
Abstract
Cerebral ischemia leads to a massive increase in cytoplasmic calcium activity resulting from an influx of calcium ions into cells and a release of calcium from mitochondria and endoplasmic reticulum (ER). It is widely believed that this increase in cytoplasmic calcium activity plays a major role in ischemic cell injury in neurons. Recently, this concept was modified, taking into account that disturbances occurring during ischemia are potentially reversible: it then was proposed that after reversible ischemia, calcium ions are taken up by mitochondria, leading to disturbances of oxidative phosphorylation, formation of free radicals, and deterioration of mitochondrial functions. The current review focuses on the possible role of disturbances of ER calcium homeostasis in the pathologic process culminating in ischemic cell injury. The ER is a subcellular compartment that fulfills important functions such as the folding and processing of proteins, all of which are strictly calcium dependent. ER calcium activity is therefore relatively high, lying in the lower millimolar range (i.e., close to that of the extracellular space). Depletion of ER calcium stores is a severe form of stress to which cells react with a highly conserved stress response, the most important changes being a suppression of global protein synthesis and activation of stress gene expression. The response of cells to disturbances of ER calcium homeostasis is almost identical to their response to transient ischemia, implying common underlying mechanisms. Many observations from experimental studies indicate that disturbances of ER calcium homeostasis are involved in the pathologic process leading to ischemic cell injury. Evidence also has been presented that depletion of ER calcium stores alone is sufficient to activate the process of programmed cell death. Furthermore, it has been shown that activation of the ER-resident stress response system by a sublethal form of stress affords tolerance to other, potentially lethal insults. Also, disturbances of ER function have been implicated in the development of degenerative disorders such as prion disease and Alzheimer's disease. Thus, disturbances of the functioning of the ER may be a common denominator of neuronal cell injury in a wide variety of acute and chronic pathologic states of the brain. Finally, there is evidence that ER calcium homeostasis plays a key role in maintaining cells in their physiologic state, since depletion of ER calcium stores causes growth arrest and cell death, whereas cells in which the regulatory link between ER calcium homeostasis and protein synthesis has been blocked enter a state of uncontrolled proliferation.
Keywords
Although the basic mechanisms underlying ischemic cell injury are not fully understood, it is widely believed that calcium plays a key role in this pathologic process. The traditional calcium hypothesis, put forward in 1981 (Hass, 1981; Siesjö, 1981), proposes that cell damage triggered by transient cerebral ischemia results from a pathologic activation of various calcium-dependent processes such as lipolysis or proteolysis, leading to secondary disturbances, including free radical formation, and finally causing damage to membranes and destruction of cell integrity. Activation of calcium-dependent processes is believed to be brought about by a marked increase in cytoplasmic calcium activity during ischemia, that is, a state of energy depletion resulting in a massive influx of calcium ions through voltage-gated and agonist-activated calcium channels and a release of calcium ions from intracellular calcium stores such as mitochondria and endoplasmic reticulum (ER).
The calcium hypothesis is based on results obtained from cell culture experiments indicating that an influx of calcium ions plays a primary role in cell injury triggered by membrane-active toxins (Schanne et al., 1979). However, with respect to the influx of calcium ions observed during a severe form of stress or exposure of cells to toxins, the in vivo situation differs fundamentally from the in vitro situation. In vivo, calcium availability is low because of the small volume of the extracellular space. In vitro, in contrast, calcium availability is high, since a small volume of cells is surrounded by a large volume of medium representing the extracellular space. Calcium antagonists failed to produce a clear neuroprotective effect in experimental models of transient global cerebral ischemia, and the slight improvements observed in studies of focal cerebral ischemia can be explained by the vasoactive properties of these drugs (for review see Hossmann, 1989).
The traditional calcium hypothesis has been modified (Kristian and Siesjö, 1996; Murphy et al., 1996), taking into account that irreversible steps of the pathologic process causing ischemic cell injury probably take place during recovery from metabolic stress. After transient cerebral ischemia, a massive uptake of calcium ions into mitochondria leads to an overloading of mitochondria with calcium ions. High intramitochondrial calcium activity is believed to induce production of free oxygen radicals and formation of mitochondrial permeability transition pores, resulting in secondary disturbances of the mitochondrial energy-producing system (Kristian and Siesjö, 1996). This concept is based mainly on the observation that cyclosporin A (CsA), an established immunosuppressant (for review see Perico and Remuzzi, 1997), which also blocks the formation of mitochondrial permeability pores (Duchen et al., 1993), has a marked neuroprotective action when its passage across the blood-brain barrier is facilitated through needle insertion (Uchino et al., 1995). However, CsA not only suppresses immune reactions and counteracts mitochondrial permeability transition, but also induces synthesis of the stress protein, glucose-regulated protein 78 (grp78) (Paslaru et al., 1994), which in itself may afford neuroprotection (see later). Furthermore, a clear cause-and-effect relation between the formation of mitochondrial permeability pores and cell damage and the interference of CsA in this process has to be established: CsA blocked apoptosis induced by 2 nmol/L thapsigargin (Tg, an irreversible inhibitor of ER Ca2+-ATPase), even when mitochondria stayed intact, as indicated by unchanged ATP level and ATP/ADP ratio (Waring and Beaver, 1996). On the other hand, CsA did not suppress apoptosis induced by 10 nmol/L Tg although, under these conditions, the mitochondrial membrane potential collapsed and ATP levels decreased markedly (Waring and Beaver, 1996). Furthermore, the observation that the immunosuppressive drugs CsA and FK506 are both neuroprotective in models of transient cerebral ischemia (Sharkey and Butcher, 1994; Uchino et al., 1995); Tokime et al., 1996), although FK506 has no effect on the membrane permeability transition, also suggests that the drug's action is independent of mitochondrial mechanisms. Finally, drugs such as CsA and FK506 suppress the immune reaction activated in T lymphocytes by an antigen receptor-generated rise in inositol 1,4,5-triphosphate (IP3) levels and IP3-induced depletion of ER calcium stores (Imboden and Stobo, 1985), indicating that IP3 formation and ER calcium release channels might be targets for drug action. In addition, activation of T lymphocytes caused a severe decrease in the expression of ER calcium pump, a response that could be inhibited by CsA (Launay et al., 1997), indicating that this drug has a direct effect on ER calcium pump expression. The role of mitochondrial dysfunction in ischemic cell injury is therefore not proven. The possibility of a causative link was further refuted by the observation that glycine, in an in vitro model simulating ischemia and reperfusion, prevented cell damage but did not inhibit mitochondrial permeability transition (Quian et al., 1997).
The possible role of disturbances of ER calcium homeostasis in neuronal cell injury caused by severe forms of stress has been overlooked until recently (Paschen, 1996; Paschen et al., 1996). The ER hypothesis was put forward because the response of neurons to a transient depletion of ER calcium stores is almost identical to their response to transient metabolic stress, implying common underlying mechanisms. This article gives a current overview of the functions of the ER and the response of cells to disturbances of ER function. It also summarizes results from experimental studies indicating that disturbances of ER function may play a critical role in the development of neuronal cell injury in acute and chronic pathologic states of the brain. Finally, the putative role of ER calcium homeostasis in maintaining the optimal physiologic conditions in which cells are balanced between a state of uncontrolled growth and a state of growth arrest and cell death are discussed.
ENDOPLASMIC RETICULUM FUNCTION AND CALCIUM HOMEOSTASIS
The ER is a subcellular compartment that plays an important role in the folding and processing of newly synthesized secretory and membrane proteins, reactions that are strictly calcium dependent (Lodish and Kong, 1990; Gosh et al., 1991; Kuznetsov et al., 1992; Lodish et al., 1992). The ER calcium activity is therefore high, lying in the lower millimolar range, that is, close to the calcium activity of the extracellular space (Hofer and Machen, 1993; Chen et al., 1996; Pozzomiller et al., 1997). Besides a high calcium activity, an oxidative environment is required in the ER compartment, since the folding of proteins is an oxidation reaction that can be hindered by compounds exhibiting a high reducing activity, such as dithiothreithol (Kuznetsov et al., 1992).
Endoplasmic reticulum calcium stores are controlled by the IP3 receptor (IP3-R); the ryanodine receptor, which, on stimulation, releases calcium from the ER; and a Ca2+-ATPase, which pumps back calcium ions against a steep concentration gradient in an energy-requiring process (Fig. 1). With respect to calcium ions, the ER is not a homogenous compartment but is divided into various subcompartments. The subcompartments have been identified and characterized according to the extent of changes in cytoplasmic calcium activity induced by pharmacologic agents known to deplete ER calcium stores (Gosh et al., 1989; Verma et al., 1990; Bian et al., 1991; Fasolato et al., 1991; Pizzo et al., 1997); one pool responds to IP3, whereas a second pool, which does not respond to IP3 under control conditions, can be transformed into a IP3-sensitive pool by guanosine triphosphate (GTP)-dependent calcium translocation. Filling of both of these pools is blocked by Tg (for review see Inesi and Sagara, 1994). The third pool contains a Tg-insensitive pump and does not respond to IP3 or GTP. When cells are exposed to the calcium ionophore A23187, all ER calcium pools are depleted. Calcium uptake into microsomes isolated from the whole brain is suppressed almost 100% by Tg plus A23187, about 90% by Tg and 40% by IP3 (Verma et al., 1990). The IP3 and ryanodine receptors have differing distributions within the forebrain: IP3-R density is high in the hippocampal CA1 subfield and the striatum, whereas ryanodine binding is high in the hippocampal CA3 subfield and dentate gyrus (Verma et al., 1992). Like GTP, the protein Bcl-2 may facilitate calcium fluxes between different ER subcompartments, as is discussed later. This idea is based on the observations that Bcl-2 is found in the ER membrane (Reed et al., 1996), that Bcl-2 overexpression helps to maintain Ca2+ levels in the ER of Tg-treated cells (Lam et al., 1994; He et al., 1997), and that Bcl-2 and Bcl-2-related proteins have been found to be capable of forming ion channels in synthetic lipid membranes (Minn et al., 1997; Schendel et al., 1997).
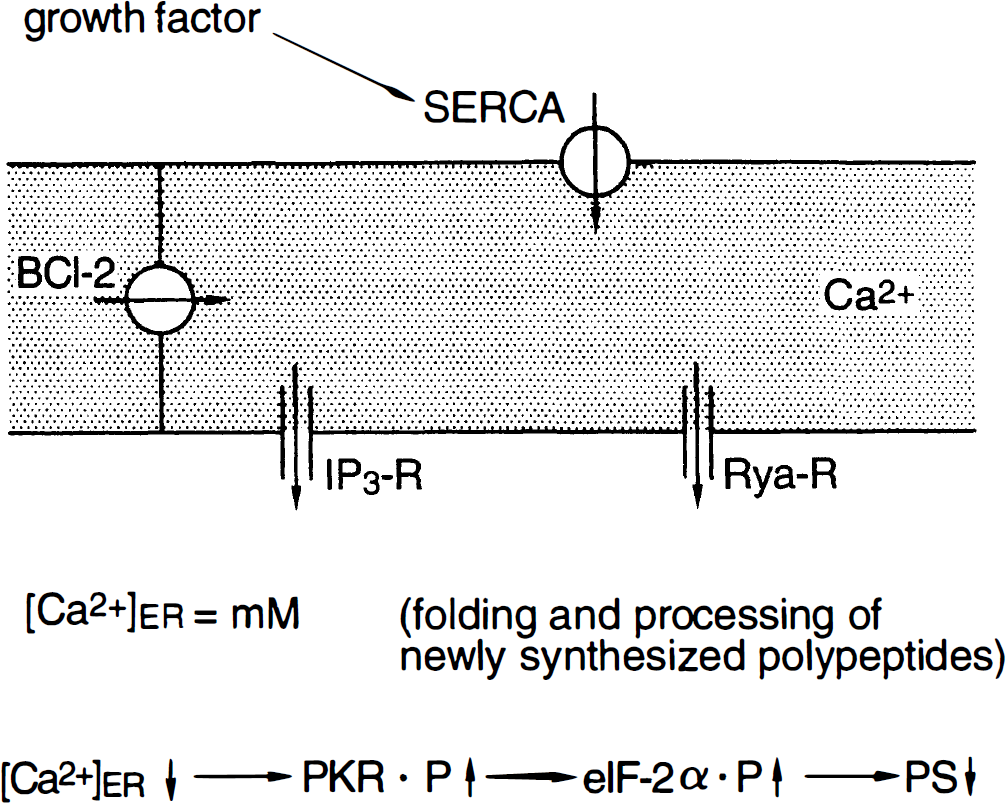
Physiologic state. Control of endoplasmic reticulum (ER) calcium homeostasis. The ER intraluminal calcium activity is in the low millimolar range. Calcium homeostasis is controlled by the inositol 1,4,5-triphosphate receptor (IP3-R) and ryanodine receptor (Rya-R), which on activation releases calcium ions from the ER, and by a Ca2+-ATPase (sarcoplasmic/endoplasmic reticulum Ca2+-ATPase [SERCA]), which pumps calcium ions back against a steep concentration gradient. Preliminary observations suggest that SERCA activity may be controlled by growth factors. With respect to calcium ions, the ER can be divided into as many as three subcompartments in which calcium release and uptake is differently regulated (see text). guanosine triphosphate facilitates calcium conductance between two of the subcompartments. Bcl-2 have a similar effect. High ER calcium activity is necessary for folding and processing of newly synthesized polypeptides. Depletion of ER calcium stores induces phosphorylation of double-stranded RNA-activated protein kinase (PKR), resulting in the phosphorylation of eukaryotic initiation factor (eIF-2α) and suppression of protein synthesis (PS).
The importance of ER calcium homeostasis for maintaining cells in their normal physiologic state is underlined by the observation that depletion of ER calcium stores by exposure to A23187 or Tg causes a block of cell division and growth arrest of cells (Gosh et al., 1991; Short et al., 1993; Waldron et al., 1994). Thus, high ER calcium activity appears to be a prerequisite for normal functioning of cells. This Tg-induced block of cell division and growth can be overcome only when Tg is washed off and the synthesis of a new ER calcium pump is activated by culturing cells in medium supplemented with high concentrations of serum. This points to an involvement of growth factors in the control of ER Ca2+-ATPase synthesis (Waldron et al., 1994). Preliminary results from our laboratory (Doutheil et al., in preparation) point to a direct effect of growth factors on ER calcium homeostasis: in primary neuronal cell cultures, serum withdrawal for 24 hours causes about a 50% decrease in ER Ca2+-ATPase activity and protein synthesis.
STRESS RESPONSE OF CELLS
Transient cerebral ischemia is a severe form of stress, causing disturbances in most biochemical and molecular-biological pathways. Cells have developed a stress response system that is highly conserved among species. If the stress response and subsequent disturbances leading to cell damage are triggered by the same signals, elucidating the basic step of the stress response should increase our knowledge of the mechanisms underlying ischemic cell injury and help to identify the disturbances that are directly related to the pathologic process. The most common response of cells to a severe form of stress is a suppression of global protein synthesis and an activation of stress gene expression. These changes have been studied in detail after transient cerebral ischemia (Kleihues et al., 1975; Cooper et al., 1977; Bodsch et al., 1985; Thilmann et al., 1986; for a review of research into gene expression see Massa et al., 1996). In the following sections, changes in protein synthesis and gene expression induced by transient cerebral ischemia are compared with those triggered by an isolated disturbances of ER function.
Protein synthesis
The pattern of ischemia-induced changes in protein synthesis has been investigated in various studies and summarized in several reviews (Kleihues et al., 1975; Cooper et al., 1977; Bodsch et al., 1985; Thilmann et al., 1986; Xie et al., 1989; Widmann et al., 1991, 1993; Bonnekoh et al., 1992; Hossmann, 1993; Hu and Wieloch, 1993; Degracia et al., 1996). Transient cerebral ischemia causes a complete cessation of protein synthesis throughout the forebrain. It then recovers in nonvulnerable brain structures such as the cerebral cortex but not in vulnerable areas such as the hippocampal CA1 subfield (Bodsch et al., 1985; Thilmann et al., 1986). Therapeutic intervention reducing ischemic cell injury (e.g., barbiturate treatment of hypothermia) did not reverse the inhibition of protein synthesis during early reperfusion but promoted the recovery of protein synthesis, even in vulnerable brain structures (Xie et al., 1989; Widmann et al., 1992), implying that it is the ability of cells to recover from this block of translation that defines the final outcome (Paschen et al., 1991). Ischemia blocks the initiation phase of protein synthesis, as indicated by an increase in the phosphorylation of the eukaryotic initiation factor, eIF-2α, suppression of the activity of the guanine nucleotide exchange factor, and disaggregation of polyribosomes (Kleihues et al., 1975; Bonnekoh et al., 1992; Hu et al., 1993; Burda et al., 1994; DeGracia et al., 1996).
It is not known whether the suppression of protein synthesis induced by transient cerebral ischemia contributes to ischemic cell injury or whether this is an epiphenomenon not directly related to the pathologic process. The observation that transient ischemia activates programmed cell death (Linnik et al., 1993; MacManus et al., 1993; Okamoto et al., 1993), an active process that can be blocked with cycloheximide, an established inhibitor of protein synthesis, argues against a direct relation. However, cycloheximide, at concentrations that only partially block protein synthesis, induces the expression and synthesis of stress genes such as Bcl-2 (Furukawa et al., 1997), implying that the protective effect of cycloheximide is brought about by activation of stress gene expression and not by suppression of protein synthesis. Furthermore, evidence shows that suppression of protein synthesis through phosphorylation of eIF-2α can mediate apoptosis directly (Srivastava et al., 1998): apoptosis induced by tumor necrosis factor-α or serum deprivation of cells could be prevented by forced expression of a mutant, nonphosphorylatable eIF-2α, whereas expression of a mutant eIF-2α (S51D), which mimics phosphorylated eIF-2α, was sufficient to induce apoptosis.
The signal transduction pathway from the signal set off by ischemia to the phosphorylation of eIF-2α has not been elucidated. Three different kinases have been identified and characterized that specifically phosphorylate eIF-2α (for review see Samuel, 1993; De Haro et al., 1996): the yeast enzyme GCN2, the heme-regulated inhibitor kinase localized predominantly in reticulocytes, and the double-stranded RNA-dependent protein kinase (PKR), which plays a key role in the defense of cells against viral infection and as a regulator of cell growth and differentiation (for reviews see Katze, 1992; Samuel et al., 1997; Gale and Katze, 1998). Notice that both tumor necrosis factor-α and serum deprivation-induced apoptosis depend on PKR activation and that cells expressing a catalytically inactive PKR are protected (Srivastava et al., 1998).
Depletion of ER calcium stores produces severe stress within cells because high intraluminal calcium levels are necessary for the folding and processing of newly synthesized proteins (see earlier). Depletion of ER calcium stores results in a suppression of protein synthesis (Fig. 2) and cell growth (Brostrom and Brostrom, 1990; Gosh et al., 1991; Short et al., 1993; Wong et al., 1993; Waldron et al., 1994; Paschen et al., 1996; Doutheil et al., 1997). Disturbances of ER calcium homeostasis induce activation of PKR (Prostko et al., 1995; Srivastava et al., 1995), which then phosphorylates eIF-2α (Prostko et al., 1992) and thus blocks the initiation process of protein synthesis, resulting in a disaggregation of polyribosomes (Fig. 3) (Wong et al., 1993; Doutheil et al., 1997). Protein synthesis also is suppressed when calcium ions are drawn out of the ER compartment by intracellular chelation (Fig. 2) (Doutheil et al., 1997), indicating that the effect on protein synthesis of agents depleting ER calcium stores is caused by a decrease in ER calcium activity and not by a corresponding increase in cytoplasmic calcium activity. Protein synthesis also can be inhibited by high cytoplasmic calcium activity (Wong et al., 1991). However, under these experimental conditions, it is the prolongation step that is suppressed, resulting in a maximal aggregation of polyribosomes (Wong et al., 1991). Furthermore, in reticulocytes, which lack the ER compartment, protein synthesis cannot be blocked by calcium chelation (Wong et al., 1991), implying that in ER-containing cells, the effect of calcium chelation on protein synthesis results from a depletion of ER calcium stores and not from a decrease in cytoplasmic calcium activity.
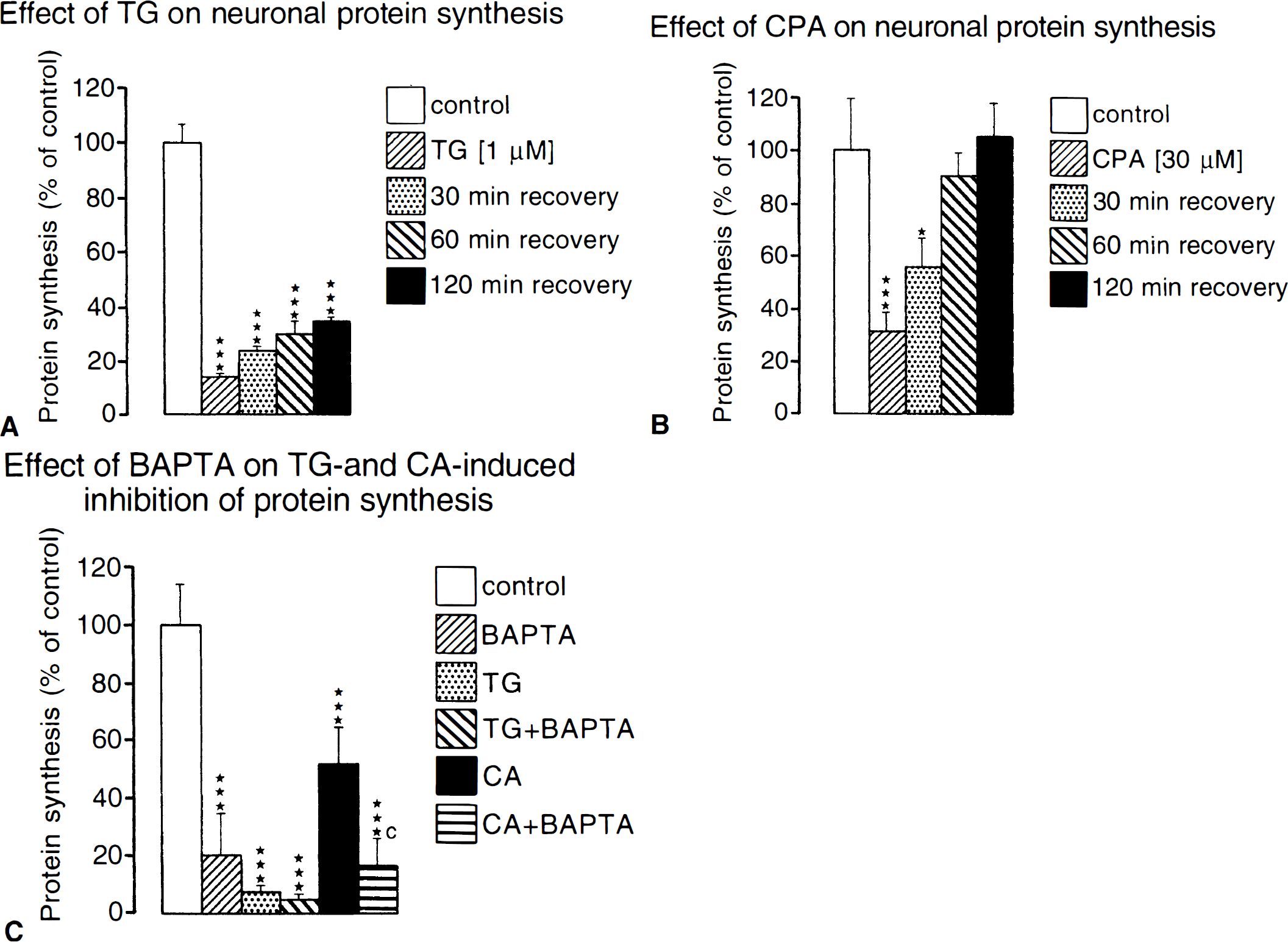
Suppression of protein synthesis induced by ER calcium pool depletion. Primary neuronal cell cultures were exposed to thapsigargin (Tg, 1 μmol/L), an irreversible inhibitor of ER Ca2+-ATPase
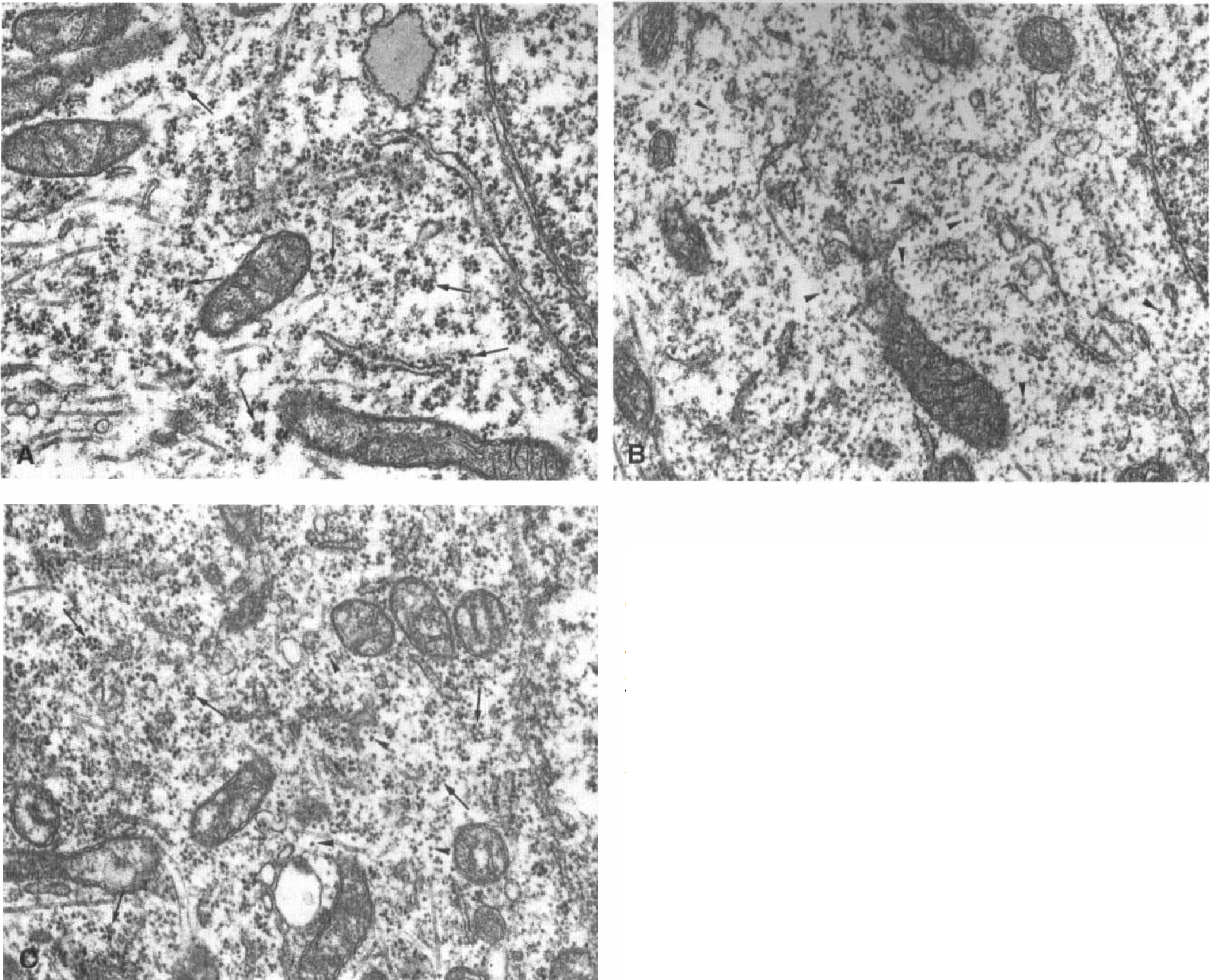
Effect of ER calcium pool depletion on ribosomal aggregation. Primary neuronal cell cultures were exposed for 30 minutes to control conditions
Gene expression
Transient cerebral ischemia activates the expression of various genes (for review see Massa et al., 1996), and the rise in cytoplasmic calcium activity during ischemia may be responsible for this stress response (Sharp and Sagar, 1994). Among these genes are those coding for grp78 and glucose-regulated protein 94 (grp94) (Wang et al., 1993; Lowenstein et al., 1994) and other ER-resident stress proteins such as hemeoxygenase-1 (Paschen et al., 1994), ER protein 72 (Paschen et al., 1998a), or the growth arrest and DNA damage-induced gene gadd153 (Paschen et al., 1998b). Since the expression of ER-resident stress proteins is specifically activated by conditions leading to perturbation of ER-specific reactions, it has been concluded that transient ischemia causes disturbances of ER function (Kuznetsov et al., 1996; Paschen, 1996).
Depletion of ER calcium stores activates the expression of several genes, including those coding for transcription factors or ER-resident stress proteins (Fig. 4) (Drummond et al., 1987; Lenormand et al., 1990; Schönthal, 1992; Li et al., 1993; Brostrom et al., 1995; Gissel et al., 1997a,b; Linden et al., 1998; Paschen et al., 1998a,b). Activation of gene expression could not be suppressed by loading cells with the calcium chelator BAPTA [tetra(acetoxymethyl)ester of 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid] (Fig. 4), which would block all possible means of raising cytoplasmic calcium activity, indicating that the effect was indeed caused by a decrease in ER calcium activity and not by a corresponding increase in cytoplasmic calcium activity (Gissel et al., 1997b; Linden et al., 1998; Paschen et al., 1998a,b). This observation is not surprising, since the expression of genes activated by a depletion of ER calcium stores also is activated in situations where ER function is disturbed without any direct changes in calcium activity, such as an inhibition of protein glycosylation or dithiothreitol-induced perturbations of the oxidative environment within the ER, which is necessary for the folding of proteins (Price and Calderwood, 1992; Brostrom et al., 1995). It can be assumed, therefore, that it is not the decrease in ER calcium activity that activates the stress response but the perturbations of ER functions resulting from a disturbance of calcium homeostasis. Interestingly, perturbations of ER function induce activation of the transcription factor NF-κB (for review see Pahl and Baeuerle, 1997), which also is activated after transient cerebral ischemia (Clemens et al., 1997).
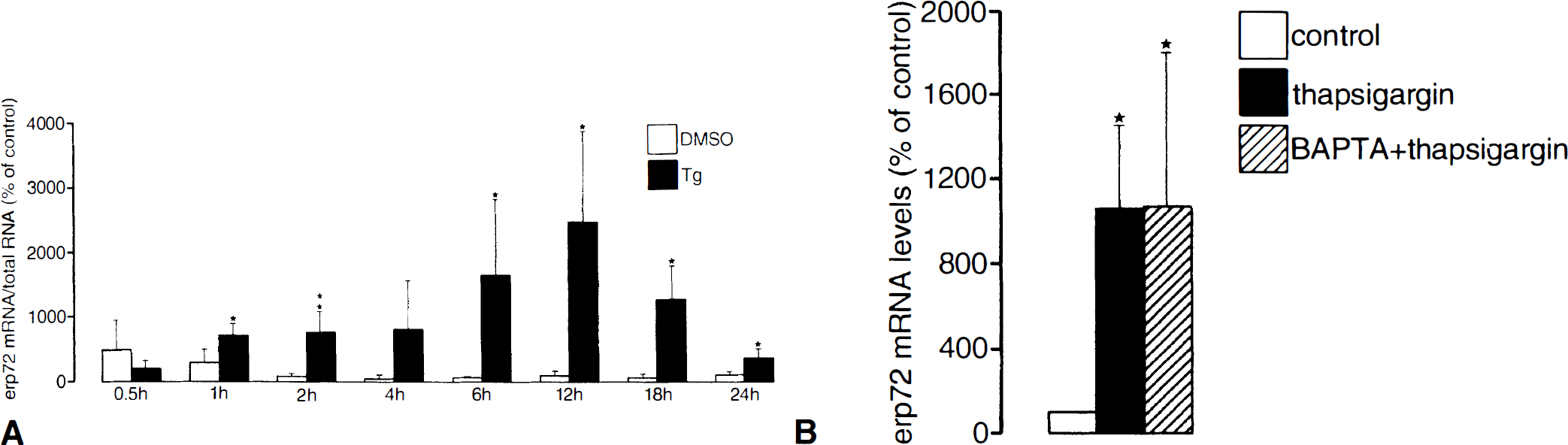
Activation of erp72 expression induced by ER calcium pool depletion
ENDOPLASMIC RETICULUM DYSFUNCTION AND ISCHEMIC CELL INJURY
Currently there is no direct evidence from in vivo experiments that ER dysfunction triggers the pathologic process underlying ischemic cell injury of neurons. However, observations from in vitro and in vivo studies indicate that ER dysfunction does play a key role in events leading to cell death, as is discussed later. Results from experiments performed in connection with acute kidney ischemia have led to the conclusion that “a key cellular lesion in ischemia is the misfolding of secretory proteins as they transit the ER” (Kuznetsov et al., 1996). Most of the available data on the role of ER dysfunction in ischemic cell injury comes from experimental studies of cerebral ischemia or myocardial ischemia (sarcoplasmic reticulum [SR] dysfunction).
Cerebral ischemia
If ER dysfunction is a causative factor in ischemic cell injury, an explanation is needed as to why certain brain structures are particularly vulnerable to transient ischemia. Further, we need to demonstrate that the stress response of cells to ER dysfunction is activated after transient cerebral ischemia, that experimental induction of disturbances of ER function causes cell damage, and that compounds known to induce or minimize ER dysfunction also aggravate or reduce ischemic cell injury.
As discussed later, the density of the IP3-R, which on stimulation gates calcium release from the ER, is particularly high in the vulnerable hippocampal CA1 subfield, whereas the immunoreactivity of the ER calcium pump is relatively low (Sharp et al., 1993; Parent and Quirion, 1994). This uneven relation between ER calcium pump and calcium release channel found in the vulnerable hippocampal CA1 subfield provides the basis for a potential imbalance between ER release and reuptake of calcium ions, particularly under pathologic conditions when cytoplasmic calcium activity is increased, because the extent of IP3-induced efflux of calcium ions from the ER is strongly affected by cytoplasmic calcium levels (Bezprozvanny et al., 1991).
As mentioned earlier, the response of cells to ER dysfunction is similar to the response induced by transient ischemia, implying common underlying mechanisms. The expression of stress genes coding for ER-resident stress proteins is activated during reperfusion after global cerebral ischemia and during focal cerebral ischemia (Wang et al., 1993; Higasi et al., 1994; Lowenstein et al., 1994; Paschen et al., 1994; Paschen et al., 1998a,b), indicating that cerebral ischemia does cause disturbances of ER function (Fig. 5). Furthermore, the observation that both after transient ischemia and after depletion of ER calcium stores, protein synthesis is suppressed by blocking of the initiation process (increased phosphorylation of eIF-2α, disaggregation of polyribosomes [Cooper et al., 1977; Bonnekoh et al., 1994; Prostko et al., 1995; Srivastava et al., 1995; DeGracia et al., 1996; Doutheil et al., 1997]) also is compatible with the supposition that transient cerebral ischemia induces disturbances of ER function.
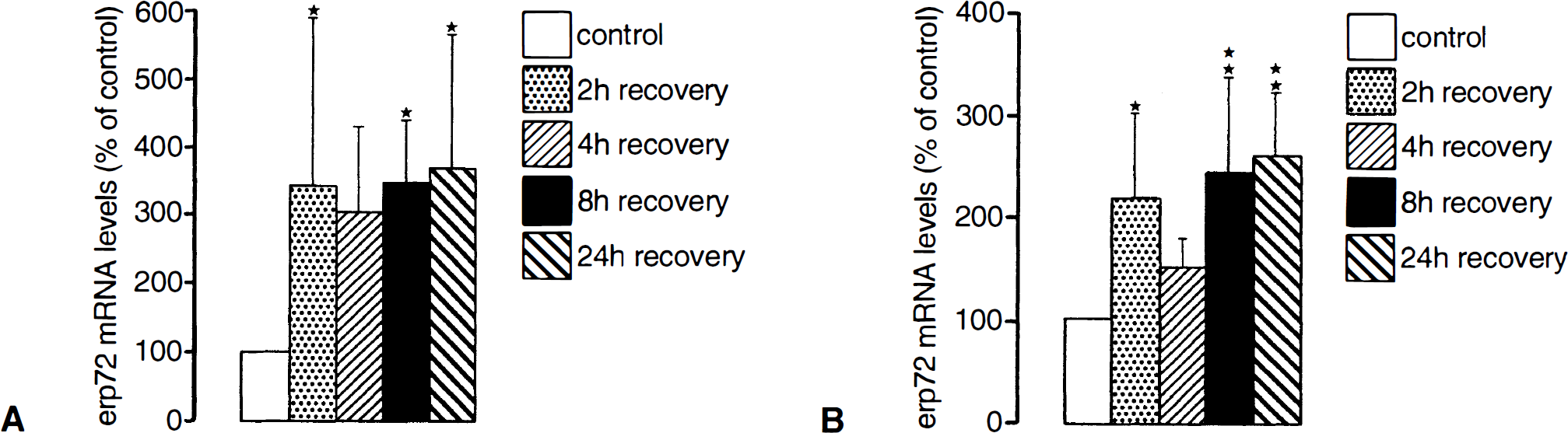
Ischemia-induced increase in erp72 mRNA levels in the cortex
Results from several experimental studies suggest that disturbances of ER calcium homeostasis play a role in the induction of neuronal cell injury caused by transient ischemia or other forms of severe stress: dantrolene, which blocks calcium release through the ER ryanodine receptor, is neuroprotective under experimental conditions (Frandsen and Schousboe, 1992; Weih and Perry, 1993; Zhand et al., 1993; Berg et al., 1995), suggesting that ER calcium pool depletion also is involved in the pathologic process. Furthermore, antagonists of group 1/5 metabotropic glutamate receptors (an activation of which results in a release of calcium ions from the ER through activation of G-protein, IP3 synthesis, and activation of IP3-R) proved to be neuroprotective, whereas agonists aggravate cell damage (Schoepp et al., 1990; Opitz and Reymann, 1991; McDonald and Schoepp, 1992; Bruno et al., 1995). Finally, intracerebroventricular infusion of group 1 metabotropic glutamate receptor agonists is sufficient to induce seizures and neuronal damage (Camon et al., 1998). Therefore, metabotropic glutamate receptors may be a suitable target for therapeutic intervention (Knopfel and Gasparini, 1996; Nioletti et al., 1996).
Several separate observations are of interest here: up-take of calcium ions into microsomes (representing the ER compartment) isolated from ischemic brains is disturbed and the extent of this disturbance correlates with the duration of ischemia (Parsons et al., 1997). However, no information is available on whether the uptake of calcium ions continues to be suppressed during reperfusion after ischemia. Also remember that the ER calcium pump is sensitive to oxidative stress (Dreher et al., 1995; Racay et al., 1995; Viner et al., 1996), which is believed to contribute to ischemic cell injury (Siesjö et al., 1995).
Further support of the suggestion that disturbances of ER function may contribute to ischemic cell injury comes from studies performed with the protein Bcl-2, which has been shown to be neuroprotective in experimental models (Zhong et al., 1993; Martinou et al., 1994; Linnik et al., 1995; Lawrence et al., 1996, 1997) and which has been linked with the development of the tolerance phenomenon in transient cerebral ischemia (Shimazaki et al., 1994). Bcl-2 is able to suppress the depletion of ER calcium stores triggered in a variety of pathologic states such as growth factor withdrawal, oxidative stress, and inhibition of the ER calcium pump (Baffy et al., 1993; Lam et al., 1994; Distelhorst et al., 1996; He et al., 1997). The Bcl-2 protein is found in the ER membrane (Reed et al., 1996), and Bcl-2 has been shown to be capable of forming ion channels in synthetic lipid membranes (Minn et al., 1997; Schendel et al., 1997), suggesting that one mode of action might involve activation of calcium fluxes within the ER. Evidence shows that the ER is divided into several subcompartments, with calcium fluxes being regulated differently in each one (Bian et al., 1994; Fasolato et al., 1991; Pizzo et al., 1997).
Myocardial ischemia
Results from experimental studies point to a role of SR dysfunction in the pathogenesis of myocardial ischemia-reperfusion injury. Detailed analysis of subcellular shifts of calcium ions occurring during myocardial ischemia and after reperfusion revealed the following pattern (Walsh and Tormey, 1988a,b): during ischemia, there was no change in calcium concentration within the different subcellular compartments studied. Shortly after onset of reperfusion, however, SR lost almost 90% of its calcium whereas mitochondrial calcium levels increased twofold. It has been shown that postischemic mechanical depression (“stunning”) is caused by damage to the SR calcium transport system (Krause et al., 1989), that SR Ca2+-ATPase activity and calcium transport remain unchanged during ischemia but are markedly lowered after reperfusion (Fukumoto et al., 1991; Zucchi et al., 1996), and that the decrease of SR Ca2+-accumulating activity by ischemia and reperfusion is more pronounced in old than in young animals (Frolkis et al., 1991). Notice that ischemia- and reperfusion-induced disturbances of calcium transport took place at the myocardial SR membrane but not on the plasma membrane (Fukumoto et al., 1991), implying a selective sensitivity of SR calcium transport system to transient ischemia. The observations that SR function is impaired on reperfusion after ischemia (Smart et al., 1997) and that SR Ca2+-ATPase is highly sensitive to oxygen free radicals (Kukreja et al., 1988) indicate that the pathologic process is mediated by radicals.
Vascular hypoxia/hypoglycemia
Vascular occlusion creates ischemic conditions in all cells within the territory supplied by the artery. Studies have therefore been carried out on the possible effects of transient ischemia on the metabolism and function of all cell types present in this tissue. After mild forms of transient ischemia, only neurons are selectively damaged, whereas nonneuronal cells are spared (Kirino, 1982). However, the regulation of cerebral blood flow is impaired during reperfusion, leading to delayed positischemic hypoperfusion (for review see Hossmann, 1997). Of interest, then, is whether a disturbance of SR calcium transport contributes to this process. It has already been shown that functional elimination of the SR compartment caused marked contraction of cerebral arteries (Asano et al., 1996), whereas hypoxia/reoxygenation impairs vasoconstrictor responses that are dependent on the release of calcium from the SR (Gao et al., 1996), indicating depletion of SR calcium stores and implying that SR dysfunction contributes to postischemic hypoperfusion.
ENDOPLASMIC RETICULUM CALCIUM HOMEOSTASIS AND APOPTOSIS
Thapsigargin, an irreversible inhibitor of ER Ca2+-ATPase, has been used in experimental studies performed on neuronal and nonneuronal cells designed to investigate the possible involvement of ER dysfunction in the initiation of apoptosis (Tsukamoto and Kaneko, 1993; Furuya et al., 1994; Kaneko and Tsukamoto, 1994; Lam et al., 1994; Takei and Endo, 1994; Waring and Beaver, 1996; Bian et al., 1997; He et al., 1997; McCormick et al., 1997; Nath et al., 1997; Preston et al., 1997; Qi et al., 1997; Takadera and Ohyashiki, 1998; Wei et al., 1998; Zhou et al., 1998). In all studies, exposure of cells to Tg triggered the process of programmed cell death. Exposure of cells to Tg causes a depletion of ER calcium stores and a corresponding increase in cytoplasmic calcium activity in two phases: a short-lasting peak resulting from the release of calcium ions from the ER, and a longer lasting plateau resulting from the capacitative influx of calcium ions needed to refuel ER calcium stores (Petersen et al., 1995). The observation that Tg induces apoptosis does not therefore clarify whether this process is initiated by the depletion of ER calcium stores or by the rise in cytoplasmic calcium activity. However, several independent observations indicate that a decrease of ER calcium activity is an important triggering event for the induction of apoptosis: i) removal of extracellular calcium augments Tg-induced apoptosis, and dexamethasone-induced apoptosis caused a partial depletion of ER calcium stores (Bian et al., 1997); ii) dantrolene, an inhibitor of the ER ryanodine-sensitive calcium-release channel, suppresses Tg-induced apoptosis, implying that ER calcium pool depletion contributes to this process (Nath et al., 1997); iii) Tg-induced apoptosis causes a decrease of ER calcium activity, a pathologic process which is prevented in Bcl-2-overexpressing cells, indicating that Bcl-2 helps to maintain ER calcium homeostasis (Lam et al., 1994); iv) evidence has been presented that sustained elevation of cytosolic calcium activity brought about by capacitive entry of calcium ions is not required for the induction of apoptosis by Tg (He et al., 1997); and v) in stage I preneoplastic cells, which are highly susceptible to apoptosis, the capacitative calcium entry at the cell membrane is diminished, resulting in decreased ER calcium pools (Preston et al., 1997). In this model, apoptosis could be blocked by raising ER calcium levels, again implying that ER calcium pool depletion is one of the factors contributing to apoptosis (Preston et al., 1997).
Several separate observations are of interest here: 1) cells deficient in IP3-R are resistant to apoptosis (Jayaraman and Marks, 1997; Sugawara et al., 1997), implying that IP3-induced calcium release from the ER plays a role in the induction of apoptosis; 2) resistance to apoptosis correlates closely with induction of ER-resident chaperons grp78/grp94, and cells undergoing Tg-induced apoptosis fail to generate the grp78/grp94 stress response (McCormick et al., 1997); 3) just as cell injury induced by transient cerebral ischemia is brought about by activation of caspase-3 (Holtzman et al., 1996; Endres et al., 1998), Tg-induced apoptosis also is accompanied by activation of caspase-3 and suppressed by caspase inhibitors such as Z-VAD-fmk or nerve growth factor (Qi et al., 1997; Namura et al., 1998; Takadera and Ohyashiki, 1998), implying common underlying mechanisms; and 4) apoptosis induced by exposure of cells to Tg is suppressed by the immunosuppressive drug CsA, depending on the Tg concentrations used (Waring and Beaver, 1996); CsA was effective in cells exposed to 2 nmol/L Tg, which caused apoptosis in the presence of intact mitochondria, but ineffective after exposure to 10 nmol/L Tg, a concentration that caused mitochondrial damage (Waring and Beaver, 1996). These observations illustrate that the neuroprotective effect of CsA observed in models of transient cerebral ischemia (for review see Kristian and Siesjö, 1998) does not necessarily arise from an inhibition of mitochondrial damage.
An interesting feature of the mechanisms triggering apoptosis is the observation that the apoptotic process activated in neurons by growth factor withdrawal is almost identical to that induced by viral infection (Allsopp et al., 1998). The response of cells to viral infection has been studied in detail (Jagus and Gray, 1994; Gale and Katze, 1997, 1998). Viruses activate the interferon system of the host cell. The basic strategy underlying the induction of the interferon system is to trigger a state of growth arrest so that infected cells cannot be forced to produce viral proteins, and to activate the process of programmed cell death so that infected cells can be safely removed. A similar pattern of apoptotic changes induced in neurons by growth factor withdrawal or viral infection suggests that the same process is activated in these two situations. Our knowledge of the response of cells to viral infection may therefore help to elucidate the mechanisms underlying the induction of apoptosis in other pathologic states, such as growth factor withdrawal or transient cerebral ischemia.
Many of the observations discussed earlier in connection with the induction of apoptosis and the possible role of disturbances of ER calcium homeostasis in this process are based on experimental studies performed on nonneuronal cells. Whether neurons react in the same way as nonneuronal cells under such conditions remains to be established.
ENDOPLASMIC RETICULUM AND DEGENERATIVE DISEASE
Disturbances of calcium homeostasis play a major role in degenerative diseases (Peterson et al., 1989; Mattson et al., 1993; Mattson, 1994; for reviews see Beal, 1995), and more than one line of evidence points to an involvement of ER in this process. In Alzheimer's disease (AD), the amyloid precursor protein is cleaved by proteases forming the amyloidogenic β-protein Aβ. Depending on the cleavage site, two different peptides are formed, Aβ(1–40) and Aβ(1–42). Even slight increases in levels of Aβ(1–42) may be sufficient to cause AD (Younkin, 1995; Hardi, 1997). The ER has been identified as the site of Aβ(1–42) formation, and the earliest event to take place in AD may be the generation of Aβ(1–42) in the ER (Hartmann et al., 1997). This observation may explain why proteins localized in the ER, such as the (mutated) presenilins, induce AD (Kovacs et al., 1996). Indeed, the amyloid precursor protein forms an immunocomplex with presenilin 2, suggesting that presenilins are involved in the processing of amyloid precursor protein molecules (Weidemann et al., 1997).
The form of prion protein transmitting prion disease also is localized on the ER: a transmembrane form on the ER membrane produced neurodegeneration in mice, from which it was concluded that aberrant regulation of protein biogenesis and changes in the topology of the ER can result in prion-induced neurodegeneration (Hedge et al., 1998). Notice that intracellular formation of Aβ is confined to neuronal cells, and that nonneuronal cells produce Aβ(1–40) and Aβ(1–42) only at the cell surface (Hartmann et al., 1997). The site of Aβ(1–42) production then may play a major role in the pathologic process culminating in AD. Aβ peptides exhibit ionophore-like properties (Engström et al., 1995) that can cause an increase in the permeability of membranes to calcium. Exposure of neurons to Aβ peptide induces a rise in cytoplasmic calcium activity and a severe suppression of carbachol-induced release of calcium ions from the ER, suggesting that the toxic effect of Aβ peptides is brought about, among other factors, by depletion of ER calcium stores (Kelly et al., 1996; Samochocki, 1998).
Several observations point to a causative link between morphologic and biochemical changes within the ER and the onset of degenerative diseases: exposure of neuronal cell cultures to Aβ results in disaggregation of polyribosomes and partial disintegration of ER (Watt et al., 1994); cells of patients with AD have lower activities of the enzyme NADH cytochrome-C reductase (which is bound to the ER membrane) but normal activities of the cell membrane-resident phosphodiesterase (Hajimohammadreza et al., 1990), suggesting a connection between ER dysfunction and AD (Zubenko et al., 1990); the agonist-induced release of calcium from internal stores is blocked by Aβ (Kelly et al., 1996) and also is reduced in patients with amytrophic lateral sclerosis (Witt et al., 1994); and AD causes a breakdown of phospholipids (Farooqi et al., 1995), which may be one of the underlying causes of ER dysfunction. Finally, dantrolene, which blocks the ER calcium release channel activated by ryanodine, reduces the degeneration of motor neurons, which are destroyed in amytrophic lateral sclerosis (Rothstein and Kuncl, 1995), indicating that ER calcium pool depletion contributes to the pathologic process causing this disease.
ROLE OF ENDOPLASMIC RETICULUM IN THE INDUCTION OF TOLERANCE
Cells subjected to transient nonlethal stress can develop tolerance to other lethal forms of stress (preconditioning). The observation that pre-exposing brains to different forms of nonlethal stress (i.e., heat-shock, transient ischemia, or spreading depression) induces tolerance and prevents ischemic cell damage (Kirino et al., 1991; Kitagawa et al., 1991; Kobayashi et al., 1995) implies that common underlying mechanisms are involved. Elucidating the mechanisms responsible for the activation of the tolerance phenomenon may further our understanding of the pathologic processes leading to ischemic cell injury.
Induction of heat-shock protein synthesis plays a major role in the tolerance phenomenon (Kirino et al., 1991). This assumption is based on the following observations: a lethal 5-minute period of transient forebrain ischemia in gerbils activates synthesis of heat-shock protein 70 (hsp70) in all brain structures except the vulnerable CA1 subfield (Kirino et al., 1991; Nowak et al., 1993); and preconditioning with mild ischemia of 2 minutes' duration induces the synthesis of hsp70 even in the CA1 subfield (Kirino, et al., 1991), and hsp70 immunoreactivity is still present in this area when, after induction of tolerance, the animal is subjected to 5-minute ischemia (Kirino et al., 1991). However, tolerance also can be activated in neurons by nonlethal heat-shock (Vogel et al., 1997), an experimental procedure which stimulates the synthesis of hsp70 in glial cells but not in neurons (Marini et al., 1990; Vogel et al., 1997), implying that hsp70 synthesis is not the main contributory factor.
The supposition that tolerance can be induced by activation of the ER-resident stress response system alone is based on the following observations: 1) ER stress protein expression induced by a nonlethal form of stress produces tolerance to lethal stress (Brostrom et al., 1990); 2) protein synthesis of cells constitutively expressing high levels grp78 is resistant to inhibition by EGTA, calcium ionophore A23187, or dithiothreitol (Brostrom et al., 1990); 3) transfection of cells with a grp78 antisense vector eliminates resistance to lethal stress (Li and Lee, 1991); and 4) deletion of grp78 is lethal to cells (Rose et al., 1989). The view that ER-resident stress protein grp78 plays a more important role in the induction of tolerance than cytoplasmic heat-shock proteins is illustrated by the following observation: trans-4,5-dihydroxy-1,2-dithiane, Tg (an irreversible inhibitor of the ER Ca2+-pump), or tunicamycin (an inhibitor of the N-linked glycosylation that takes place in the ER), three drugs that enhance the expression of the ER chaperons grp78 and grp94 (Liu et al., 1997), protect cells from iodoacetamide toxicity, whereas a prior heat-shock did not (Liu et al., 1997). These results are based on studies performed exclusively on nonneuronal cells, but no evidence suggests that, with regard to the role of these genes in coding for ER-resident stress proteins, neurons react differently to nonneuronal cells. Preliminary results from our laboratory suggest that neuronal protein synthesis can accommodate to disturbances of ER calcium homeostasis by a transient ER calcium pool depletion (Fig. 6). The tolerance induced in neurons by a transient EGTA exposure was, however, clearly less compared with that found in stable nonneuronal cells (Brostrom et al., 1990).
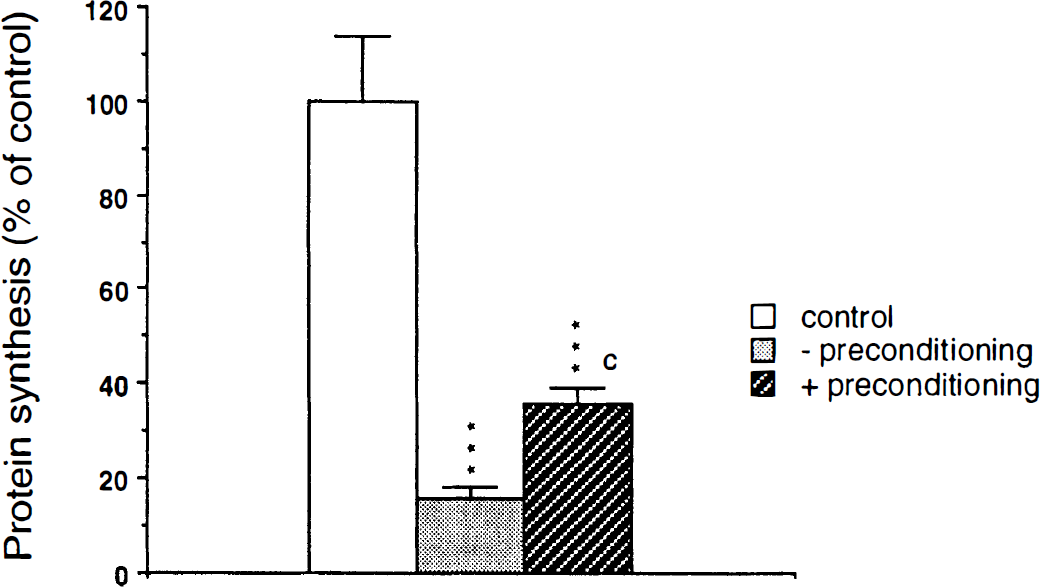
Suppression of protein synthesis induced by ER calcium pool depletion. Induction of tolerance by transient ER calcium pool depletion. Primary neuronal cell cultures were exposed for 150 minutes to calcium-containing (− tolerance) or calcium-free buffer supplemented with 1 mmol/L EGTA (+ tolerance), followed by 5 hours of recovery. Cells then were exposed for 30 minutes to calcium-free medium supplemented with EGTA, and protein synthesis was evaluated in the same medium. Control cells were incubated in calcium-containing medium throughout the experiment. After transient exposure of cells to calcium-free medium, suppression of protein synthesis induced by a depletion of ER calcium stores was significantly less pronounced, indicating an accommodation of protein synthesis to stress. Data are presented as means ± SD. Statistically significant differences between protein synthesis of control cultures and cultures exposed to calcium-free medium are indicated by ***P < 0.001, and between protein synthesis of cultures exposed to calcium-free medium with or without prior preconditioning are indicated by c P < 0.001 (ANOVA, followed by Scheffe's F-test).
IS THE ENDOPLASMIC RETICULUM SENSITIVE TO TRANSIENT METABOLIC STRESS?
To prove that disturbed ER calcium homeostasis is involved in the development of cell damage induced by ischemia, it is important to establish whether this intracellular compartment is particularly sensitive to transient metabolic stress. Furthermore, it is necessary to know why transient cerebral ischemia causes significant damage to vulnerable brain structures such as the hippocampal CA1 subfield but relatively little injury to nonvulnerable structures such as the neocortex. Ischemiainduced disturbances of ER calcium homeostasis could be triggered by two different mechanisms: by an excess release of calcium ions from the ER, or by an inhibition of the calcium-ATPase necessary for the re-uptake of calcium ions into the ER. As discussed earlier, the neuroprotective effects produced by antagonists of group 1/5 subtypes of metabotropic glutamate receptors (blocking of IP3 formation) and of the ryanodine receptor suggest that after ischemia, an imbalance arises between the release and re-uptake of ER calcium.
The IP3-R density varies considerably in different forebrain structures, as revealed by 3H[IP3] binding, with the highest density occurring in the vulnerable hippocampal CA1 subfield (Sharp et al., 1993; Parent and Quirion, 1994). The immunoreactivity of ER calcium ATPase, on the other hand, is lower in the hippocampal CA1 subfield than in the CA2 to CA4 subfields (Sharp et al., 1993). It can therefore be assumed that the vulnerable hippocampal CA1 subfield is a forebrain region particularly prone to an imbalance between ER release and reuptake of calcium. This imbalance has been illustrated by Verma et al. (1992), who used frozen tissue sections of rat brain to trace the uptake of calcium ions into the ER and the distribution of intracellular calcium pools mobilized by IP3 (Verma et al., 1990): when calcium uptake was investigated in the presence of IP3, a net uptake was still detectable in the hippocampal CA2 to CA4 subfields but not in the CA1 subfield, indicating that under these experimental conditions, IP3-induced calcium release from the ER exceeded calcium uptake into the ER. This imbalance may be aggravated even further after transient ischemia, when the calcium uptake capacity of ER is diminished (Parson et al., 1997).
It is not known whether, after transient cerebral ischemia, disturbances of ER calcium homeostasis occur in all the cells in tissue subjected to transient reductions in perfusion. For a better understanding of the pathologic process involved, it is important to know whether the ER calcium homeostasis in vulnerable cells such as neurons is particularly sensitive to a transient form of stress. Another possible reason for the selective vulnerability of neurons is that the capacity to recover from disturbances of ER calcium homeostasis is less developed in neurons than in nonneuronal cells. The observation that Tg dose-dependently causes pan-necrosis when injected into the brain of perinatal rats (Silverstein and Nelson, 1992) suggests that all of the different types of cells in the brain are equally sensitive to an irreversible inhibition of ER Ca2+-ATPase. It can be assumed, therefore, that irreversible disturbances of ER calcium homeostasis are induced only in cells destined to die after ischemia. Under more damaging conditions (i.e., when the duration of ischemia is prolonged or induction of damage aggravated by hyperthermia or hyperglycemia (Siesjö and Smith, 1991; Lundgren et al., 1994; Kristian and Siesjö, 1996) irreversible disturbances of ER calcium homeostasis also may be induced in cells that are spared after mild forms of metabolic stress.
CONTROL OF CELL GROWTH AND DEATH BY ENDOPLASMIC RETICULUM CALCIUM HOMEOSTASIS
Evidence has been presented that disturbances of ER calcium homeostasis may be lethal to cells. As discussed earlier, depletion of ER calcium stores induces a state of growth arrest and cell death (Fig. 7A). At first sight, it is difficult to understand the physiologic role of the suppression of protein synthesis induced by ER calcium pool depletion because under physiologic conditions, calcium ions also are released from the ER in various states of cell activation: on activation, extracellular glutamate levels are increased, resulting in an IP3-induced ER calcium release. Indeed, exposing hippocampal slices to glutamate causes a moderate suppression of protein synthesis (Djuricic et al., 1994). However, the calcium spikes resulting from an activation of the ER IP3-R and the suppression of protein synthesis probably are only short-lasting phenomena (Djuricic et al., 1994; for a review on calcium release see Ogden, 1996), which may not affect protein synthesis significantly over a longer period of time. In addition, calcium ions are taken up by the ER, and intraluminal calcium levels increase immediately after activation (Pozzomiller et al., 1997). Furthermore, the temporary suppression of the initiation process of protein synthesis may facilitate translation of certain mRNA species, since during polyribosomal disaggregation, all mRNA molecules bound to the ribosomes and involved in the translational process are released. In the physiologic state, when temporary ER calcium pool depletion has caused a block of protein synthesis at the initiation step, the re-initiation of protein synthesis may be more closely connected with activation processes than the suppression of protein synthesis itself. On the other hand, evidence indicates that it is not the ER calcium pool sensitive to activation of the IP3-R that controls protein synthesis (Preston and Berlin, 1992; Alcázar et al., 1997), suggesting that the calcium store sensitive to ryanodine receptor activation may play a more prominent role in this process.
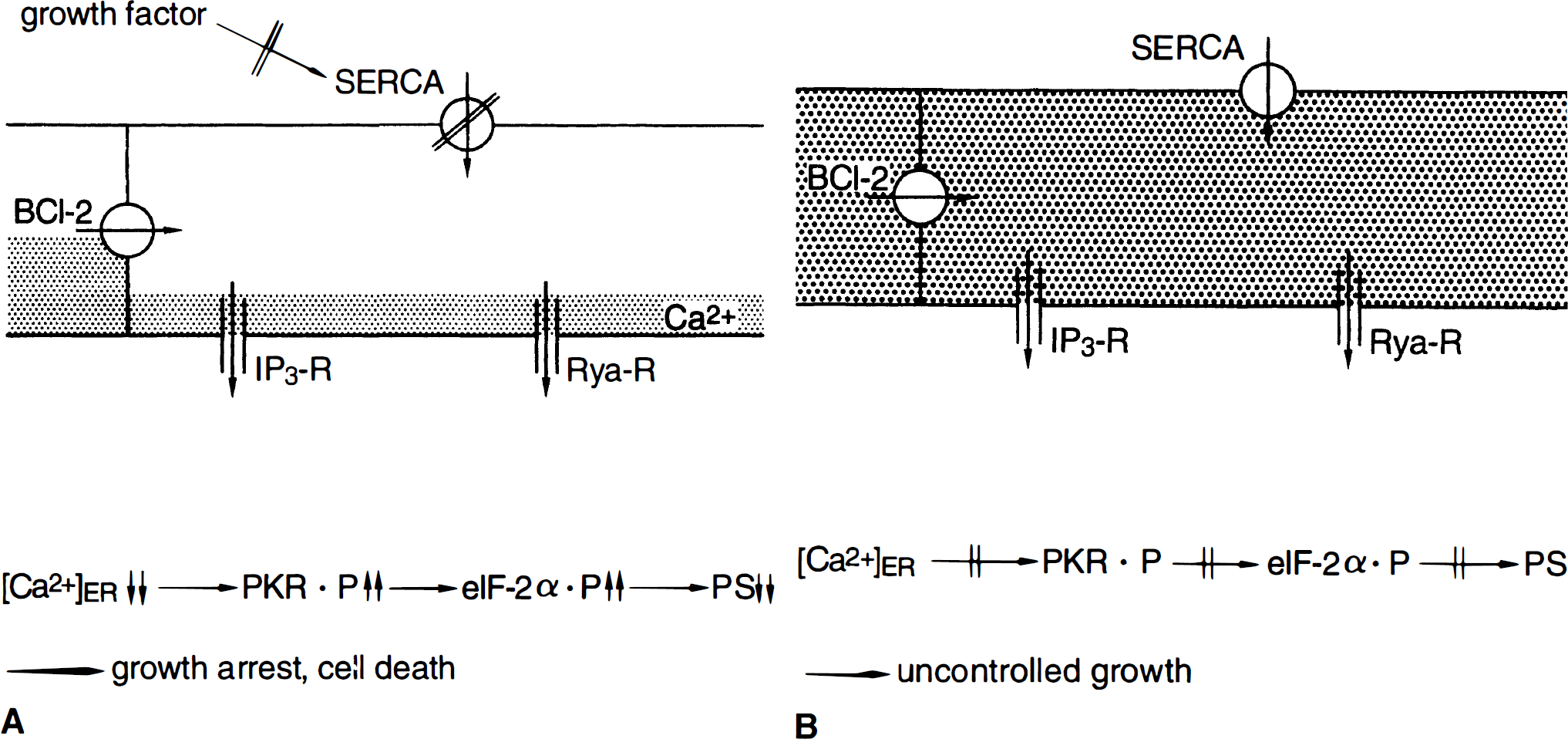
Regulatory link between ER calcium homeostasis and protein synthesis.
The control of protein synthesis by ER calcium homeostasis may not only be an important means of keeping cells balanced between the physiologic state and a state of growth arrest and cell death, but also between the physiologic state and a state of uncontrolled growth (Fig. 7B). Manipulations that sever the regulatory link between ER calcium homeostasis and translational initiation (suppression or mutation of PKR, or mutation of eIF-2α at 51Ser to 51Ala, which cannot be phosphorylated) induce a transition from a state of controlled to a state of uncontrolled proliferation (Meurs et al., 1993; Koromilas et al., 1992; Barber et al., 1994; Mundschau and Faller, 1994; Donze et al., 1995). It can be concluded, therefore, that the potentially negative control of protein synthesis by ER calcium homeostasis plays an important role in stabilizing cells in a physiologic state, and that cells in which this regulatory link is blocked will enter a state of uncontrolled growth (Fig. 8).
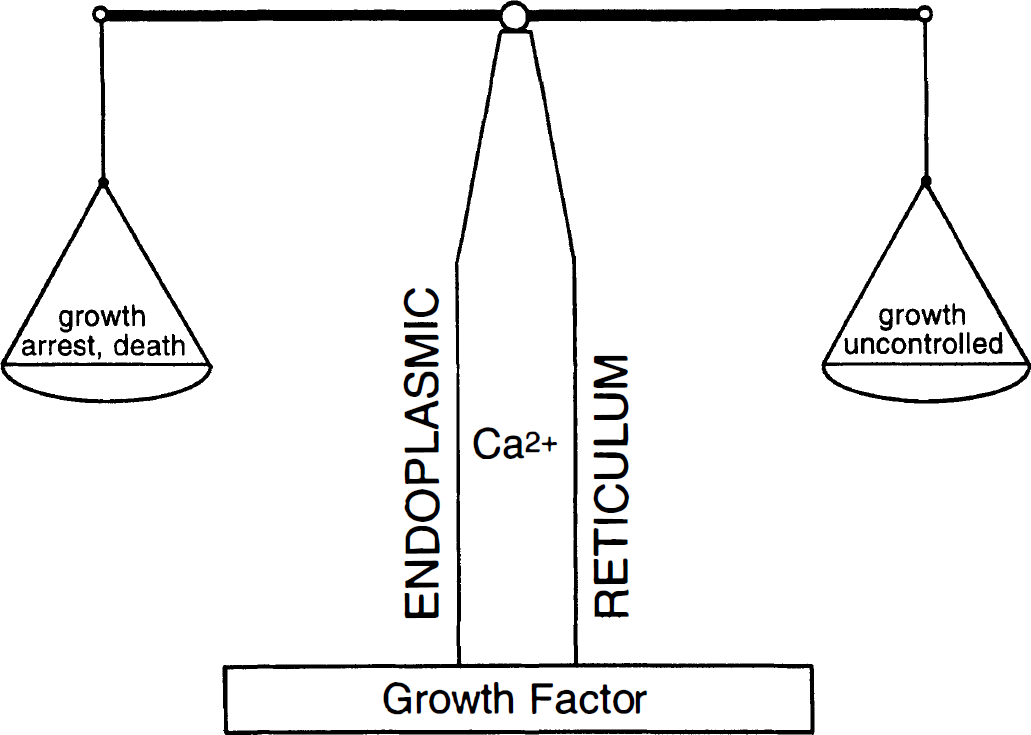
Diagram illustrating the putative role of ER Ca2+ homeostasis in maintaining cells in a physiologic state. In the physiologic state, ER calcium homeostasis is maintained and the regulatory ink between ER calcium pools and protein synthesis is intact. Growth factors help cells to keep this system in balance. In pathologic states, ER calcium pools are depleted, resulting in growth arrest and, eventually, cell death. On the other hand, when the regulatory link between ER calcium pools and protein synthesis is blocked, cells are turning into a state of uncontrolled growth.
CONCLUSION
In this review, evidence has been presented that disturbances of ER function may contribute significantly to cell injury: depletion of ER calcium stores without any parallel increase in cytoplasmic calcium activity is sufficient to induce cell death. In addition, many observations from in vitro and in vivo studies indicate that disturbances of ER function play a role in the pathologic process leading to cell death. Depending on which cell types are affected and whether calcium homeostasis or protein processing of ER/SR is the primary target, ER dysfunction may result in apoptosis or necrosis, heart failure, vasoconstriction, or amyloid plaque formation (Fig. 9). The molecular mechanisms underlying ER dysfunction remain to be established. Knowledge of these mechanisms will improve the chances of finding therapeutic strategies that will interfere directly with the pathologic process itself. Therapeutic interventions could be directed against the ER or downstream processes that are influenced by ER dysfunction, such as PKR or PKR-dependent reactions like protein synthesis or the expression of those genes triggered directly or indirectly by activated PKR. In this endeavor, there may be something to be learned from the strategies adopted by cells to combat viral infection or by viruses to overcome the antiviral defense mechanisms set up in infected cells (Galvan and Roizman, 1998). The finding that the pattern of apoptotic changes induced in primary cultures of sensory neurons by growth factor deprivation and viral infection are identical implies common underlying mechanisms (Allsopp et al., 1998) and makes this line of research highly promising.
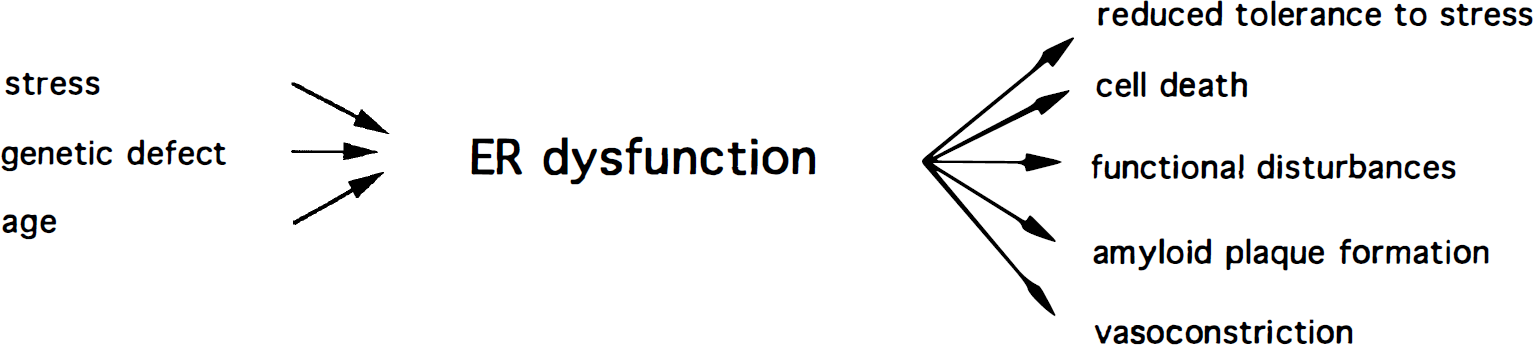
Diagram summarizing the possible effects of endoplasmic/sarcoplasmic reticulum (ER/SR) dysfunction. Depending on the molecular basis of disturbances and on the cells affected, ER/SR dysfunction may induce various patholog ic changes.