
Abstract
Transforming growth factor-α (TGF-α) is a ligand for the epidermal growth factor (EGF) receptor (EGFR), and is more abundant than EGF in the brain. The authors studied whether administration of exogenous TGF-α into the brain can protect neurons against ischemia in a model of permanent middle cerebral artery (MCA) occlusion in the rat, and whether any effect of TGF-α was mediated by EGFR by administering 4,5-dianilinophthalimide (DAPH), a protein-tyrosine kinase inhibitor with high selectivity for EGFR. Rats received either TGF-α (10 or 25 ng), DAPH (100 ng), DAPH plus TGF-α (25 ng), or vehicle in the ipsilateral first ventricle. Drugs were administered twice: 30 minutes before and 30 minutes after MCA occlusion, and infarct volume was evaluated 24 hours later. Transforming growth factor-α at the dose of 25 ng caused a statistically significant reduction of infarct volume (60%) in relation to ischemic rats administered vehicle. This reduction was no longer seen when TGF-α was administered in combination with DAPH. The present results show that TGF-α can protect neurons from ischemic damage, and that this effect is mediated by EGFR. It is suggested that activation of EGFR-mediated intracellular signalling pathways contributes to the survival of neural cells susceptible to ischemic injury.
Administration of exogenous growth factors, such as brain-derived neurotrophic factor acting through the high-affinity receptor tyrosine kinase, TrkB (Schabitz et al., 1997), and ciliary neurotrophic factor (Kumon et al., 1996), can protect the brain against focal ischemia. Likewise, many growth factors have been shown to exert a protective effect in transient forebrain ischemia. In this latter condition, it has recently been reported that epidermal growth factor (EGF) protects neuronal cells (Peng et al., 1998). Here we examined whether exogenous transforming growth factor-α (TGF-α), a growth factor that binds the EGF receptor (EGFR), was able to exert any protective effect in a model of permanent focal ischemia in the rat, and whether EGFR activation was involved.
Transforming growth factor-α is a 5 kDa polypeptide showing 35% homology to EGF (Marquardt et al., 1983). Transforming growth factor-α also mediates its biological activity upon binding the EGFR with high affinity, induces tyrosine phosphorylation of the receptor, and has strong mitogenic activity for epithelial and mesenchimal cells (Massagué, 1983; Reynols et al., 1983; Derynck, 1988). Both TGF-α and EGF are constitutively expressed in the developing and adult brain (Kudlow et al., 1989; Kaser et al., 1992; Lazar and Blum, 1992; Ma et al., 1992; Seroogy et al., 1993; Ferrer et al., 1995,1996), although TGF-α is more abundant than EGF in the CNS (Lazar and Blum, 1992; Kaser et al., 1992; Seroogy et al., 1993). In the adult brain under physiologic conditions, TGF-α is found in neurons and some glial cells (Seroogy et al., 1993; Ferrer et al., 1995, 1996), whereas EGFR is essentially located in neurons (Gómez-Pinilla et al., 1988; Kaser et al., 1992; Kornblum et al., 1995; Ferrer et al., 1996, Planas et al., 1998). However, EGFR is highly induced under conditions involving glial reactivity (Gómez-Pinilla et al., 1988; Nieto-Sampedro et al., 1988; Ferrer et al., 1996). In particular, EGFR is expressed in reactive astrocytes and microglia/macrophages after transient focal ischemia in the rat brain (Planas et al., 1998). Today, the role and mechanism of action of TGF-α and EGFR in neural cells remains to be clarified. Transforming growth factor-α is a soluble factor that is released through enzymatic cleavage from a larger precursor protein anchored in the cell membrane (Massagué, 1990; Derynck, 1992). Transforming growth factor-α released to the extracellular space as a soluble factor is bioactive by acting on the membrane-bound EGFR, causing paracrine stimulation. An autocrine stimulation is also feasible, because TGF-α and EGFR colocalize in many neurons (Ferrer et al., 1996). In addition, several lines of evidence suggest that noncleaved, pro-TGF-α interacts with EGFR on a neighboring adjacent cell, or the same cell, and exerts a juxtacrine effect (Wong et al., 1989; Anklesaria et al., 1990; Massagué et al., 1990). Exogenous TGF-α administered into the brain would be expected to diffuse and act on membrane-bound EGFR. This mode of action would mimic, to some extent, paracrine stimulation of EGFR by endogenous soluble TGF-α.
METHODS
Rats and Surgery
Adult male Sprague-Dawley rats (280 to 320 g body weight) obtained from Iffa-Credo (Lyon, France) were kept under a 12-hour light-dark cycle and allowed free access to food and water. Animal work was conducted in compliance with the Spanish legislation on “Protection of Animals used for Experimental and other Scientific Purposes”, and in accordance with the Directives of the European Community on this subject. Surgery was performed under halothane anesthesia. One day before ischemia, guide cannulae were implanted stereotaxically into the ipsilateral ventricle at the following coordinates: 0.3 mm posterior, 1.2 mm lateral, 3.2 mm ventral to bregma (Paxinos and Watson, 1986). Focal cerebral ischemia was produced by intraluminal occlusion of the middle cerebral artery (MCA) following the method of Longa et al. (1989), with modifications (as in our case MCA occlusion was permanent). Briefly, rats were anesthetized with halothane, tracheotomized orally, and kept ventilated. Body temperature was maintained at 37.5°C during surgery. Permanent ischemia was produced by introducing a 26-mm long, 3/0 nylon monofilament, blunted at the tip, through the internal carotid artery to the level where the MCA branches out. After surgery, rats were allowed to recover spontaneous breathing and were kept for 24 hours in their cages with free access to food and water. Rats were killed at 24 hours after MCA occlusion, the brain was rapidly frozen, and 20 µm-thick coronal sections were cut in a cryostat. Sections collected at 1-mm intervals were stained with cresyl violet for image analysis.
Drugs and Treatments
Human recombinant TGF-α (Oncogene Science Calbiochem, AMS Biotechnology, Madrid, Spain) was prepared following the instructions of the manufacturer, and finally dissolved in 0.1 M phosphate-buffered saline yielding pH 7. Phosphate-buffered saline was administered as the vehicle solution in the control group. The EGFR inhibitor 4,5-dianilinophthalimide (DAPH) was purchased from RBI (Köln, Germany), and was dissolved in phosphate-buffered saline. Rats were given vehicle (phosphate-buffered saline) (n = 7), 10 ng TGF-α (n = 7), 25 ng TGF-α (n = 7), 100 ng DAPH and 25 ng TGF-α (n = 5), and 100 ng DAPH (n = 6). Treatments were administered into the ipsilateral ventricle twice: 30 minutes before and 30 minutes after MCA occlusion.
Evaluation of infarct volume
Infarct volume was evaluated by means of an image analyzing system (AIM, Image Research, Canada). The whole area of infarct, which at 24 hours is apparent by marked pallor in cresyl violet stained sections, was measured in each section, and then data from all sections were integrated. In addition, contralateral and ipsilateral hemisphere areas were measured, and the difference between ipsilateral and contralateral areas in each section was used to calculate the volume of brain swelling. Measures were performed by one of the authors who was blind to the treatment of each particular rat. Statistical analysis of data was performed using Mann-Whitney U-test, a nonparametric test for comparison of mean group values. The relationship between the two parameters that were calculated for each rat, i.e. infarct volume and volume of swelling, was evaluated with simple regression analysis.
RESULTS
Rats were randomly assigned to one of the following treatment groups: control (vehicle), 10 ng TGF-α, 25 ng TGF-α, 25 ng TGF-α plus DAPH, and DAPH. Treatment was administered to the right ventricle 30 minutes before and 30 minutes after permanent occlusion of the ipsilateral MCA. No differences in behavior were observed between groups. Infarct volume was evaluated at 24 hours. Infarct affected the ipsilateral cortex and striatum, although rats receiving 25 ng TGF-α showed comparatively smaller infarcts, especially in the cortex. This latter effect was no longer observed after administration of TGF-α plus DAPH. Infarct volume was calculated by measuring infarct area in serial coronal sections and then integrating all measures. Statistical comparison of data demonstrated that rats administered 25 ng TGF-α had infarcts that were 60% smaller than controls (P < 0.02) (Fig. 1). Also, rats receiving the dose of 10 ng TGF-α showed a mean infarct volume 30% smaller than that of controls (Fig. 1), but this effect was not statistically significant. The protective effect of 25 ng TGF-α was eliminated by coadministration of TGF-α plus the EGFR protein-tyrosine kinase inhibitor DAPH (Fig. 1). Comparison of infarct volume between rats administered 25 ng TGF-α and those receiving 25 ng TGF-α plus DAPH showed significant differences (P < 0.02). Rats given DAPH showed no differences in relation to controls (Fig. 1).
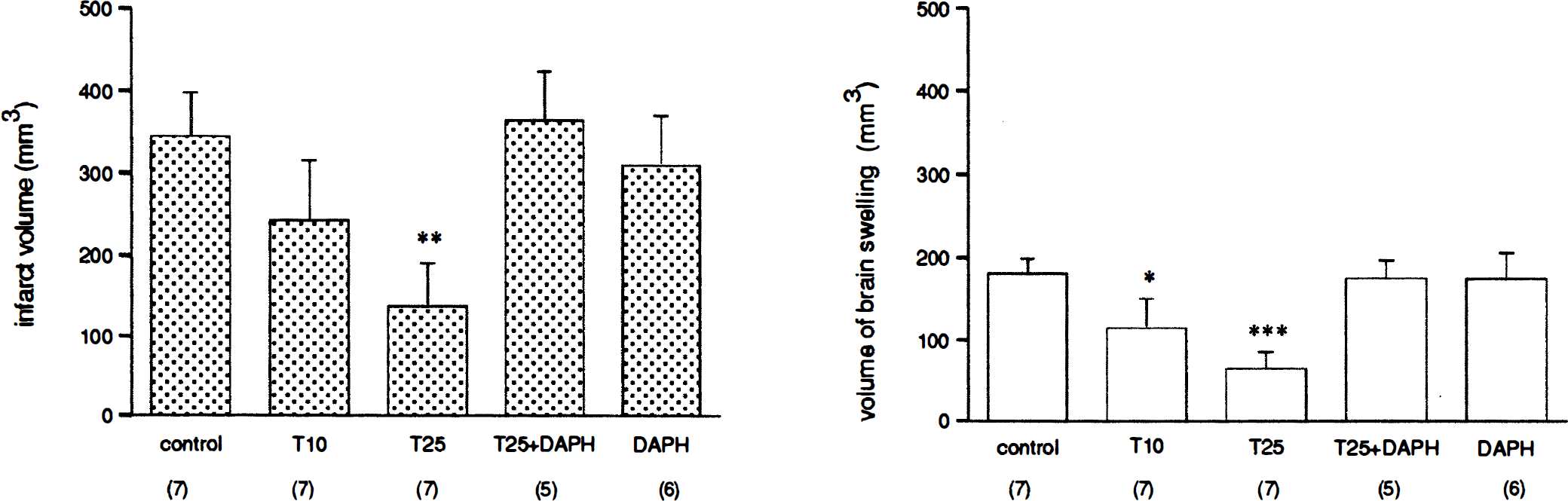
Histograms showing infarct volume (mm3)
An index of the extent of edema in terms of volume of swelling (mm3) was calculated by measuring for each section the area of the whole ipsilateral and contralateral hemispheres, then subtracting the contralateral from the ipsilateral brain side, and integrating areas. The ipsilateral ischemic hemisphere was larger than the contralateral for all groups (Fig. 1), indicating that quite large edemas had been formed within 24 hours of permanent occlusion of the MCA. However, less swelling than controls was detected in the two groups of rats receiving TGF-α. Swelling was reduced by 36% (P < 0.05) and 64% (P < 0.005) in rats receiving 10 and 25 ng, respectively. The effect of 25 ng TGF-α on brain swelling was also eliminated by coadministration of DAPH (P < 0.001) (Fig. 1). Linear regression analysis considering all rats showed a significant linear correlation (P < 0.001, r = 0.95, slope = 0.463) indicating proportionality between infarct volume and volume of swelling (Fig. 2). Significant correlation between these two parameters was also found within each treatment group (P < 0.01).
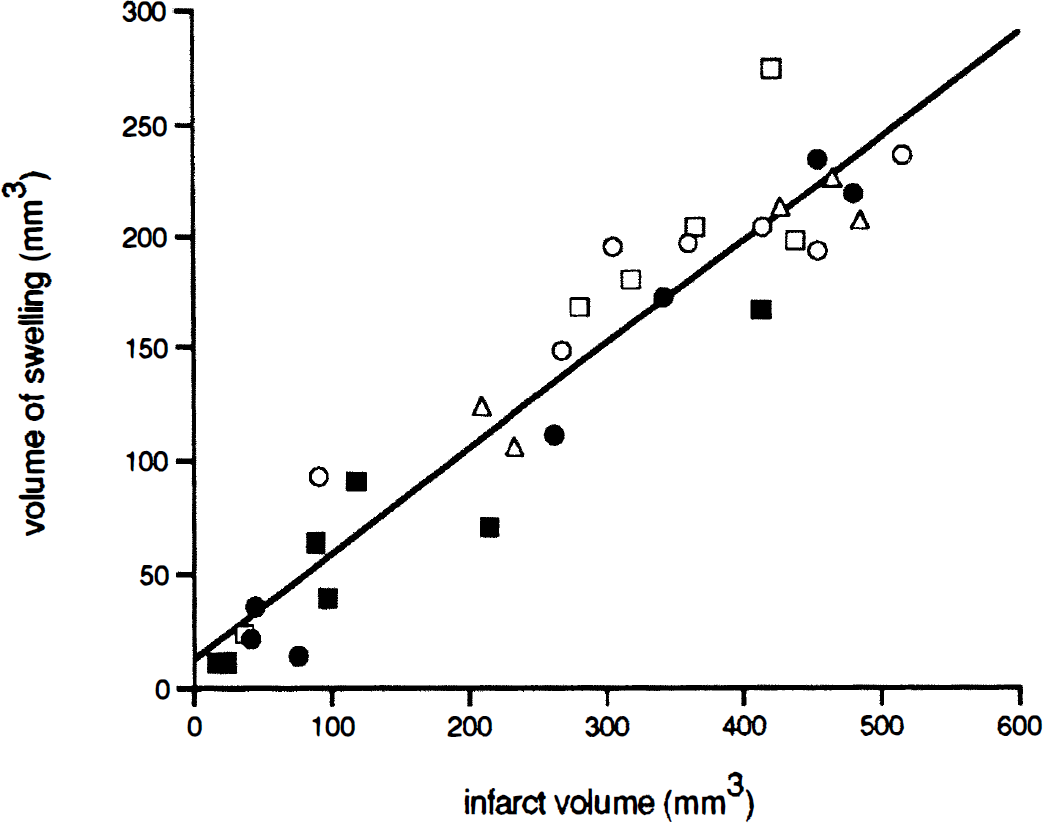
Infarct volume (X axis) and volume of brain swelling (Y axis) show a linear correlation (P < 0.001), as determined by simple regression analysis. The pair of values (X, Y) for each rat belonging to the different treatment groups were considered in the analysis. Rats were treated with: ○ vehicle (control); • 10 ng TGF-α (T10); △ 25 ng TGF-α (T25); ▲ 25 ng TGF-α plus DAPH (T25+DAPH); or ' DAPH. Correlation coefficient = 0.95; slope = 0.46. Points represent individual animals. DAPH, 4,5-dianilinophthalimide; TGF-α, transforming growth factor-α.
DISCUSSION
Growth factors promote growth and differentiation of cells in culture, and many previous studies have shown that administration of different trophic factors can offer protection to the brain against ischemic injury (e.g., Kumon et al., 1996; Schabitz et al., 1997). Here we studied the effect of intraventricular administration of TGF-α to rats subjected to permanent MCA occlusion. Transforming growth factor-α reduced infarct volume to 60% of that of ischemic controls at 24 hours after permanent occlusion of the MCA. Transforming growth factor-α is a ligand for EGFR, a membrane receptor with intrinsic tyrosine kinase activity. We have inhibited EGFR protein-tyrosine kinase with a drug, DAPH, that is highly selective toward EGFR (Buchdunger et al., 1994; Furet et al., 1995; Yen and Scoong, 1996). The results show that the reduction of infarct volume caused by 25 ng TGF-α is completely eliminated when TGF-α is administered in combination with DAPH. This strongly suggests that the protective effect of TGF-α is mediated by EGFR. In parallel to reducing infarct volume, TGF-α caused a dose-dependent decrease in brain swelling. Swelling and infarct volumes were strongly correlated, showing that the extent of swelling is proportional to the extent of infarction, as previously observed (Jacewicz et al., 1990; Kusumoto et al., 1992).
The present results are in agreement with a recent study showing that intraventricular infusion of EGF in the gerbil protects hippocampal neurons against ischemia, possibly through inhibition of free radical neurotoxicity and lipid peroxidation (Peng et al., 1998). Transforming growth factor-α has also been shown to protect striatal neurons against excitotoxic injury induced by quinolinic acid (Alexi et al., 1997). Also, an EGFR-dependent pathway is involved in the protective effect of chromaffin granules on mesencephalic dopaminergic neurons (Krieglstein and Unsicker, 1997). Other studies have concluded that TGF-α acts as a general neuronal survival factor, affecting cholinergic and GABAergic neurons (Mazzoni and Kenigsberg, 1996). Transforming growth factor-α is normally expressed in neurons and glia of the adult brain (Ferrer et al., 1995), but whether basal TGF-α can exert any neuroprotective effect in ischemia is unknown. Here, DAPH, which is expected to prevent effects of basal TGF-α, did not produce any substantial modification of infarct volume in relation to controls. Nevertheless, loss of basal TGF-α has been detected within the ischemic area at 6 hours after MCA occlusion (Ferrer et al., 1996). This would hamper TGF-α-dependent neuroprotective action after ischemia, and thus prevent DAPH from exacerbating ischemic damage. However, the possibility that increases in infarct volume were not detected here cannot be ruled out because controls already show very large infarct volumes.
Epidermal growth factor receptor is expressed in neurons and glial cells in the developing brain, and mainly in neurons in the adult brain (Gómez-Pinilla et al., 1988; Kaser et al., 1992; Kornblum et al., 1995; Ferrer et al., 1996, Planas et al., 1998). After transient MCA occlusion, EGFR is highly induced in reactive astrocytes and microglia/macrophages by 3 to 4 days after ischemia (Planas et al., 1998). After permanent MCA occlusion, the first signs of glial reactivity (i.e. increase in glial fibrillary acidic protein indicating astrogliosis, and neutrophil infiltration) are seen at 12 hours (Clark et al., 1994). Therefore, exogenous TGF-α administered in this study probably acts on neuronal membrane-anchored EGFR because neuronal protection must take place during the first hours after MCA occlusion before marked glial reactivity is apparent. Neurons within the MCA territory, i.e. lateral cortex and striatum, are rich in EGFR (Planas et al., 1998), and are thus susceptible to the action of exogenous TGF-α. Ligand-binding to EGFR can activate intracellular signal transduction pathways involving proteins of the family of signal tranducers and activators of transcription (STATs) (Darnell et al., 1994; Ruff-Jamison et al., 1994; Ihle, 1996). On stimulation, STATs can translocate to the nucleus and activate transcription, thus mediating a cellular response to changes in the environment. We have previously reported the presence of STAT proteins in the developing and adult rat brain (Planas et al., 1996; 1997a,b). Whether or not STAT proteins become activated after stimulation of the EGFR by exogenous TGF-α and mediate neuronal protection remains to be established.
In brief, the present results show that TGF-α can protect neurons from ischemic damage and that this effect is mediated by EGFR. It is suggested that activation of EGFR-mediated intracellular signaling pathways promotes survival of neural cells susceptible to ischemic injury.
Footnotes
Acknowledgment
The authors thank Ms. A. Ramirez for excellent technical assistance.