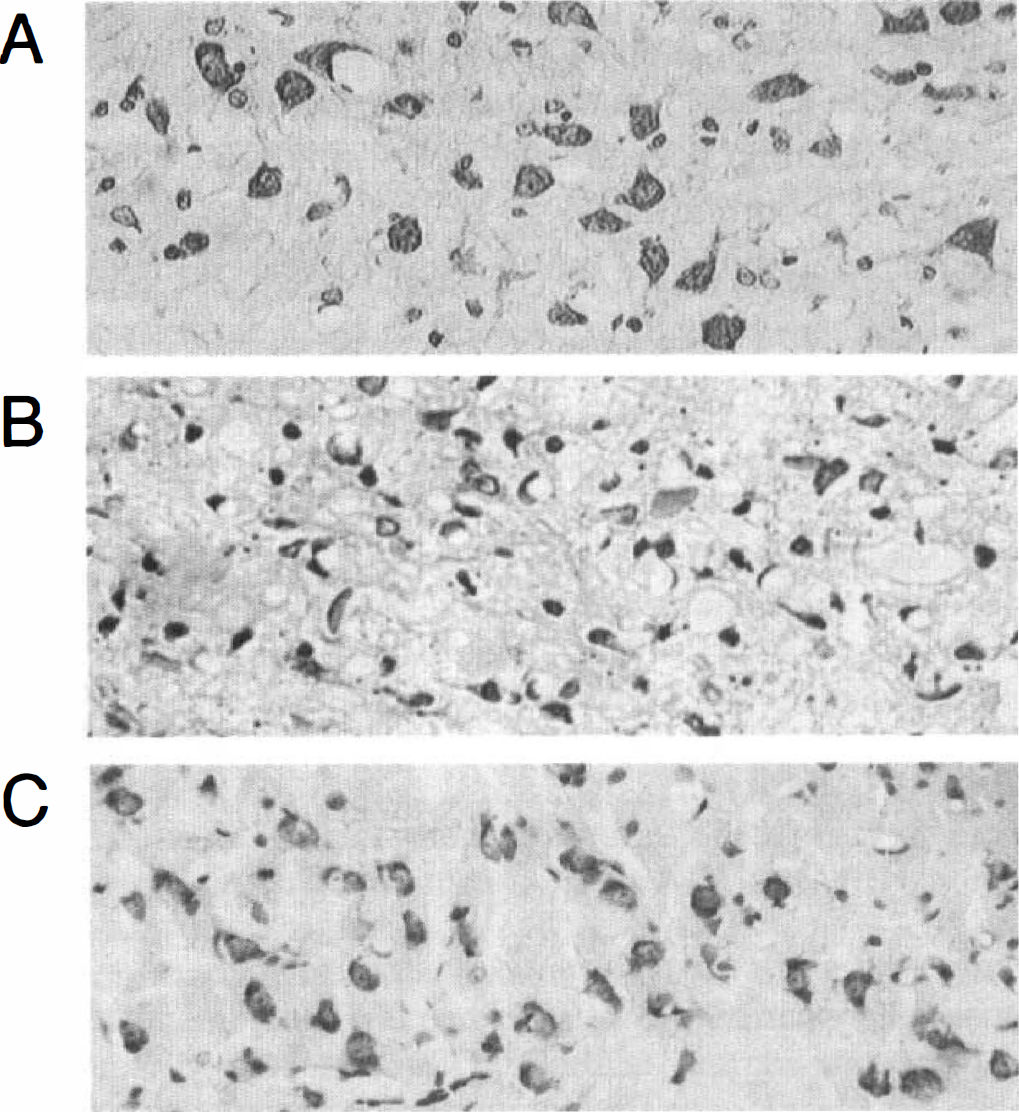
Bright-field photomicrographs of the left ventro-posterior thalamic nucleus.
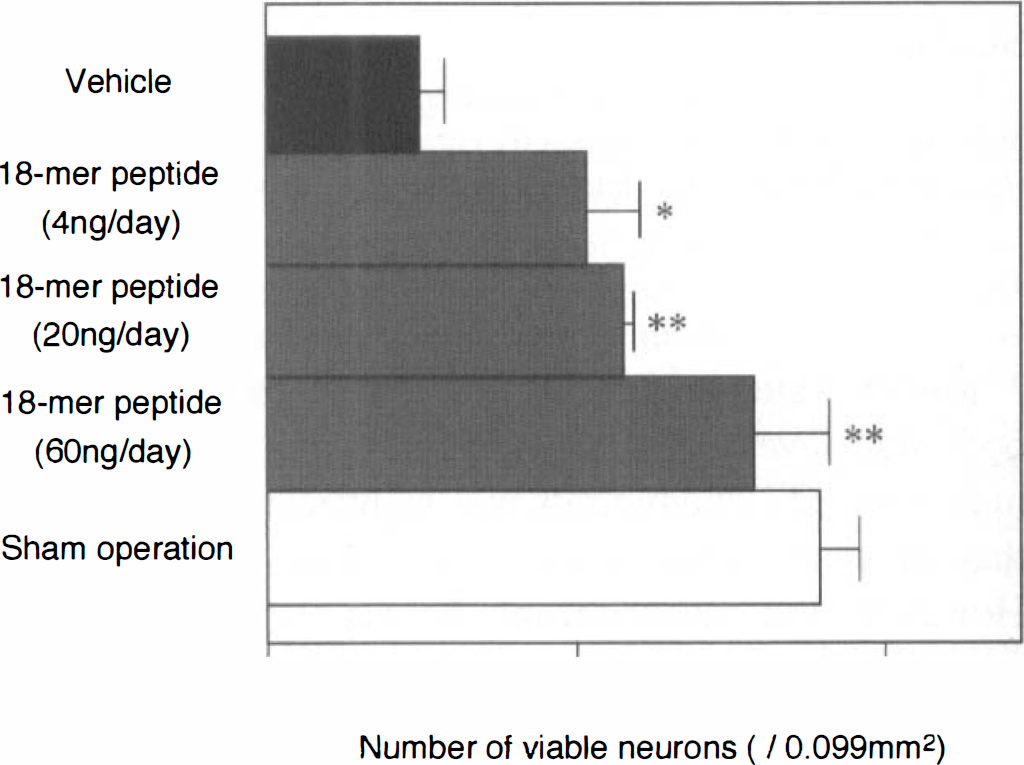
Number of viable neurons in the ventro-posterior thalamic nucleus with or without 18-mer peptide treatment. The number of viable thalamic neurons in 18-mer peptide-treated rats was significantly larger than that in vehicle-infused rats, and thus the peptide treatment attenuated the secondary thalamic degeneration in dose-dependent manner. **P < 0.01 and *P < 0.05, significantly different from the value of the vehicle-induced ischemic rats.
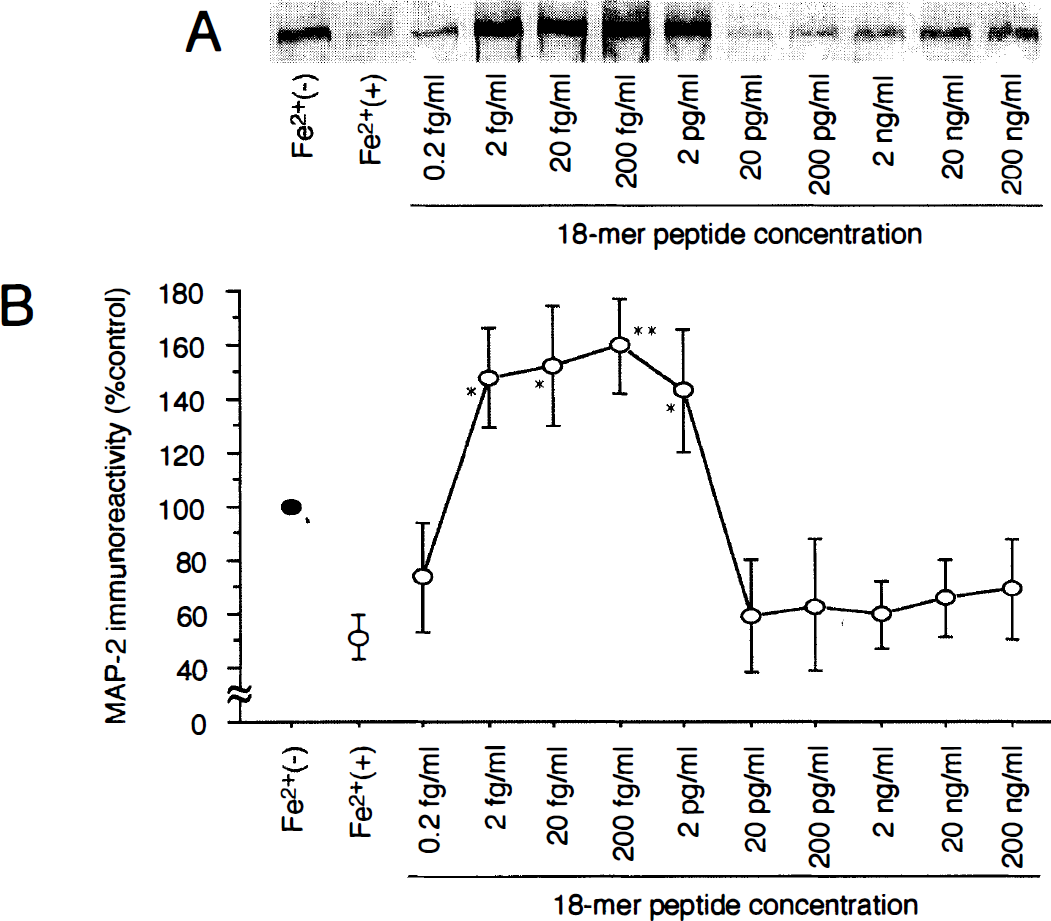
To characterize the neuroprotective action of the 18-mer peptide in vitro, the peptide was applied to cultured cortical neurons exposed to or not exposed to FeSO4. The neuronal viability was investigated by using western blotting with a monoclonal antibody specific to MAP2 in neurons (Fig. 5). MAP2 was apparently detected in the control cortical neurons [Fig. 5A, lane Fe2+(−)] but was hardly detected in FeSO4-treated neurons [Fig. 5A, lane Fe2+(+)]. When the 18-mer peptide was added to the culture medium beforehand, FeSO4-induced neuron death was prevented at a rather narrow range of concentration between 2 fg/mL and 2 pg/mL (Fig. 5A). The neuroprotective effect of the 18-mer peptide against the lethal oxidative injury caused by FeSO4 was quantitatively evaluated by densitometric analysis of the immunoreactive bands (Fig. 5B). The results showed the neurotrophism of the 18-mer peptide at concentrations of 2 fg/mL to 2 pg/mL. Furthermore, FeSO4-induced lipid peroxidation as estimated by TBARS concentration was significantly attenuated by pretreatment with 2 to 200 fg/mL of the 18-mer peptide (Fig. 6). The higher concentrations of the peptide did not reduce the production of TBARS by FeSO4. The effective concentrations of the 18-mer peptide for reduction of lipid peroxidation were similar to those of the peptide as determined by the densitometric analysis of MAP2-immunoreactive bands.
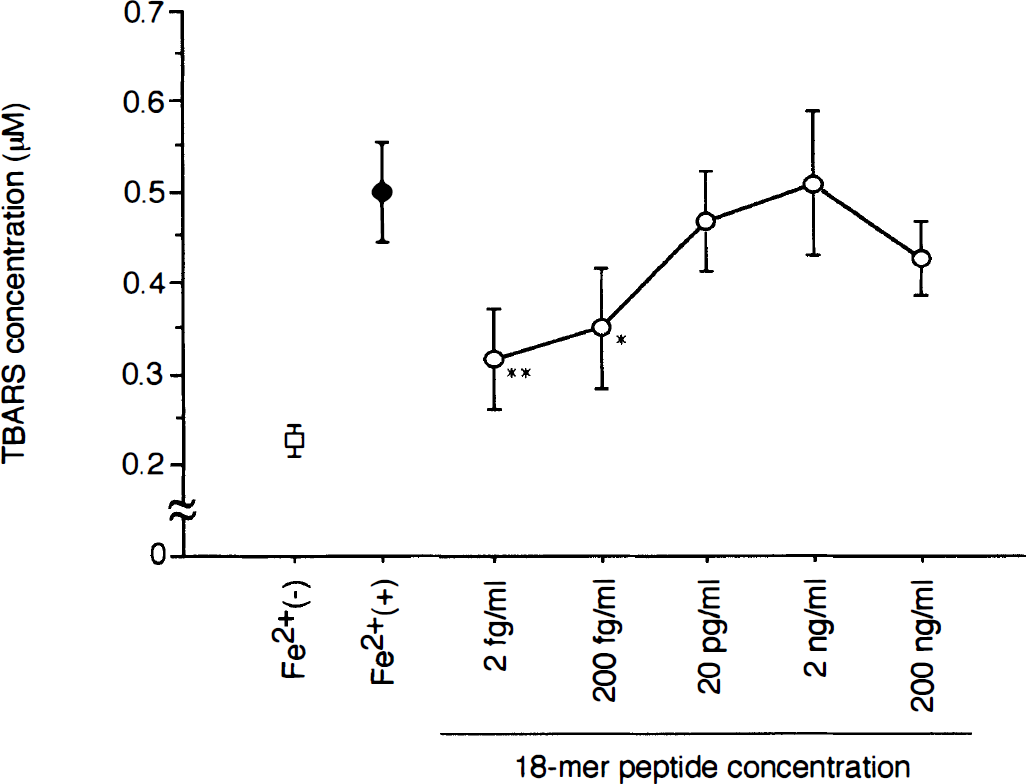
Lipid peroxidation in neurons as evaluated by TBA-reactive substances (TBARS) concentration. TBARS was increased by treating neuronal culture with FeSO4. However, pretreatment with the 18-mer peptide at the concentrations of 2 fg/mL and 200 fg/mL significantly suppressed the generation of TBARS. **P < 0.01 and *P < 0.05, were significantly different from the control. Data from 4 separate experiments are expressed as mean ± SD.
Neuronal damage induced by hypoxic stress was assessed by measuring the activity of LDH released from damaged neurons into culture medium (Fig. 7). In the control neuron culture without hypoxic stress or 18-mer peptide treatment, the ratio of LDH activity to that in the Tween-20-treated culture was 2.5 ± 0.4%. Exposure of cortical neurons to a hypoxic environment increased the ratio of LDH activity up to 17.8 ± 2.3%. Treatment with the 18-mer peptide at concentrations of 200 pg/mL to 20 ng/mL significantly attenuated the hypoxic damage as evaluated by the LDH assays (Fig. 7).
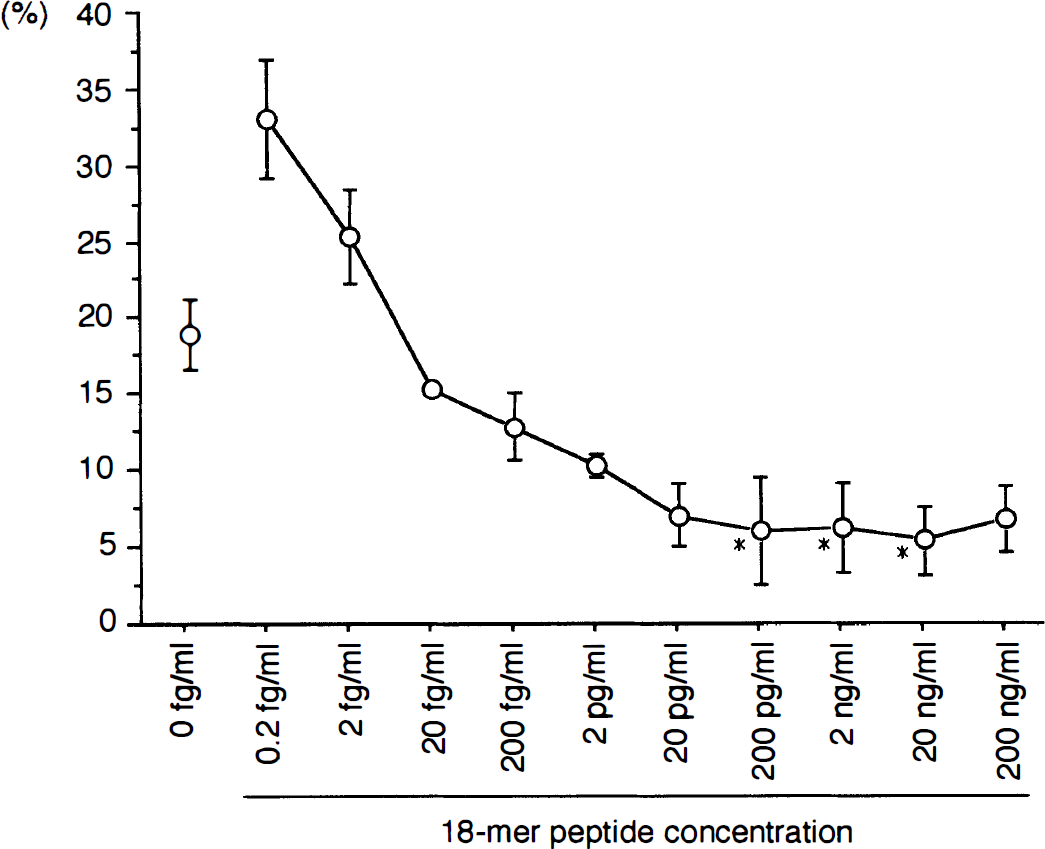
The ratio of lactate dehydrogenase (LDH) released from neurons loaded with hypoxic stress to that from tween-20-treated neurons. The LDH release measured by the activity was significantly suppressed when the neurons were pretreated with the 18-mer peptide at concentrations between 200 pg/mL and 2 ng/mL (*P < 0.05 versus control value). Data from 6 separate experiments are expressed as mean ± SD.
Fig. 8A shows the sensorgrams of a cortical synaptosome fraction to the blank chip and 44-mer peptide-immobilized chip on the BIAcore system. Binding response to the 44-mer peptide comprising the 18-mer peptide was expressed by subtracting the response to the blank chip from that to the 44-mer peptide-immobilized chip. Fig. 8B shows changes in binding response to synaptosome and membrane fractions. Both the cortical and the thalamic membrane fractions exhibited no significant changes in binding to the 44-mer peptide at the 1st and 7th days after MCA occlusion. Binding of the cortical synaptosome to the 44-mer peptide significantly declined at the 1st day after MCA occlusion and returned to a level close to the control value at the 7th day, whereas the synaptosome fraction of the thalamus did not show any changes in binding to the peptide in response to MCA occlusion.
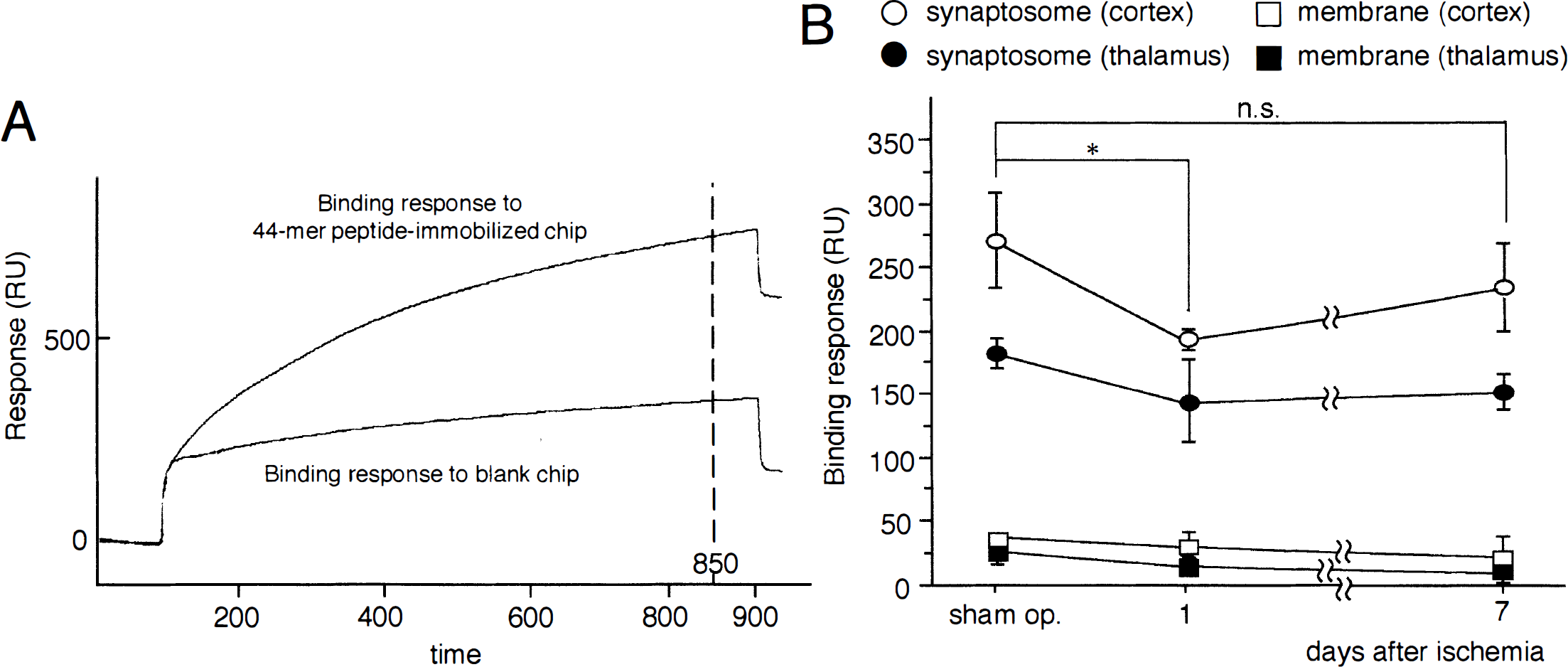
Characterization of the binding of cortical synaptosome and membrane fractions to a saposin C fragment (44-mer peptide sequence) comprising the 18-mer peptide by the BIAcore system.
DISCUSSION
In developing the water maze task for the first time, Morris (1981) reported that experimental animals with a hippocampal lesion exhibit place navigation disability. However, Okada et al. (1995) showed the occurrence of long-term spatial cognitive impairment (i.e., place navigation disability) in rats with MCA occlusion. We also showed that occlusion of the unilateral MCA above the rhinal fissure and distal to the striate branches in SP-SH rats causes place navigation disability (Zhang et al., 1998). Thus, several brain regions including the hippocampus and the cerebral cortex may be responsible for place navigation ability. Kumon et al. (1996) demonstrated that permanent occlusion of the MCA distal to the striate branches in SP-SH rats causes place navigation disability that is ameliorated by cerebroventricular infusion of CNTF. It is speculated at this moment that the amendment of place navigation disability is associated with a reduction by CNTF in the size of the primary ischemic lesion within the temporoparietal cortex. Like CNTF (Kumon et al., 1996), the 18-mer peptide infused into the cerebroventricles of MCA-occluded rats ameliorated the ischemia-induced place navigation disability and cortical infarction in the present study. However, one cannot exclude the possibility that thalamic neurons rescued by the 18-mer peptide infusion from secondary degeneration contribute to the amendment of place navigation disability in MCA-occluded rats. In support of this speculation, patients with cortical infarction after MCA occlusion develop dementia in association with the retrograde thalamic degeneration (Tamura et al., 1991). The thalamus may act as a relay center to convey sensory information necessary for the acquisition and/or maintenance of place navigation ability. If this is the case, the thalamus is likely to control the place-navigating behavior of rats together with the cerebral cortex and hippocampus. We speculate that multiple brain nuclei such as the hippocampus, cerebral cortex, and the thalamus, which constitute complex neural circuits, are responsible for the occurrence of learning behavior, as suggested by Okada et al. (1995).
Upon permanent occlusion of the unilateral MCA distal to the striate branches, a part (ischemic core) of the temporoparietal cortex supplied by the MCA alone soon enters irreversible cerebral infarction. In contrast, an area surrounding the ischemic core (ischemic penumbra), even though blood supply from the MCA is terminated, begins to have a compensatory but insufficient blood supply possibly from dilated branches of the anterior and posterior cerebral arteries. It is likely that in the ischemic penumbra, where termination and insufficient resumption of the blood supply take place, oxygen free radicals are generated to damage neurons in situ. If the blood-brain barrier is disrupted in the ischemic penumbra, leukocytes in the peripheral blood and/or tissue macrophages may also act as generators of oxygen free radicals (Matsuo et al., 1995). In support of this speculation, Imaizumi et al. (1990) reported that superoxide dismutase-entrapped liposome attenuates cerebral infarction in rats with permanent occlusion of the MCA, suggesting that oxygen-derived free radicals are, at least in part, responsible not only for ischemia/reperfusion injuries (Siesjö, 1981; Siesjö and Bengtsson, 1989) but also for brain damage in rats with permanent MCA occlusion. In the present culture experiments, the 18-mer peptide at low concentrations (2 fg/mL to 2 pg/mL) protected cortical neurons against oxidative injuries caused by the free radical-producing agent FeSO4. On the other hand, the higher concentrations of the peptide (200 pg/mL to 20 ng/mL) attenuated neuronal damage elicited by hypoxic insult. Therefore, we speculate that at early postischemic periods when the concentration of the 18-mer peptide in brain extracellular spaces is <2 pg/mL, the peptide acts as an antioxidant agent, and that with accumulation of the peptide in the extracellular spaces, it protects cortical neurons against chronic hypoxic stress. However, this speculation must be verified in the future by a sensitive assay for the 18-mer peptide in the MCA-occluded brain.
Recently, ceramide has been shown to prevent neuronal apoptosis induced by deprivation of nerve growth factor (Ito and Horigome, 1995). Since saposin C activates sphingolipid hydrolase (acid β-glucosidase) in lysosome, ceramide is likely to be produced as a result of sphingolipid metabolism. However, the 18-mer peptide, even though being a fragment of saposin C that activates acid β-glucosidase, does not act as a co-factor for the hydrolysis of glucosylceramide by the enzyme (Qi et al., 1996), and thus the possibility may be excluded that the 18-mer peptide rescues ischemic neurons through ceramide generation.
The molecular mechanisms underlying the neurotrophism of the 18-mer peptide are unknown at present. O'Brien et al. (1994) have postulated the presence of a high-affinity prosaposin receptor on the surface of a neuroblastoma cell line, and prosaposin induces mitogen-activated protein kinase phosphorylation in PC12 cells (Campana et al., 1996) and in Schwann cells and oligodendrocytes (Hiraiwa et al., 1997). These findings support the notion that the 18-mer peptide also exerts a biological action through binding to a specific prosaposin receptor. The present BIAcore analysis demonstrated that binding of the cellular membrane fraction to the 44-mer peptide fragment that composes the 18-mer peptide was lower than that of the synaptosome fraction. This binding was eliminated by digestion of the synaptosome with trypsin (data not shown). These findings suggest that prosaposin receptor chiefly resides in the synaptosome. Furthermore, the binding response of the cortical synaptosome fraction to the 44-mer peptide significantly declined at the 1st day after ischemia and recovered to the control level by the 7th day after ischemia. Therefore, in the early postischemic periods, endogenous prosaposin may be induced to bind to the receptor for neuronal survival, causing a transient decline in prosaposin receptor. Another possibility is that prosaposin receptor is down-regulated transiently after MCA occlusion as a result of a decrease in total protein synthesis (Mies et al., 1991) and subsequently up-regulated in surviving neurons. Molecular cloning and characterization of prosaposin receptor would clarify the uncertain point of the present study.
Footnotes
Acknowledgements
The authors thank Drs. H. Tanabe, A. Sano, K. Kondoh, Y. Kotani (Department of Neuropsychiatry, Ehime University), and S. Matsuda (Department of Anatomy, Ehime University) for the generous provision of the synthetic 18-mer peptide and for their fruitful suggestions and encouragement throughout this work. The authors also thank D. Shimizu and K. Kameda in the Central Research Laboratory at Ehime University School of Medicine for their excellent technical assistance.