
Abstract
One approach for developing targeted stroke therapies is to identify the neuronal protective and destructive signaling pathways and gene expression that follow ischemic insult. In some neural injury models, the transforming growth factor-beta family member activin can provide neuroprotective effects in vivo and promote neuronal survival. This study tests if activin supports cortical neurons after ischemic challenge in vitro and if signals after cerebral ischemia involve activin in vivo. In a defined cell culture model that uses hydrogen peroxide (H2O2)-free radical stress, activin addition maintained neuronal survival. H2O2 treatment increased activin mRNA twofold in surviving cortical neurons, and inhibition of activin with neutralizing antibodies caused neuronal death. These data identify activin gene changes as a rapid response to oxidative stress, and indicate that endogenous activin acts as a protective factor for cortical neurons in vitro. Similarly, after transient focal cerebral ischemia in adult mice, activin mRNA increased at 1 and 4 h ipsilateral to the infarct but returned to control values at 24 h after reperfusion. Intracellular activated smad signals were detected in neurons adjacent to the infarct. Activin was also increased after 2 h of 11% hypoxia. Activin mRNA increased at 1 h but not 4 or 24 h after hypoxia, similar to the time course of erythropoietin and vascular endothelial growth factor induction. These findings identify activin as an early-regulated gene response to transient ischemia and hypoxia, and its function in cortical neuron survival during oxidative challenge provides a basis to test activin as a potential therapeutic in stroke injury.
Keywords
Introduction
Ischemic stroke is a devastating disease that ranks third among all causes of death and leaves many permanently disabled in the United States, (Thom et al, 2006), yet targeted clinical therapies for stroke remain elusive. Cerebral blood vessel occlusion results in oxygen and glucose deprivation that leads to rapid, irreversible necrotic death within the central region of injured tissue. Surrounding the necrotic core, the ischemic penumbra receives limited blood flow and contains cells that pathologically increase intracellular calcium, glutamate, and reactive oxygen species (ROS) promoting additional cell death through apoptosis (for a review, see (Dirnagl et al, 1999). Neural responses in the penumbra include increases in gene expression associated with oxygen homeostasis (Bergeron et al, 1999; Matrone et al, 2004), angiogenesis (Hayashi et al, 1997; Marti et al, 2000; Zhang et al, 2000, 2002), and inflammation (Fujimura et al, 1999; Schneider et al, 1999; Herrmann et al, 2005). There is significant interest in identifying the functional role of these endogenous signals because they represent targets for treatment options.
Rodent experimental models such as middle cerebral artery occlusion (MCAO) followed by reperfusion stroke model (Connolly et al, 1996) or nonlethal exposure to hypoxia have revealed potential molecular mechanisms involved in neuroprotection. In mice, transient MCAO damages much of the ipsilateral hemisphere with the infarct predominantly located in cortical and striatal regions and variable infarcts affecting hippocampal, substantia nigral, and thalamic areas (Kanemitsu et al, 2002; Carmichael, 2005). By contrast, exposure of rodents to a brief, nonlethal hypoxic episode can provide some resistance to a subsequent severe insult such as stroke (Miller et al, 2001; Sharp et al, 2004). Genetic changes occurring within the cortex in stroke and hypoxia models have identified some of the activated signals as potential neuroprotectants including erythropoietin (Epo) (Prass et al, 2003) and vascular endothelial growth factor (VEGF) (Wick et al, 2002).
Growing evidence suggests that the transforming growth factor beta (TGF-beta) family member activin plays a role in neuroprotection and represents a novel potential therapeutic target. Activin supports survival of neuron-like P19 cells (Schubert et al, 1990), as well as dopaminergic (Krieglstein et al, 1995) and hippocampal neurons (Iwahori et al, 1997) in vitro. In vivo, activin mRNA is strongly upregulated in the hippocampus within hours of excitotoxic injury (Tretter et al, 1996) and neonatal hypoxic-ischemic injury (Wu et al, 1999), and added activin administration can increase neuronal survival in some of these models (Hughes et al, 1999; Wu et al, 1999; Tretter et al, 2000). Although these data suggest that responses to central nervous system injury include activin increases, the effects on cortical neurons and role after transient focal ischemia are not well understood.
Activin binds to selective heteromeric cell surface serine/threonine kinase receptors to activate smad2 and smad3 intracellular signals (Massague, 1996; Massagué, 1998). After activation, smads assemble into a complex with smad4, translocate into the nucleus and affect gene transcription (Heldin et al, 1997). Phosphorylated smad2 and smad3 can be used as indicators of cellular response to activin or TGF-beta ligands after injury.
In this study, activin is tested for its ability to promote survival of cortical neurons and protect these cells from hydrogen peroxide (H2O2) application, and endogenous activin is assessed after transient focal ischemia and hypoxia. Real-time polymerase chain reaction (PCR) is used to detect increases in activin mRNA and these changes are compared with the temporal pattern of neurovascular genes Epo and VEGF that are known to be early responders to ischemia or hypoxia. Additionally, activated smad protein levels were assayed and cell localization studied within the brain. These data suggest that cerebral ischemia injury and nonlethal hypoxia involve endogenous activin signals in neurons and that activin protects against H2O2-induced neuronal death.
Materials and methods
Embryonic Cortical Culture
Timed-pregnant Sprague—Dawley rats with pups at embryonic day 18 (E18, Zivic Miller, Pittsburgh, PA, USA) were used for cortical cell preparation as described (Perrone-Bizzozero et al, 1986). Cerebral cortices were dissected and meninges were removed in Hank's buffered salt solution (Gibco HBSS; Invitrogen, Carlsbad, CA, USA). Cells were grown on 0.1 mg/mL poly-
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde for 1 h at room temperature (RT), washed in phosphate-buffered saline (PBS) and blocked for 1 h in dilution buffer (0.4% Triton X-100, 0.4% Tween, and 3% bovine serum albumin/PBS) before reacting with antibodies. Cells were incubated with mouse anti-TuJ1I antibody (1:50, Sigma) or mouse anti-glial fibrillary acidic protein (GFAP) (1:100; Jackson Immunoresearch, West Grove, PA, USA) diluted in the same buffer overnight at 4°C. After washes, appropriate secondary antibody conjugated with Cy3 or Cy2 (1:300, Jackson Immunoresearch, USA) was applied for 1.5 h at RT, followed by washing and mounting.
In some studies, 10 μmol/L 5-bromo-2′-deoxyuridine was added for 1 h to assay DNA synthesis for cell proliferation. Cells were fixed with 95% methanol/5% acetic acid for 12 min in -20°C and permeabilzed with 2 mol/L HCl. After PBS washes, rat anti-5-bromo-2′-deoxyuridine (1:5, Harlan Sera-Lab, Hophyrst Lane, England, UK) was applied in PBS/0.4% Tween at 4°C overnight. After rinses, secondary goat anti-rat fluorescein isothiocyanate (1:100, Jackson Immunoresearch) was applied for 1.5 h at RT, washed and coverslips were mounted in 2% n-propyl-gallate to prevent fading.
At least 200 cells/well were quantified for each treatment in triplicate wells and were used to obtain a mean observation for each study. At least three independent cell isolations were performed. Unpaired t-test for two-group comparisons and one-way analysis of variance followed by Bonferroni/Dunn post hoc test for multiple group comparisons was applied (Statview 4.1 software; Abacus Concepts, Berkeley, CA, USA). Data are presented as mean ± s.e.m.
Ischemia Model
Adult, male CD-1 mice (5 to 7 weeks of age; 28 to 35 g; Harlan, Indianapolis, IN, USA) were used in MCAO experiments. All animals were housed in cages and maintained on a 12 h light/dark cycle, and the experimental protocols with animals were reviewed and performed in accordance with the Institutional Animal Care and Use Committee at Case Western Reserve University.
The monofilament model for middle cerebral artery occlusion was used to induce transient, focal ischemia in mice as previously described (Connolly et al, 1996; Endres et al, 1999). Mice were anesthetized with 2% halothane in 30% oxygen and 70% nitrous oxide. Temperature was maintained at 37°C ± 0.1°C with a heating lamp linked to a rectal thermometer (YSI; Yellow Springs, OH, USA). After skin incision and left carotid artery isolation, the tip of a 15 cm flame polished 6-0 monofilament nylon suture (Ethicon; Somerville, NJ, USA) was introduced into the external carotid lumen through a small perforation. After 1 h MCAO, animals were reanesthetized and the suture was removed. The time of filament removal was considered the onset of reperfusion. Animals were included if they met criteria for surgical time (under 35 mins), absence of visible hemorrhage at the base of the brain, or at the MCA internal carotid artery (ICA) junction and presence of modified neurobehavior tests (Connolly et al, 1996). All animals were screened for spontaneous movement without deficit (grade 0), asymmetric forepaw outstretch (grade 1), forelimb flexion contralateral to injured hemisphere and internal rotation (grade 2), circling towards paretic side or spinning to right (grade 3); animals exhibiting grades 2 to 3 after stroke were included. Of the 73 animals that underwent MCAO surgery for these experiments, 14 animals were excluded because of failure to meet surgical criteria, death before analysis or the presence of a clot on inspection and 16 animals were excluded because of a behavior grade of 0 to 1 immediately after surgery.
For sham surgeries, mice were anesthetized as above and a midline incision was made. To determine the appropriate sham control, two procedures were performed: either a temporary suture was placed around the common carotid artery for 60 min, animals reanesthetized and suture was removed or animals had a midline incision with no manipulation of the arteries. The ipsilateral hemispheres for both sham procedures were taken for real-time PCR analysis and activin mRNA was quantified. There was no difference in activin mRNA between the two groups (data not shown). In the present studies, the sham used to evaluate mRNA changes consisted of a midline incision with no artery manipulation.
Infarct volume: After killing, brains were collected and four, 2 mm coronal sections were collected using a mouse brain matrix (WPI, Sarasota, FL, USA) as described (Tureyen et al, 2004). Sections were incubated in 2% 2,3,5-triphenyltetrazolium chloride solution dissolved in normal saline at 37°C. Sections were kept in 4% paraformaldehyde at 4°C overnight and images were taken on a flat-bed camera stand with a digital camera. Infarct area was quantified using OpenLab 3.5 image analysis system (Improvision; Lexington, MA, USA). To minimize artefact produced by cerebral swelling, infarct volume was determined by delineating the area of the contralateral hemisphere and the nonaffected ipsilateral hemisphere (area without pallor). Infarct calculations were made by the formula: percentage of (contralateral hemisphere—noninfarcted ipsilateral hemisphere)/contralateral hemisphere and volume determined from four sections obtained as described (Endres et al, 1999).
Hypoxia induction: Sublethal hypoxia has been shown to protect against subsequent ischemic insult in mice (Miller et al, 2001). Mice were exposed to 11% O2 for 2 h by use of sealed normobaric chambers with oxygen concentrations monitored and regulated by nitrogen gas (Biospherix, Redfield, NY, USA).
RNA Isolation and cDNA Synthesis
RNA was isolated from cultured cells or mouse MCAO brains at 1 or 24 h of reperfusion for reverse transcription-PCR analysis. Isolation of RNA from cultured cells was performed according to the manufacturer's protocol (Qiagen, Valencia, CA, USA). Fresh frozen brain hemispheres were homogenized in 0.5 μg/mL ice-cold Trizol reagent (Invitrogen) and extracted in chloroform as described (Xu et al, 2005). Extracted RNA was treated with DNase to remove genomic DNA contamination using the DNA-free kit (Ambion, Austin, TX, USA) according to the manufacturer's instructions and confirmed by testing in real-time PCRs with all sets of the primers. No valuable Ct was detected.
First-strand cDNA synthesis was performed as described (Xu et al, 2005). Any contamination of the reagents was tested by omitting RNA in the RT reactions followed by real-time PCR with ribosomal-18s primers. No valuable Ct was detected.
Quantitative Real-Time PCR
SYBR green PCR reaction was performed using an iCycler (Bio-Rad laboratories, Hercules, CA, USA). Two microliters of reverse transcribed reaction was added to 25 μL PCR reaction mixture based on SYBR Green PCR buffer. Amplification protocol for activin, epo, hypoxia-inducible factor-1a (HIF-1a), and 18s were as follows: cycle 1: 95°C for 3 mins, cycle 2: 95°C for 15 secs, 60°C for 45 secs, repeat cycle 2 for 40 cycles. For VEGF: cycle 1: 95°C for 3 mins; cycle 2: 95°C for 30 secs, 61.9°C for 40 secs, 72°C for 40 secs, repeat cycle 2 for 40 cycles. Melting curves did not detect primer dimers and all primers have PCR amplification efficiencies of 90% to 100%. Product specificity was confirmed by 2% agarose gel electrophoresis. All samples were run in triplicates for each experiment and each sample was amplified in two to three independent experiments. PCR reaction without cDNA template was used as negative control. The Ct of each gene from each sample was normalized against that of glyceraldehyde 3-phosphate dehydrogenase for cell culture and 18s for MCAO studies. Fold changes of RNA levels were calculated by 2−ΔΔCt method (Livak and Schmittgen, 2001), in which the relative changes of genes of interest in the experimental group was calculated as the ratio of normalized data over control group. Statistical data are presented as mean ± s.e.m.
The following PCR primers were used: rat activin BA subunit: forward, 5′tgtgaacagtgccaggaga3′, reverse 5′agaaagacggaagtgacgga3′ 102 bp fragment (Becker et al, 2003); rat glyceraldehyde 3-phosphate dehydrogenase: forward, 5′tcaaggctgagaatgggaag3′, reverse 5′tactcagcaccagcatcacc3′ 103 bp fragment; mouse activin BA subunit: forward, 5′tgtgagcagtgccaggagag3′, reverse 5′tccgtcactccatctttct3′ 102 bp fragment (Becker et al, 2003); mouse EPO: forward 5′atgtcgcctccagataccac3′, reverse 5′ctctcccgtgtacagcattc3′ 119 bp fragment; mouse HIF-1a subunit: forward 5′gaaatggcccagtgagaaaa3′, reverse 5′cttccacgttgctgacttga3′ 119 bp fragment; mouse common VEGF forward: 5′aacgatgaagccctggagtg3′, reverse VEGF164 subunit 5′gacaaacaaatgctttctccg3′ 205 fragment (Ploplis et al, 2004); mouse ribosomal-18s: forward 5′aaacggctaccacatccaag3′, reverse 5′cctccaatggatcctcgtta3′ 155 bp fragment, glyceraldehyde 3-phosphate dehydrogenase or 18s were used as internal controls.
Western Blot Analyses
MCAO brain tissues were collected after 24 h of reperfusion and lysed on ice in 9 × volume of radioimmuno-precipitation assay (RIPA) buffer (20 mmol/L Tris, 150 mmol/L NaCl, 1 mmol/L NaVn, 10 mmol/L NaF, 1 mmol/L ethylenediaminetetraacetic acid (EDTA), 1 mmol/L ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid, 1% triton, 0.1% sodium dodecyl sulfate, 0.5% deoxycholic acid) and 1/10 volume protease inhibitor cocktail (Sigma). Samples were prepared and analyzed for sodium dodecyl sulfate-polyacrylamide gel electrophoresis on a 10% separating gel and transferred to nitrocellulose for approximately 180 V/h with the following modifications (Cruise et al, 2004). Nonspecific sites were blocked for 1 h at room temperature in 5% nonfat dry milk, 0.05% Tween 20 in TBS (blocking solution) before incubation overnight at 4°C in primary antibody diluted in blocking solution: rabbit anti-psmad 2/3 (1:250; Cell Signaling, Danvers, MA, USA). After washes in 0.05% Tween 20 in TBS, membranes were incubated 1 h at room temperature in appropriate peroxidase-conjugated secondary antibody diluted in blocking solution: goat anti-rabbit peroxidase immunoglobulin G (1:2500, Upstate Biotechnology, Lake Placid, NY, USA). Membranes were washed in Tween 20 in TBS before visualizing proteins using a chemiluminescent substrate for horseradish peroxidase (SuperSignal West Pico, Pierce Chemicals, Rockford, IL, USA). Primary antibody was omitted in control studies and specific signal was absent. To estimate protein changes, membranes were stripped in 100 mmol/L β-mercaptoethanol, 2% sodium dodecyl sulfate, 62.5 mmol/L Tris-HCL for 30 mins at 50°C. Membranes were washed in Tween 20 in TBS, incubated in blocking solution for 1 h at room temperature, and incubated overnight at 4°C in mouse anti-Smad2 antibody (1:1,000, Santa Cruz Biotechnology, Santa Cruz, CA, USA) or goat anti-actin antibody (1:500, Santa Cruz Biotechnology). Membranes were washed in Tween 20 in TBS and followed by incubation with peroxidase-linked secondary antibody in blocking solution (goat anti-mouse peroxidase, 1:2500, Jackson Immunoresearch), and washed and visualized as described above. Flat-bed densitometry was used to quantify the reaction product of bands at the appropriate molecular weight, normalized to loading, and expressed in arbitrary units.
Immunohistochemistry: A 2 mm coronal section was collected from MCAO and sham mice at the region of infarct using a mouse brain matrix (WPI, Sarasota, FL, USA) and fixed in 4% paraformaldehyde in 4°C overnight. Mouse brains were cyrosectioned at 20 um, mounted on slides and stored in -20°C. One section for each stroked animal was taken for cresyl violet staining. For immunohistochemistry, slides were washed in PBS and blocked for 2 h at RT in dilution buffer (0.4% Triton X-100, 3% bovine serum albumin in PBS). Sections were incubated in primary antibody rabbit anti-pSmad2/3(1:300, Santa Cruz) diluted in above buffer at 4°C overnight. After PBS rinses, sections were incubated in biotinylated donkey anti-rabbit (1:300; Jackson) antibody in 0.4% Triton X-100, 3% bovine serum albumin/PBS, for 1.5 h followed by streptavidin Cy3 antibody (1:850, Jackson Immunoresearch). For double-label studies, mouse anti-NeuN (1:100, Chemicon Temecula, CA, USA), mouse anti-GFAP (1:100, Chemicon), mouse anti-ED1 (1:300, Chemicon), and rabbit anti-factor VIII (1:200, Sigma) were used. For fluorescence visualization, donkey anti-rabbit or donkey anti-mouse Cy2 (1:200, Jackson) were used. DAB detection of pSmad2/3 was carried out using Vectastain DAB kit (Vector Labs; Burlingame, CA, USA), according to the manufacturer's protocol. For determination of infarct border, images of cresyl violet and DAB were taken using a Leica (Nussloch, Germany) DMR microscope and a Hamamatsu (C4742-12G04; Hamamatsu, Shizuoka, Japan) digital CCD camera. The distance from dorsal midline to the infarct border as determined by pallor on cresyl violet-stained sections was measured by OpenLab 3.1.5. The same distance was calculated on a parallel pSmad2/3 DAB image and an infarct borderline was demarcated. Omission of the primary antibody revealed no specific staining.
Results
Activin is a Survival Factor for Embryonic Rodent Cortical Neurons In Vitro
Activin was found in initial reports to support neuronal survival (Krieglstein et al, 1995; Iwahori et al, 1997) or neural differentiation (Belecky-Adams et al, 1999; Timmer et al, 2002) in vitro, but its activity in embryonic cortical neurons is not clear. In this study, the embryonic cortical cultures prepared were overwhelmingly comprised of TuJ1-positive neurons with few GFAP-positive astrocytes present (Figure 1A). Because embryonic cortical neurons grown in serum-free medium are known to require additional survival factors for maintenance of cell numbers over time (Ricart and Fiszman, 2001), cortical cell number was tested in defined medium. To learn if activin was a survival factor, cortical neurons were maintained overnight to allow equal cell attachment and then treated with varied concentrations of activin for 1 day and cells counted. In control cultures, approximately half the cortical cells died between 24 and 48 h without supplements. The addition of 1 or 5 ng/mL activin had no effect on neuronal survival; however, the addition of either 10 or 25 ng/mL sustained most cortical neurons in vitro during the 24 to 48 h test period (Figure 1B).
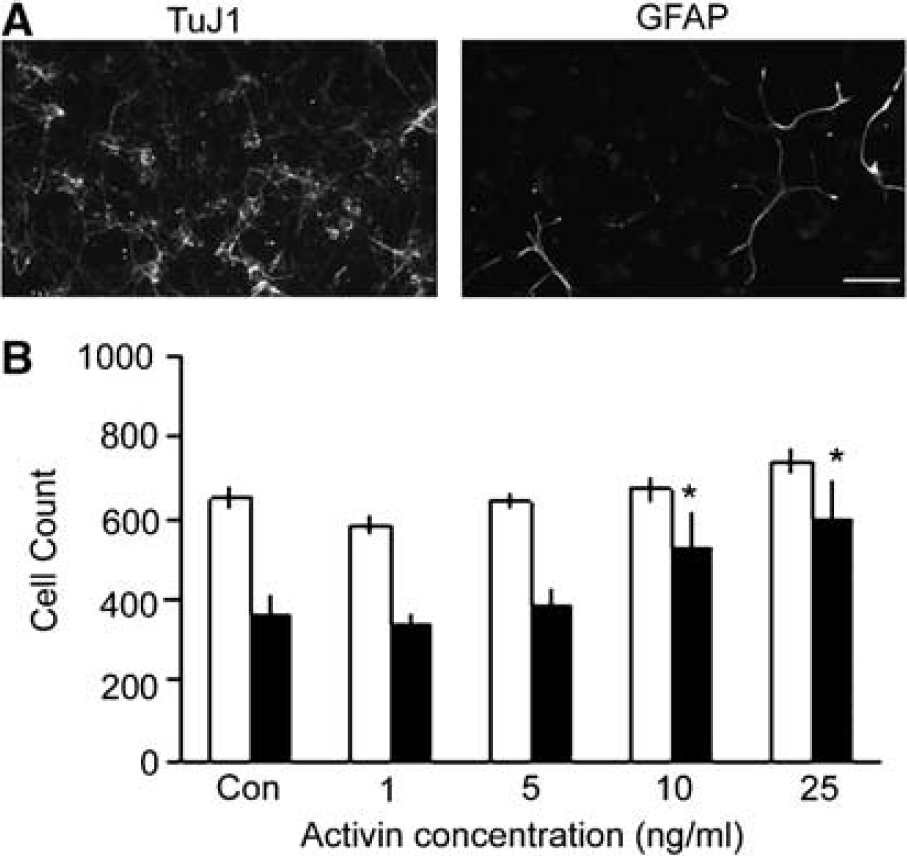
Activin maintains cortical neuron number in vitro. (
To test the possibility that cell proliferation contributed to sustained cell numbers, the thymidine analog 5-bromo-2′-deoxyuridine was added at 24 h with or without 25 ng/mL activin and 5-bromo-2′-deoxyuridine-labeled cells counted at 48 h. There was no increase in cell proliferation with the addition of activin (10.7% ± 1.0%) compared with control cultures (17.9% ± 0.6%), but rather cell proliferation decreased in activin-treated cultures in three independent experiments. Indeed, others have noted survival effects on neurons with no increase in cell proliferation (Krieglstein et al, 1995), or that activin promotes neuronal differentiation in a PC12-derived cell line with a concomitant reduction in cell division (Jornvall et al, 2001). These data in combination show that 25 or 50 ng/mL activin maintains embryonic cortical neuron survival in vitro.
Activin Protects Neurons Against Hydrogen Peroxide-Induced Death
Reactive oxygen species are key mediators of the damage observed in a variety of central nervous system injuries including stroke (Saito et al, 2005) and this effect can be partially mimicked in vitro with the addition of H2O2. An H2O2-induced oxidant injury screening assay has been an effective means to identify neuroprotective compounds (Sarang et al, 2002) and was adapted in these studies. Treatment of cortical cells in defined medium with H2O2 for 24 h reduced cell numbers as expected, with 75 μmol/L H2O2 treatment reducing cell number by half (Figure 2A). This concentration was used in subsequent studies to examine activin effects on H2O2-induced neuronal death.
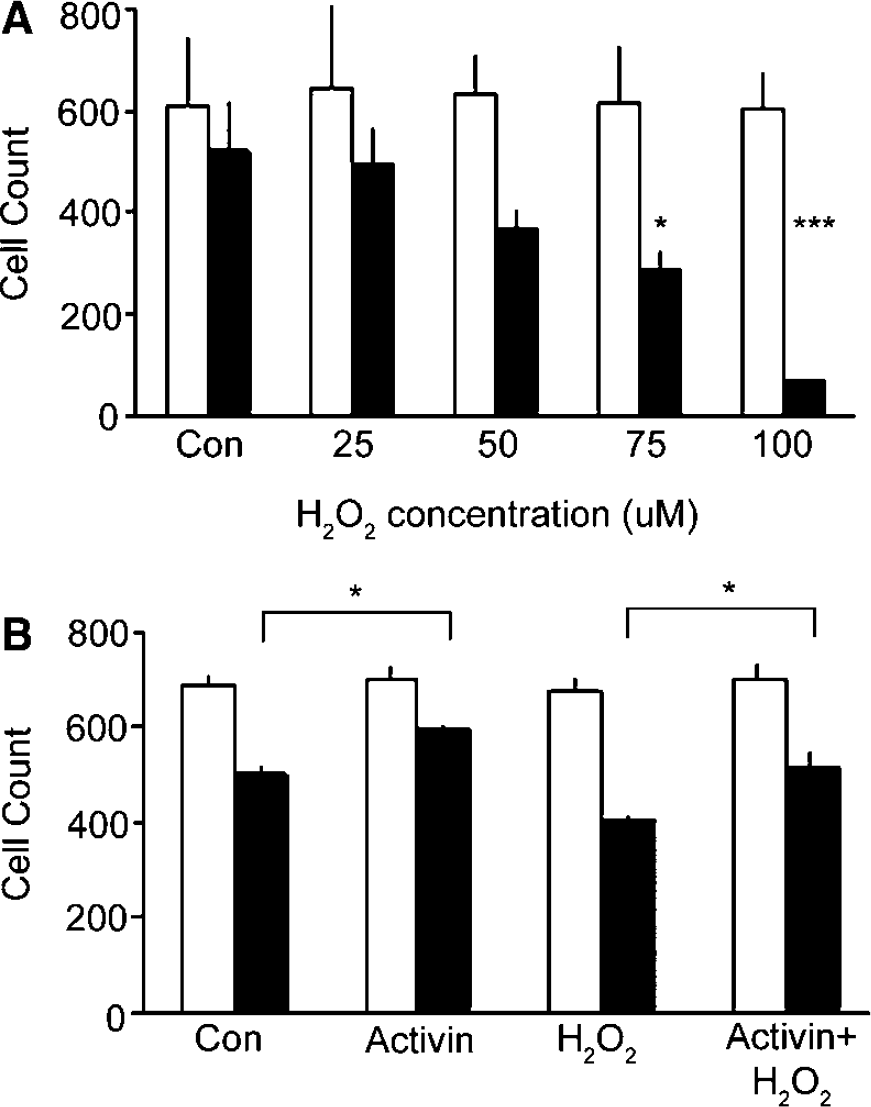
H2O2 and activin effects on cortical neurons in vitro. Cell counts at 24 h after plating before treatments show similar plating density (light bars). (
To test if 25 ng/mL activin protected against H2O2 challenge, cortical neurons were allowed to attach, then treated with activin for 24 h before adding H2O2 at 48 h, and cells were counted at 72 h. The addition of 25 ng/mL activin alone significantly sustained more cortical neurons than control from 24 to 72 h time period as expected from its survival effects seen above (Figure 1B and Figure 2B). As expected, H2O2 treatment resulted in fewer cells than controls, because of the known toxic effects of oxidative damage. However, in cultures treated with activin and then H2O2 insult, there was a significant rescue of cortical cell numbers at 72 h (Figure 2B), showing that activin addition protected cortical neurons against H2O2 insult.
In these studies, while many neurons died, other neurons survived despite the H2O2 concentration administered. One hypothesis to explain their continued survival is that H2O2 stimulated the remaining neurons to produce endogenous protective factors such as activin. Two assays were performed to test the molecular changes in surviving neurons after H2O2 challenge. In the first analysis, activin mRNA levels were examined by quantitative real-time PCR after addition of H2O2. In these studies, cortical neurons were allowed to attach overnight and then 75 μmol/L H2O2 was added. Activin mRNA levels were assayed at 1, 4, 12, and 24 h after H2O2 addition (Figure 3A). While no increases were seen at early time points, at 12 and 24 h after H2O2, activin mRNA levels increased more than twofold compared with control. These data suggest that at least some neurons respond to H2O2 with activin mRNA synthesis.
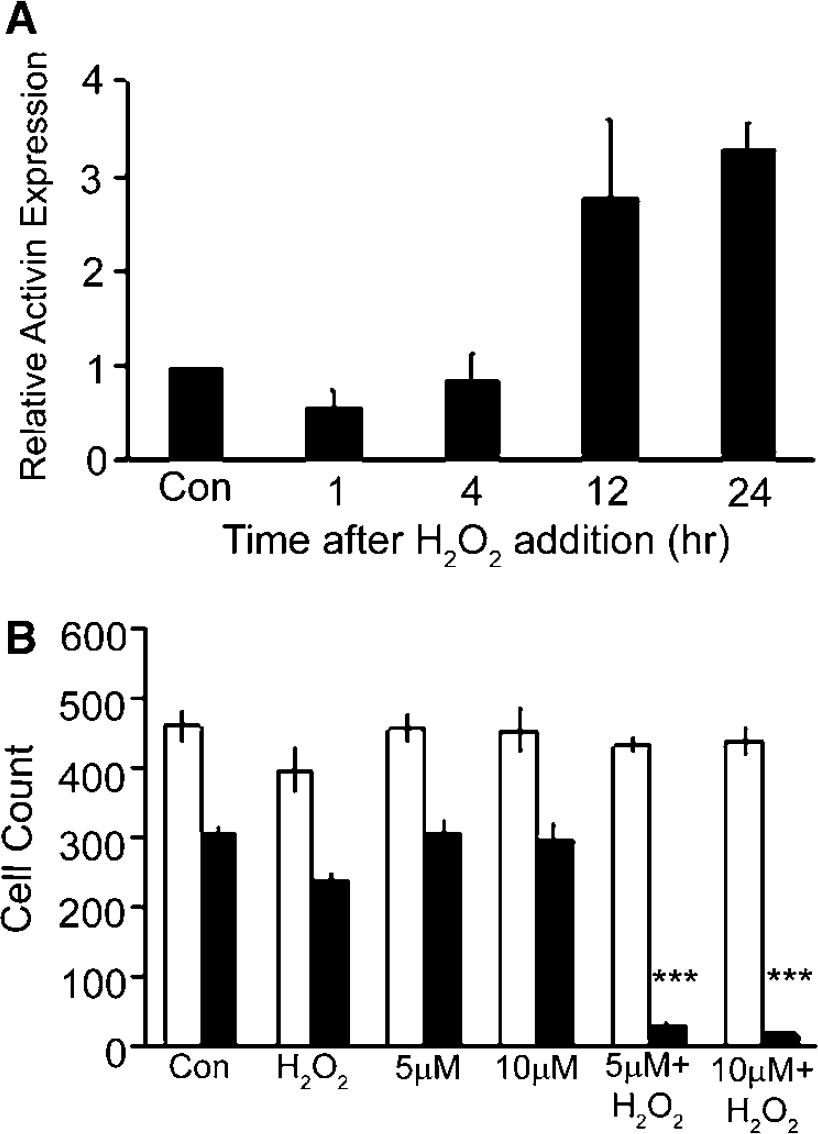
Activin mRNA increases after H2O2 challenge and activin protein inhibition increases H2O2-induced neuronal death. (
In a second analysis, the functional role of endogenous activin was assayed. The goal of this study was to test if endogenous activin made by neurons accounted for neuronal survival. Neutralizing antibodies that block activin binding to its receptor were used to inhibit activin biologic effects. The addition of the antibody alone at 5 to 10 μmol/L had no effect on neuronal survival, as expected, because the reagent was not toxic, nor was there activin in the cell culture. However, the addition of neutralizing antibody in combination with H2O2 resulted in robust cell death that exceeded cell loss with H2O2 alone (Figure 3B). Together these data support the idea that cortical neurons make activin in response to H2O2 and this activin supports neuronal survival. These observations lead to the hypothesis that after injury, some cortical neurons make activin that functions to support their survival. This notion was tested in two systems, transient focal cerebral ischemia and hypoxia, described below.
Activin mRNA is Increased after Transient Focal Cerebral Ischemia
These mechanistic observations in vitro led us to examine if activin and activin-mediated signals were increased after ischemic injury in vivo. Activin was assayed in ipsilateral and contralateral hemispheres after 1 h MCAO in adult mice. Transient focal ischemia resulted in reproducible infarct volumes (55% ± 3% at 24 h, n = 11 in control studies) in CD-1 mice as measured by 2,3,5-triphenyltetrazolium chloride staining. To investigate if activin expression changed after ischemic injury, activin mRNA levels were assayed in both ipsilateral and contralateral hemispheres and compared with sham control animals in quantitative real-time PCR assays. Because VEGF mRNA has been shown to increase as early as 1 h in the ipsilateral hemisphere after transient MCAO in the rat (Hayashi et al, 1997), the VEGF164 isoform was used in these experiments as a positive control for genetic changes after ischemia. Activin mRNA increased almost threefold in the ipsilateral hemisphere at 1 h of reperfusion and was significantly higher in the ipsilateral than the contralateral hemisphere (Figure 4A). As expected, VEGF mRNA also increased at 1 h in the ipsilateral hemisphere and increases were pronounced in the ipsilateral hemisphere (Figure 4B). At 24 h of reperfusion, however, activin mRNA expression had returned to control levels as had VEGF164 mRNA (Figure 4A and Figure 4B). These data suggest that transient cerebral ischemic injury in vivo produces a rapid, local, and transient activin mRNA increase.
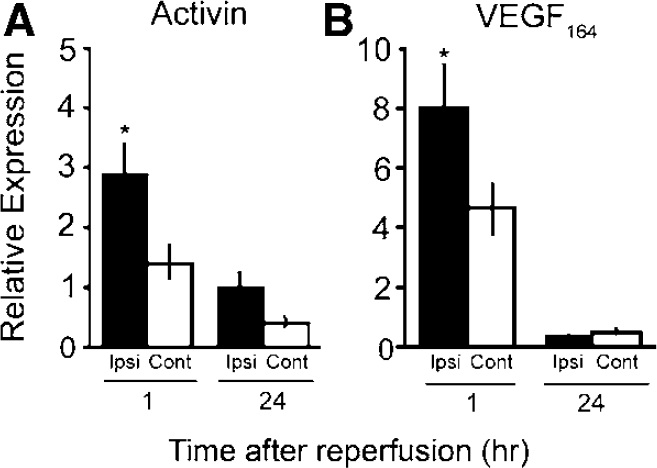
Activin and VEGF164 mRNA changes after transient focal cerebral ischemia in mice. Activin or VEGF164 mRNA changes were measured 1 or 24 h after MCAO in ipsilateral (ipsi) and contralateral (cont) hemispheres relative to sham animals. (
Focal Cerebral Ischemia Increased Activin Signaling
To test if activin mRNA increases seen after MCAO corresponded with cell to cell signals associated with activin in the brain, Western immunoblot analysis was used to quantify phosphorylated smad2/3 protein levels. At 24 h after MCAO, psmad2/3 was consistently increased in the ipsilateral hemisphere as compared with the contralateral hemisphere or control brains (Figure 5A). These data are consistent with the activation of activin receptors in the ipsilateral hemisphere, although the ligand that initiated this signal is not clear.
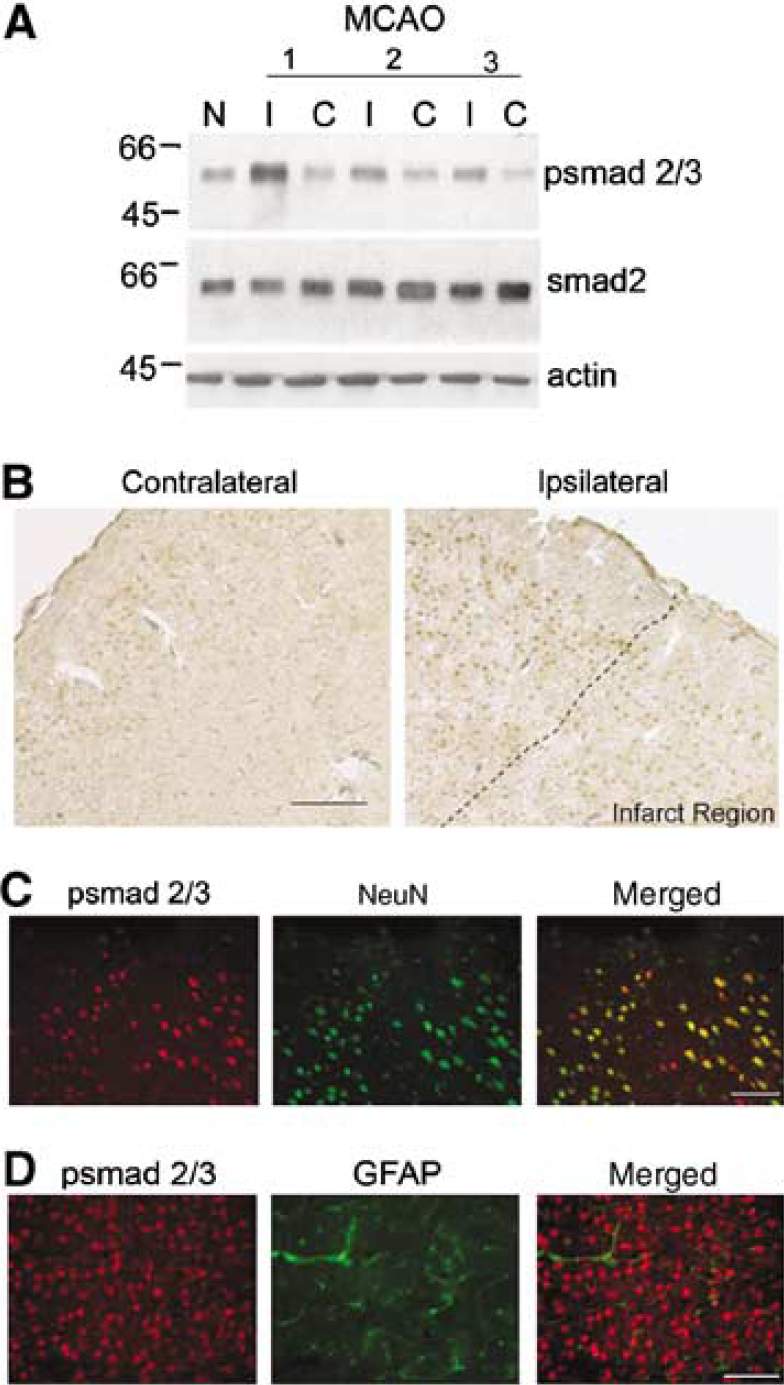
Smad 2/3 protein was activated in neurons after focal ischemia. (
To identify which cells may respond to activin signals after MCAO, immunohistochemisty was used to examine psmad2 immunoreactive cells. Psmad2/3 immunoreactivity was present in nuclei predominantly in the ischemic hemisphere with intense staining in the cortical regions surrounding the infarct (Figure 5B). Psmad2/3-positive cells were confirmed to be neurons as they colabeled with the neuron-specific marker NeuN (Figure 5C). Activated smad 2/3 responses were not observed in GFAP-positive glial cells (Figure 5D), ED1-positive macrophages, or Factor VIII-positive endothelial cells at this time point (data not shown). These data suggest that activated smad2 signals are increased in close proximity to the infarcted region after MCAO and that these responses occur principally in neurons.
Activin mRNA Increases after Brief Hypoxia
Studies of transcriptional changes induced by hypoxia have identified potential pathways involved in neuroprotection (Sharp et al, 2004), and one advantage of this experimental model is that robust genetic changes occur throughout the brain. To learn if brief hypoxia induces activin expression, adult mice were exposed to 2 h of 11% hypoxia followed by ambient air for 1, 4, or 24 h, and mRNA levels for activin were assayed by quantitative realtime PCR. Again, because both VEGF and Epo mRNA have previously been reported to increase transiently after hypoxia (Marti and Risau, 1998; Bernaudin et al, 2002; Grimm et al, 2002; Prass et al, 2003), these two markers were used as positive controls to ensure that the hypoxia regimen resulted in established VEGF and Epo cerebral mRNA changes. Activin mRNA levels increased four fold at 1 h but not at 4 or 24 h (Figure 6A). VEGF164 isoform as well as Epo mRNA levels (Figure 6B and Figure 6C) also showed characteristic increases at 1 h after hypoxia, but returned to control values at 4 h. There was no significant change in hypoxia-inducible factor 1a (HIF-1a) mRNA levels at any time after this short hypoxic regimen (Figure 6D), consistent with other reports indicating that HIF-1 activation occurs predominantly through protein stabilization rather than mRNA induction (Semenza, 2001). These data suggest that activin mRNA increases occur rapidly but transiently after decreased oxygen concentrations in the brain.
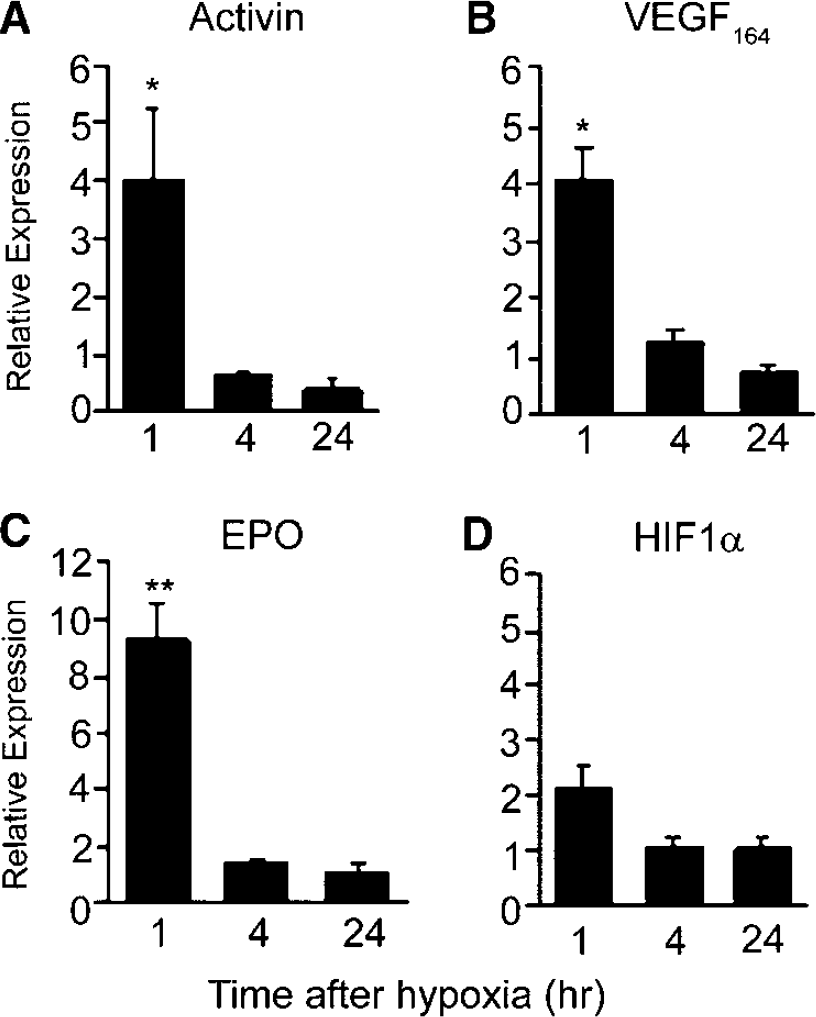
mRNA changes after hypoxic exposure. Mice were exposed to 2 h of 11% O2 and whole brain mRNA assayed at 1 to 24 h as indicated. Normoxic control animals were treated in parallel and analyzed at the same time points. Quantitative realtime PCR reveals increases in mRNA levels for (
Discussion
Neuronal responses to ischemic injury or hypoxia are heterogeneous and depend in part on their proximity to the injured region and the time after the initial insult. This report shows for the first time that cortical neurons after stress increase activin mRNA, and that activin biological function is important for neuronal survival after H2O2 oxidative stress in vitro. An interesting characteristic of this response is that activin appears to function as a paracine or autocrine factor to rescue neurons. Cell culture studies suggest that endogenous activin signals preserve some neurons after injury and offer the possibility that additional exogenous activin may aid survival. Studies in vivo indicate that one early and transient neuronal response to ischemic injury and hypoxia is an increase in activin and potentially its intracellular signals. In combination, these data place activin mRNA induction and protein function in the early phases of neuronal responses after injury. These fundamental observations lead to future studies to determine what signals initiate activin after neural injury and how interventions based on activin application may be used in vivo. These observations are discussed below.
Activin appears to be made by neurons (Florio et al, 2000; Bottner et al, 2006) and acts on neurons (this report), suggesting that injury may induce a paracrine signaling event. This report shows that 10 or 25 ng/mL activin retained cortical neurons in defined medium. These concentrations are comparable with earlier reports showing the trophic properties of activin in vitro in which 10 or 30 ng/mL activin supported hippocampal neurons (Iwahori et al, 1997) and 5 or 20 ng/mL activin supported midbrain dopaminergic neurons (Krieglstein et al, 1995). However, not all cortical neurons were maintained with activin addition in vitro. One possibility is that activin supports the survival of a distinct subset of cortical neurons while others are unable to respond. A previous report examining the effect of activin administration in vivo after a quinilonic acid lesion showed the robust survival of striatal cholinergic projection and interneurons, but not subsets of GABAergic neurons (Hughes et al, 1999). Variations in neural responsiveness to activin may be because of differences in receptor expression (Cameron et al, 1994; Funaba et al, 1997) or intracellular mechanisms (Zhang et al, 2005) activated by activin in neuronal phenotypes. One recent report suggests that activin mRNA and signaling, as well as behavioral improvements are also noted after antidepressant activity (Dow et al, 2005), but the mechanism of action in this case is not known. It will be interesting to learn whether activin addition after ischemic injury in vivo can maintain survival of all cortical neuronal types or only subpopulations and if the latter proves true, whether the salvage of particular subset of neurons leads to functional behavioral improvements.
An important caveat to this interpretation is embryonic neurons in vitro may not be an appropriate model for adult neurons in vivo. Embryonic neurons are dependent on the continual presence of growth factors for their survival while adult neurons may require trophic support only in times of injury (Benn and Woolf, 2004). Activin mRNA and protein are present at basal levels throughout the adult brain including cortical regions (Roberts et al, 1996) with significant upregulation of activin mRNA, its protein and activin signals in adult neurons can be detected within hours after injury (our data; Tretter et al, 1996; Dow et al, 2005; Bottner et al, 2006). Because activin administration protects adult hippocampal neurons from excitotoxic kainic acid and quinolic acid addition in vivo (Hughes et al, 1999; Tretter et al, 2000), it seems likely that increased amounts will improve neuronal survival after ischemic injury.
Activin administered before H2O2 addition maintained neuronal survival. Oxidative injury is mediated through the production of reactive oxygen species (ROS) and is linked to a variety of neural injury states including ischemia (Saito et al, 2005). In other studies of E16 to E17, rat cortical neurons exposed to 100 μmol/L H2O2, cell death was induced via apoptotic mechanisms (Whittemore et al, 1994; Whittemore et al, 1995). In our cultures, cell death occurred in an H2O2 concentration-dependent fashion with more cortical neurons maintained after activin administration, consistent with previous reports (Ricart and Fiszman, 2001). Interestingly, activin has also been shown to protect against 1-methyl-4-phenyl 1,2,3,6-tetrahydropyridine (MPTP)-induced neurotoxicity in dopaminergic neurons (Krieglstein et al, 1995) in which the toxic metabolite MPP+ leads to increased intracellular ROS (Nicotra and Parvez, 2002). This potential capacity for activin to protect neurons from ROS-induced toxicity makes it an attractive molecule for study in a variety of injury paradigms that involve free radical production.
One cellular response to oxidative stress includes activin, which acts as a neuronal survival factor. It is important to note that simply eliminating activin with neutralizing antibody did not affect neuronal survival in the present cell culture studies. Only after H2O2 addition was activin mRNA, and presumably protein, increased in these cultures. The addition of antibody and H2O2 resulted in substantial neuronal death supporting the notion that for some cells, endogenous activin becomes critical when cells are exposed to an insult. Clarifying how activin restricts ROS-induced death could be valuable in predicting when after injury activin effects would be most effective therapeutically and for which types of central nervous system damage.
Activin mRNA increased after ischemia in vivo and occurred with a temporal profile similar to VEGF164 (Hayashi et al, 1997; Mu et al, 2003). Both genes had no detectable increases at 24 h after ischemia, confirming some reports that indicate activin mRNA is increased rapidly and transiently after focal injuries (Tretter et al, 1996). This time course for activin mRNA increase in vivo does not wholly correlate with our observations of activin expression in neuronal cultures after H2O2 addition. The multiple cellular events initiated by ischemic stroke and the involvement of non-neuronal cells not present in our cultures are likely to be contributing factors to the timeline for activin mRNA increases in vivo. Conceivably, activin is released as an early central nervous system stress signal with smad activation occurring within the day (our data; Dow et al, 2005). While the present report shows transient and early activin mRNA changes, a recent study examining activin mRNA using the related permanent MCAO in adult, female rats (Bottner et al, 2006) illustrated that permanent loss of blood flow produced a robust, longer lasting activin response. Similar observations between transient and permanent occlusions have been made for VEGF mRNA (Mu et al, 2003) and may indicate that activin induction and signaling events are closely associated with the neurovascular unit and reintroduction of blood flow. Currently, it is unclear what molecular signals initiate activin induction after in vivo injury.
Other reports suggest that activin increases occur within neurons close to the injury site (Tretter et al, 1996; Bottner et al, 2006) and this spatial pattern, as well as the present mechanistic cell culture studies suggest that neurons both make and respond to activin stimulation. After transient ischemia, phosphorylated smad2/3 intracellular signals are increased within the ischemic hemisphere and are seen within neurons that lie in close proximity to the infarcted region. A comparison of activin expression in other reports and our own data with psmad2/3 indicate that there are some neurons within the penumbral region that neither produce activin nor respond to the ligand. It is not clear from these assays whether these neurons may be activated at different times after ischemic injury or if they are a population of neurons that are unresponsive to activin signaling.
While our data suggest that activin initiates the observed phosphorylated smad2/3 increase, TGF-beta also signals through smad2/3 and may account for some of this activation (Gomes et al, 2005). Transforming growth factor-beta1 levels are enhanced after cerebral ischemia, agonists reduce infarct volumes after ischemia and antagonists show increased neuronal damage (see review (Dhandapani and Brann, 2003). Increases in TGF-beta1 mRNA develop ipsilateral to the infarct within 6 h of reperfusion and remains high for 72 h after transient MCAO ischemia in rats (Lehrmann et al, 1998). Because TGF-beta1 also has effects on neurons after injury (Zhu et al, 2002), the precise role of TGF-beta1 and activin ligands on neurons will need to be assayed in ischemia possibly through genetic inactivation of the activin ligand or type 1-specific receptors in neuronal cells.
Activin increases after hypoxic stress provide important insights into molecular changes after injury, and have been suggested as useful biomarkers of neonatal hypoxic stress in infants. Studies of neonatal cord blood indicate that increased activin protein levels may provide a reliable marker of perinatal hypoxia (Perrone et al, 2002; Bracci et al, 2006). In our hands, decreased oxygen capacity initiates activin mRNA increases in mice. A similar regimen of 2 h of 11% hypoxia provides neuroprotection from MCAO in adult mice (Miller et al, 2001), and leads us to speculate that activin may be involved in hypoxic preconditioning, but this notion requires further study.
Interestingly, activin mRNA increases after hypoxia follow a similar time course to VEGF (Marti and Risau, 1998) and Epo (Grimm et al, 2002; Prass et al, 2003). Both of these genes show an oxygen concentration-dependent rise in expression linking their transcriptional activation to systems that sense oxygen homeostasis, such as HIF-1-mediated pathways. The temporal dynamics of VEGF and Epo, both of which are HIF-1 targets, and the time course observed for activin mRNA suggest that activin induction may be downstream of HIF-1a activation. Additional support for this idea was obtained from microarray studies using endothelial cells exposed to either 1% oxygen or transfected with a constitutively active HIF-1a construct that showed increases in activin gene expression (Manalo et al, 2005). Clearly, there are other transcriptional factors involved in hypoxia regulation such as CREB (Gao et al, 2006), activating protein 1 (Kapinya et al, 2000), and nuclear factor-κB (Liu et al, 2005) which could also be regulators of activin expression.
Rigorous testing is required to determine whether activin is an HIF-1a target gene and whether activin provides neuronal support for neurons after insult. Previous studies in vivo have shown that exogenous activin protects hippocampal neurons after kainic acid lesions when acting in combination with basic fibroblast growth factor (bFGF) lending support to the concept of activin functioning as a protective agent in acute brain injury (Munz et al, 2001). Similarly, in vitro mechanistic studies in cortical neurons in this report lend credence to the likelihood that activin will support neuronal survival after an ischemic insult. However, any therapeutic potential of exogenous or endogenous activin after an injury must now be formally tested in an adult stroke model, combined with behavioral outcome testing. The present studies provide the understanding of activin mRNA spatiotemporal changes after injury that justifies this important next step.
Footnotes
Acknowledgements
We are grateful to Drs Carolina Maier and Pak Chan at Stanford University for their assistance in developing the mouse MCAO model.