
Abstract
Cells respond to conditions associated with endoplasmic reticulum (ER) dysfunction with activation of the unfolded protein response, characterized by a shutdown of translation and induction of the expression of genes coding for ER stress proteins. The genetic response is based on IRE1-induced processing of xbp1 messenger RNA (mRNA), resulting in synthesis of new XBP1proc protein that functions as a potent transcription factor for ER stress genes. xbp1 processing in models of transient global and focal cerebral ischemia was studied. A marked increase in processed xbp1 mRNA levels during reperfusion was observed, most pronounced (about 35-fold) after 1-h occlusion of the right middle cerebral artery. The rise in processed xbp1 mRNA was not paralleled by a similar increase in XBP1proc protein levels because transient ischemia induces severe suppression of translation. As a result, mRNA levels of genes coding for ER stress proteins were only slightly increased, whereas mRNA levels of heat-shock protein 70 rose about 550-fold. Under conditions associated with ER dysfunction, cells require activation of the entire ER stress-induced signal transduction pathway, to cope with this severe form of stress. After transient cerebral ischemia, however, the block of translation may prevent synthesis of new XBP1proc protein and thus hinder recovery from ischemia-induced ER dysfunction.
Keywords
A key feature of the brain pathobiochemistry triggered by transient cerebral ischemia is a suppression of protein synthesis (Kleihues et al., 1975; Cooper et al., 1977). In vulnerable neurons this is an irreversible process, whereas in resistant cells protein synthesis recovers during reperfusion (Bodsch et al., 1985; Thilmann et al., 1986). The ability of neurons to restore normal rates of translation thus defines the final outcome after ischemia. It has therefore been assumed that shutdown of translation and the inability of vulnerable neurons to restore normal protein synthesis rates after ischemia contributes to the pathologic process culminating in ischemic cell injury (Hossmann, 1993).
After transient cerebral ischemia, protein synthesis is suppressed at the initiation step, as indicated by phosphorylation of the alpha subunit of the eukaryotic initiation factor (eIF2α) and disaggregation of polyribosomes (Cooper et al., 1977; Bonnekoh et al., 1992; Burda et al., 1994; DeGracia et al., 1996; Althausen et al., 2001). The only eIF2α kinase identified so far as being phosphorylated and thus activated after transient cerebral ischemia is the PKR-like endoplasmic reticulum (ER) kinase (PERK) (Kumar et al., 2001). PKR-like endoplasmic reticulum kinase is a kinase specifically activated under conditions associated with ER dysfunction (Harding et al., 1999), implying that transient cerebral ischemia indeed causes disturbance of ER functions as suggested earlier (Paschen, 1996; Paschen and Doutheil, 1999; Paschen and Frandsen, 2001).
A major function of the ER is the folding and processing of membrane and secretory proteins (Lodish and Kong, 1990; Gosh et al., 1991; Kuznetsov et al., 1992; Lodish et al., 1992). The importance of this process for the correct functioning of cells is indicated by the observation that blocking of these reactions is lethal to cells (for a review see Kaufman, 1999). To cope with conditions associated with ER dysfunction, cells have developed a highly conserved stress response, known as the unfolded protein response (UPR), which is activated when unfolded or misfolded proteins accumulate in the ER lumen. The unfolded protein response is characterized by an activation of two ER membrane-resident proteins, PERK and IRE1. After being activated, PERK induces phosphorylation of eIF2α (Harding et al., 1999), whereas the endonuclease IRE1 induces processing of xbp1 messenger RNA (mRNA) (Shen et al., 2001; Yoshida et al., 2001; Calfon et al., 2002). In nonstressed cells, xbp1 mRNA is translated into 33-kd X-box protein 1 (XBP1). In cells with disturbed ER function, when xbp1 mRNA is processed by activated IRE1, a short sequence of 26 bases is cut out of the coding region of xbp1 mRNA. This leads to a shift of the open reading frame of the message, resulting in the formation of a new 54-kd processed XBP1 protein (XBP1proc), which translocates into the nucleus and functions as an active transcription factor inducing expression of ER stress genes such as grp78 and grp94 (Calfon et al., 2002).
Here we describe activation of xbp1 processing after transient cerebral ischemia. Since protein synthesis is severely suppressed after ischemia, activation of xbp1 processing was not followed by a similar increase in mRNA levels of ER stress genes. Block of translation may thus be a key factor limiting postischemic recovery of neurons from ischemia-induced ER dysfunction.
MATERIALS AND METHODS
Animal experiments
C57 black mice weighing 23 to 27 g or male Wistar rats weighing 200 to 250 g were used for this study. Animals were housed under diurnal conditions and were allowed free access to water and food. Animal experiments were carried out according to the National Institutes of Health guidelines for the care and use of laboratory animals and were approved by the local authorities. Focal cerebral ischemia was induced in mice by occluding the right middle cerebral artery (MCA) using the intraluminal filament technique, as described elsewhere (Hata et al., 1998). Animals were anesthetized with 1% halothane in 70% N2O and 30% O2. An 8–0 nylon monofilament coated with silicon resin was introduced through a small incision into the right common carotid artery and was advanced 9 mm distal to the carotid bifurcation for occlusion of the right MCA. Body temperature was controlled throughout the experiments and kept at 37 ± 0.5°C using a feedback-controlled heating system. Middle cerebral artery occlusion–induced changes in cortical blood flow and the extent of reperfusion were verified by laser-Doppler flowmetry. After 1-h MCA occlusion and an additional 1, 3, or 6 h of reperfusion, animals were reanesthetized and brains were frozen in situ as described earlier (Pontén et al., 1973).
Ischemia-induced changes in protein synthesis were evaluated by measuring the incorporation of radioactively labeled leucine into brain proteins. Forty-five minutes before the animals were killed, they were given l-[4,5–3H]leucine (150 μCi/animal, specific activity 150 Ci/mmol; Amersham Buchler, Braunschweig, Germany). After in situ freezing, coronal sections of 20 μm thickness were cut in a cryostat at −20°C. Sections were washed for 30 minutes in perchloric acid solution (0.3 mmol/L)/EGTA (1 mmol/L) to remove label that had not been incorporated into proteins. Radioactivity incorporated into proteins was measured autoradiographically by exposing sections to tritium-sensitive film (Hyperfilm 3H; Amersham Buchler) for 2 weeks.
Transient global cerebral ischemia was induced in rats anesthetized with halothane in 70% N2O and 30% O2, using the four-vessel occlusion model (Pulsinelly and Brierley, 1979) with modifications (Schmidt-Kastner et al., 1989). After 30 minutes of ischemia, brains were recirculated for periods of 2, 4, 8, or 24 h. At the end of the experiments, animals were reanesthetized, and brains were frozen in situ (Pontén et al., 1973).
Cell culture
Primary neuronal cell cultures were prepared from brains of embryonic Wistar rats at days 15 to 17 of gestation, essentially as described elsewhere (Paschen et al., 1996). Cortices were dissected out and the tissue was dissociated by incubation with trypsin and DNAse I for isolation of neurons. Cells were suspended in minimal essential medium, supplemented with 5% horse serum, 30 mmol/L glucose, 2 mmol/L glutamine, and 10 IU/mL penicillin, and 10 ng/mL streptomycin (Gibco BRL, Eggenstein, Germany). Cells were plated on polyethyleneimine-coated dishes and were used for experiments after 10 d in vitro.
To investigate xbp1 processing under conditions known to be associated with severe ER stress, cultures were exposed to thapsigargin (Tg, 1 μmol/L; Calbiochem-Novabiochem, Bad Soden, Germany), a specific and irreversible inhibitor of ER Ca2+-ATPase (Inesi and Sagara, 1994), or to the Tg solvent dimethyl sulfoxide (0.1%). In additional experiments, cultures were exposed to the proteasome inhibitor MG-132 and to the proteasome inhibitor I (20–100 μmol/L; Calbiochem-Novabiochem) to investigate whether proteasome inhibition activates xbp1 processing. At the end of experiments, cultures were washed with phosphate-buffered saline solution, the medium was completely aspirated, and cultures were stored at −80°C before being used for analysis.
Analysis of messenger RNA levels
Brains were removed in a cold temperature cabinet set at −20°C. Tissue samples were excised from the right and left hemisphere from the territory supplied by the MCA (mice experiments) or from the cerebral cortex, striatum, and hippocampus (rat experiments). Total RNA was isolated from brain or tissue cultures samples using the acid guanidinium thiocyanate-phenol-chloroform extraction technique (Chomczynski and Sacchi, 1987). Total RNA was reverse-transcribed into complementary DNA (cDNA) using the RNAse H-reverse transcriptase reaction (Gibco BRL) and a mixture of random hexamers (50 ng) and oligo(dT) (500 ng) as primers. For analysis of xbp1-processing, a polymerase chain reaction (PCR) product of 601 bp was amplified spanning the 26 bases cut out in the processed xbp1 mRNA by the endonuclease IRE1 activated under ER stress conditions. A site specific for the restriction enzyme Pst I is present in the sequence excised by IRE1. The extent of xbp1 processing can thus be investigated by Pst I restriction analysis of the xbp1 PCR product. Polymerase chain reaction products were incubated with Pst I (MBI Fermentas, St. Leon-Rot, Germany) for 5 h at 37°C using an excessive amount of the enzyme (18 U), and restriction fragments were separated by agarose gel electrophoresis. The following xbp1 primers were used: upper strand primer: 5′-AAACAGAGTAGCAGCTCAGACTG-3′, and lower strand primer: 5′-GGATCTCTAAAACTAGAGGCTTGGTG-3′. To verify the sequence of PCR products left intact after Pst I incubation, restriction digests were separated by agarose gel electrophoresis, bands were cut out from gels, and DNA was isolated using the geneclean kit (Quiagene, Hilden, Germany). Polymerase chain reaction products were subcloned into the pCR II vector using the TA cloning kit (Invitrogen NV, Leek, The Netherlands). The cDNA of the insert was sequenced using the dideoxynucleotide chain termination method.
Ischemia-induced changes in mRNA levels of grp78, grp94, growth arrest and DNA damage-inducible gene (gadd)153, and hsp70 were evaluated by quantitative PCR, essentially as described elsewhere (Gissel et al., 1997b; Mengesdorf et al., 2001). Polymerase chain reactions were run in the presence of internal standard cDNA, which had a nonmatching sequence in the middle of the molecule [derived from glutamate receptor subunit 5 (GluR5) or from gadd153 (for hsp70)] and the sequence of interest in the last bases at each end. Isolation of total RNA and synthesis of cDNA was performed as described previously. The following primers were used: for grp78 (Parfett et al., 1989): upper strand primer: 5′-GTTCTGCTTGATGTGTGTCC-3′ and lower strand primer: 5′-TTTGGTCATTGGTGATGGTG-3′; for grp94 (Sorger and Pelham, 1987): upper strand primer: 5′-TCCCCCTTAATGTTTCCCGTG-3′ and lower strand primer: 5′-TAGCCCTTCTTCAGAAGCCTC-3′; for gadd153 (gene bank accession number: U30186): upper strand primer: 5′-TCAGATGAAATTGGGGGCAC-3′ and lower strand primer: 5′ TTTCCTCGTTGAGCCGCTCG-3′; and for hsp70 (Longo et al., 1993): upper strand primer: 5′-TGCTGACCAAGATGAAGG-3′ and lower strand primer: 5′-AGAGTCGATCTCCAGGC-3′. For quantitative evaluation, the cDNA samples used as templates for PCR were diluted (if necessary) so as to bring the optical density of bands derived from the sample and internal standard cDNA within the same range.
Ischemia-induced changes in processed xbp1 mRNA levels were evaluated by quantitative PCR using processed xbp1-specific primers. Polymerase chain reactions were run in the presence of defined amounts of internal standard cDNA. The following set of primers was used for analysis of xbp1 mRNA levels (GenBank accession numbers: AF 027963 and AF 443192): upper strand primer: 5′-CTGAGTCCGCAGCAG-3′, lower strand primer: 5′- GGATCTCTAAAACTAGAGGCTTGGTG-3′. The internal standard used for analysis of processed xbp1 mRNA levels had a nonmatching internal sequence (derived from rat GluR5 cDNA [see: Paschen and Djuricic, 1994]) and processed xbp1 sequence (see above) at each end. The internal standard cDNA could therefore be amplified together with processed xbp1 cDNA derived from samples, using the same set of primers but yielding a smaller amplification product. The internal standard cDNA was produced using three different consecutive PCR reactions, essentially as described elsewhere (Gissel et al., 1997b). In short, in the first reaction, the sequence of rat GluR5 was amplified from brain cDNA using GluR5-specific primers (Paschen and Djuricic, 1994). The product was used as template for the second PCR reaction with primers where the first bases had processed xbp1 sequence and the last bases the GluR5 sequence. The product was taken as template for the third PCR reaction using the processed xbp1 primers.
The final PCR products derived from the third PCR reaction and from tissue cDNA amplified with processed xbp1 primers were subcloned into the pCR II vector (Invitrogen NV), bacteria were transfected, and isolated plasmids containing internal standard or processed xbp1 cDNA sequences as inserts were taken to construct a standard curve. Plasmids were mixed to obtain concentration ratios from 32:1 to 0.063:1 (nonprocessed to processed xbp1 cDNA), and these mixtures were taken as templates for PCR. Amplification products were separated on a 2% agarose gel supplemented with ethidium bromide, gels were transilluminated with ultraviolet light, and bands were photographed. Optical densities (OD) of bands were evaluated by image analysis, and a standard curve was constructed by relating the OD ratios of bands [ln(OD nonprocessed xbp1/OD processed xbp1 × 10)] to the concentration ratios of templates [ln(concentration of plasmid containing nonprocessed xbp1 cDNA as insert/concentration of plasmid containing processed xbp1 cDNA as insert)]. This standard curve was used to quantify changes in processed xbp1 mRNA levels.
Immunoblotting
Ischemia- or Tg-induced changes in levels of processed XBP1 (XBP1proc) protein and ischemia-induced changes in the phosphorylation state of the eukaryotic initiation factor eIF2α were evaluated by Western blot analysis. Tissue or cell culture samples were homogenized in lysis buffer supplemented with protease inhibitors, essentially as described recently (Mengesdorf et al., 2002a). Twenty micrograms of protein extracts were loaded on a 10% or 15% Tris-HCl sodium dodecylsulfate–polyacrylamide gel. After electrophoresis, bands were transferred to polyvinylidene difluoride membranes (Hybond; Pharmacia Biotech). After blotting, membranes were incubated with a mouse monoclonal antibody specific for XBP1 (Santa Cruz Biotechnology, Heidelberg, Germany) or with an eIF2α-P specific antibody raised in rabbits (Research Genetics, Huntsville, AL, U.S.A.) for 16 h at 4°C, followed by incubation with the secondary antibody (anti-mouse/sheep horseradish peroxidase conjugates or anti-rabbit/goat horseradish peroxidase conjugates; Pharmacia Biotech) for 1 h at room temperature. Bands were visualized using the ECL Western blot analysis system (Pharmacia Biotech).
Statistical analysis
Data are presented as means ± SD, with n = 4 independent samples per group for transient global cerebral ischemia experiments, and n = 5 independent samples per group for transient focal cerebral ischemia experiments. Statistically significant differences between control and experimental groups were evaluated by ANOVA, followed by Fisher's protected least-significant difference test (transient global cerebral ischemia experiments). Changes in gene expression and xbp1 processing evaluated in samples taken from the MCA territory of both hemispheres from mice subjected to transient occlusion of the right MCA were evaluated by Student's t-test for paired samples. A probability of 95% was taken to indicate a significant difference.
RESULTS
Endoplasmic reticulum dysfunction-induced activation of the processing of xbp1 mRNA is characterized by excision of a 26-base sequence from the coding region of xbp1 mRNA, encompassing a Pst I restriction site (Calfon et al., 2002). Changes in xbp1 processing can therefore be evaluated by reverse transcription of xbp1 mRNA into cDNA, and PCR amplification of xbp1 cDNA across these 26 bases, followed by restriction analysis of the PCR product with Pst I. According to the assay described by Calfon et al. (2002), the fraction of xbp1 PCR product derived from nonprocessed xbp1 mRNA is completely cut into two fragments upon Pst I incubation, whereas the PCR product derived from processed xbp1 mRNA is left intact. To investigate whether this assay system is suitable for neuronal cells, we have evaluated this approach using primary neuronal cell cultures exposed to Tg, 1 μmol/L, a specific irreversible inhibitor of ER Ca2+-ATPase (Fig. 1). Blocking the neuronal ER calcium pump with this irreversible inhibitor of ER Ca2+-ATPase is thought to induce severe ER stress and thus elicit the maximal ER stress response. In control cultures, PCR with xbp1-specific primers yielded one band of 601 base pairs (Fig. 1 A, arrow) that was completely cut by Pst I, suggesting the absence of detectable amounts of processed xbp1 mRNA (Fig. 1B, arrows). In cultures exposed to Tg, a second faster moving band of 575 base pairs appeared in gels suggesting Tg-induced activation of xbp1 processing (Fig. 1, arrowhead). In cultures exposed to Tg followed by 12 or 24 h of recovery, the 575-bp band was much more prominent than the 601-bp band, indicating almost complete processing of xbp1 mRNA. After Pst I incubation of PCR products derived from cultures exposed to Tg, we observed two bands left intact, the expected band of 575 bp and a second band of 601 bp (Fig. 1B, arrowhead). Sequencing of both bands revealed that the faster moving band exhibited the sequence of processed xbp1, whereas the slower moving band appeared to have nonprocessed xbp1 sequence (data not shown).
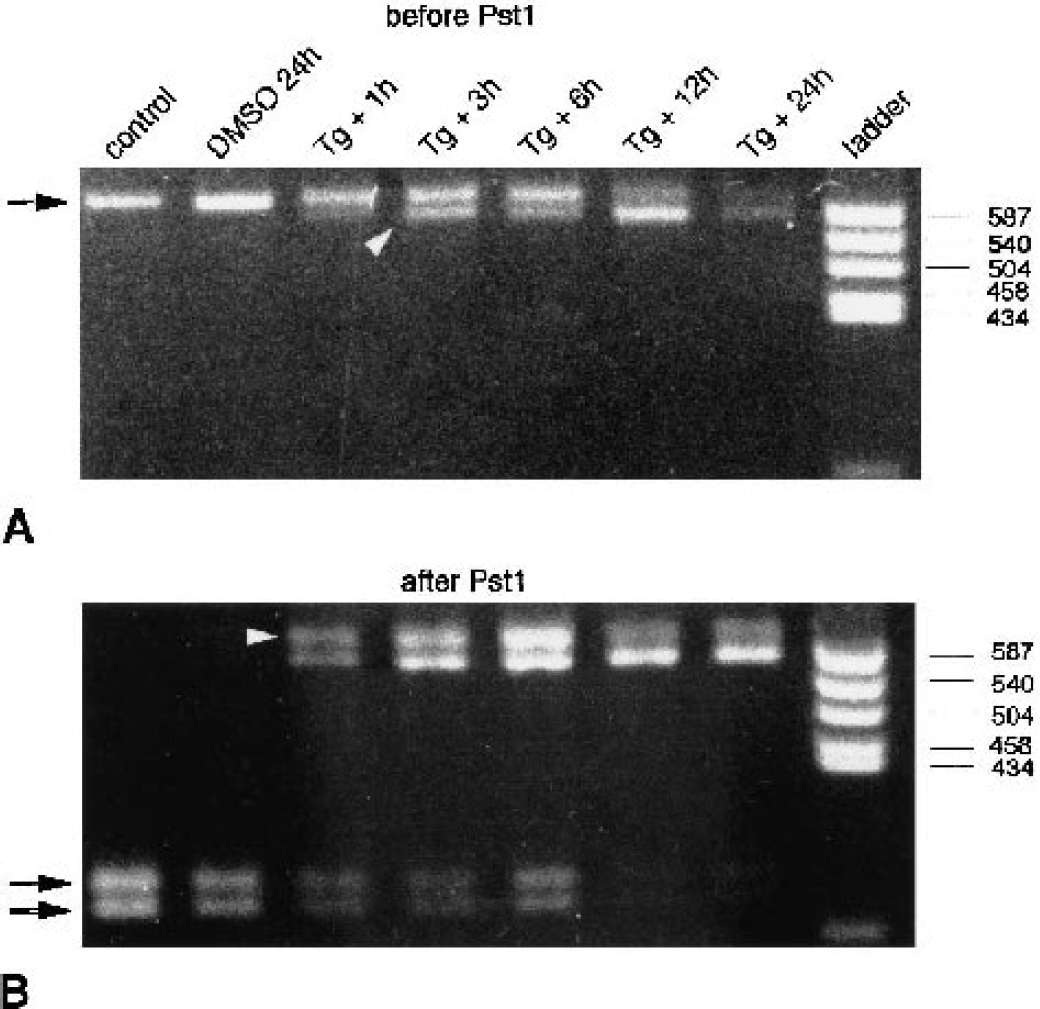
Analysis of xbp1 processing in neuronal cultures exposed to endoplasmic reticulum (ER) stress. Primary neuronal cell cultures were exposed to thapsigargin (Tg, 1 μmol/L), an irreversible inhibitor of ER Ca2+-ATPase. Processing of xbp1 messenger RNA (mRNA) in cultures exposed to Tg was evaluated by polymerase chain reaction (PCR) amplification of xbp1 complementary DNA across the processed sequence and analysis of the PCR product with the restriction enzyme Pst I. PCR products are illustrated before
The appearance of two bands of xbp1 PCR products left intact after Pst I restriction analysis of samples in which xbp1 mRNA was partially processed hindered the correct evaluation of the extent of xbp1 processing because we do not know how much of the PCR product derived from processed xbp1 cDNA was present in the slower moving band left intact after Pst I incubation. Thus, the processed xbp1 assay system described by Calfon et al. (2002) did not work reliably in our neuronal system. We have therefore set up a direct PCR assay to quantify changes in levels of processed xbp1 mRNA (Fig. 2). The 5′ PCR primer was designed so as to allow processed xbp1 cDNA to be preferentially amplified in a mixture of processed and nonprocessed xbp1 cDNA (Figs. 2A and 2B). For these experiments, we have used control tissue cultures and cultures exposed to Tg. The best results were obtained with a 5′ primer sequence containing 15 bases upstream and only one base downstream of the processed 26 bases (Fig. 2A and 2B, primer P#4). Different concentration ratios of plasmids, with processed and nonprocessed xbp1 cDNA as inserts, were used to create a standard curve (Figs. 2C and 2D), which was then taken to quantify ischemia-induced changes in levels of processed xbp1 mRNA. Values given on top of Fig. 2C indicate the concentration ratios of plasmids (relative concentration of plasmid containing nonprocessed xbp1 cDNA as insert/relative concentration of plasmid containing processed xbp1 cDNA as insert) that were used as templates for PCR reactions.
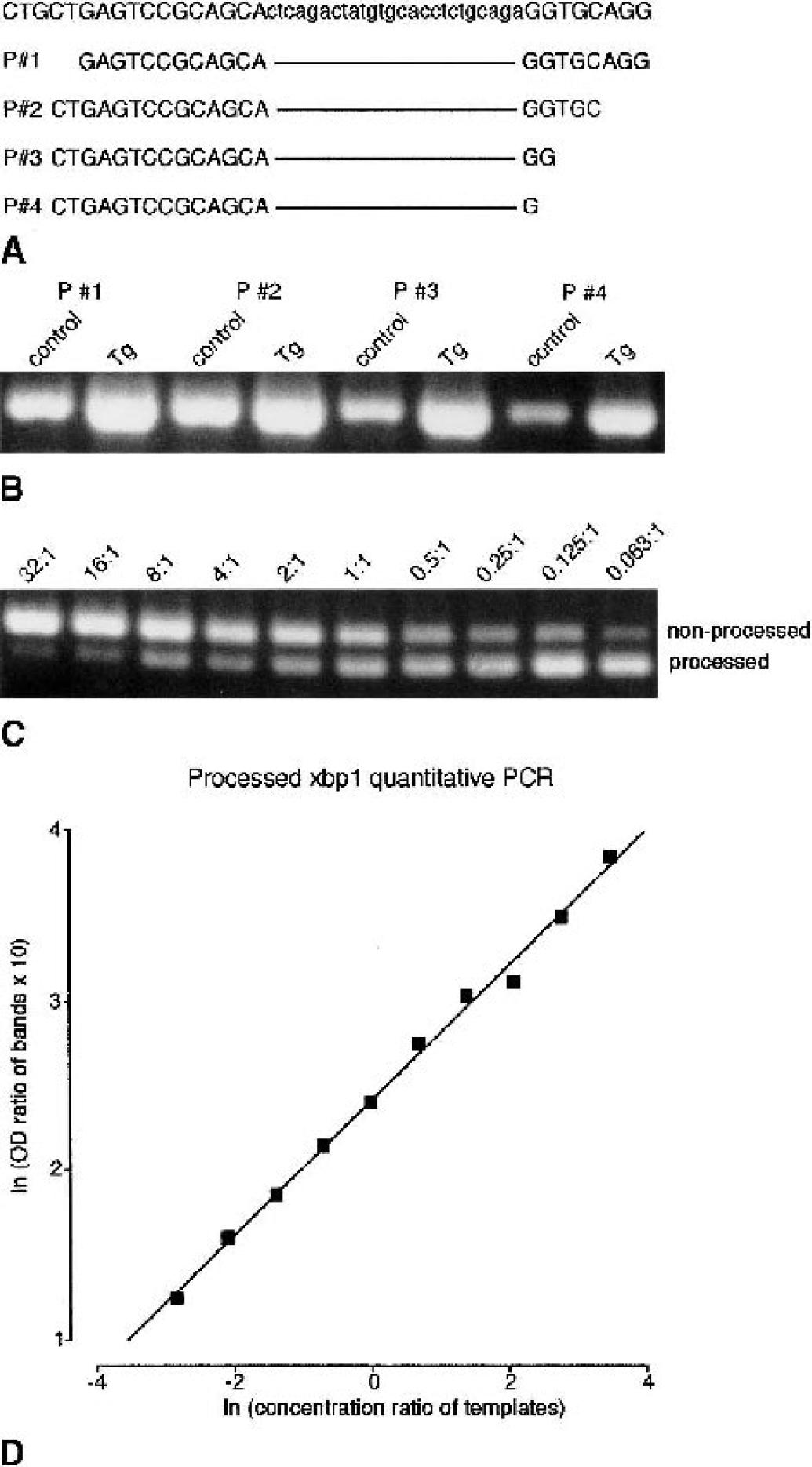
Processed xbp1 quantitative polymerase chain reaction (PCR). To measure changes in processed xbp1 messenger RNA (mRNA) levels directly, a quantitative PCR approach was set up. First, a set of different 5′ primers was used
Ischemia-induced changes in processed xbp1 mRNA levels are illustrated in Figs. 3A and 4A, and quantitative data are presented in Figs. 3B–D and 4B. In rat brains subjected to 30 minutes of forebrain ischemia and 2 h of reperfusion, processed xbp1 mRNA levels rose to about 2,500%, 2,600%, and 2,100% of control in the cerebral cortex, hippocampus, and striatum, respectively (Fig. 3B–D). In the cerebral cortex, processed xbp1 mRNA levels stayed high after 4 h of reperfusion, and declined to values about 280 and 300% above control levels 8 and 24 h after ischemia. In the hippocampus and striatum, levels had already declined after 4 h of reperfusion.
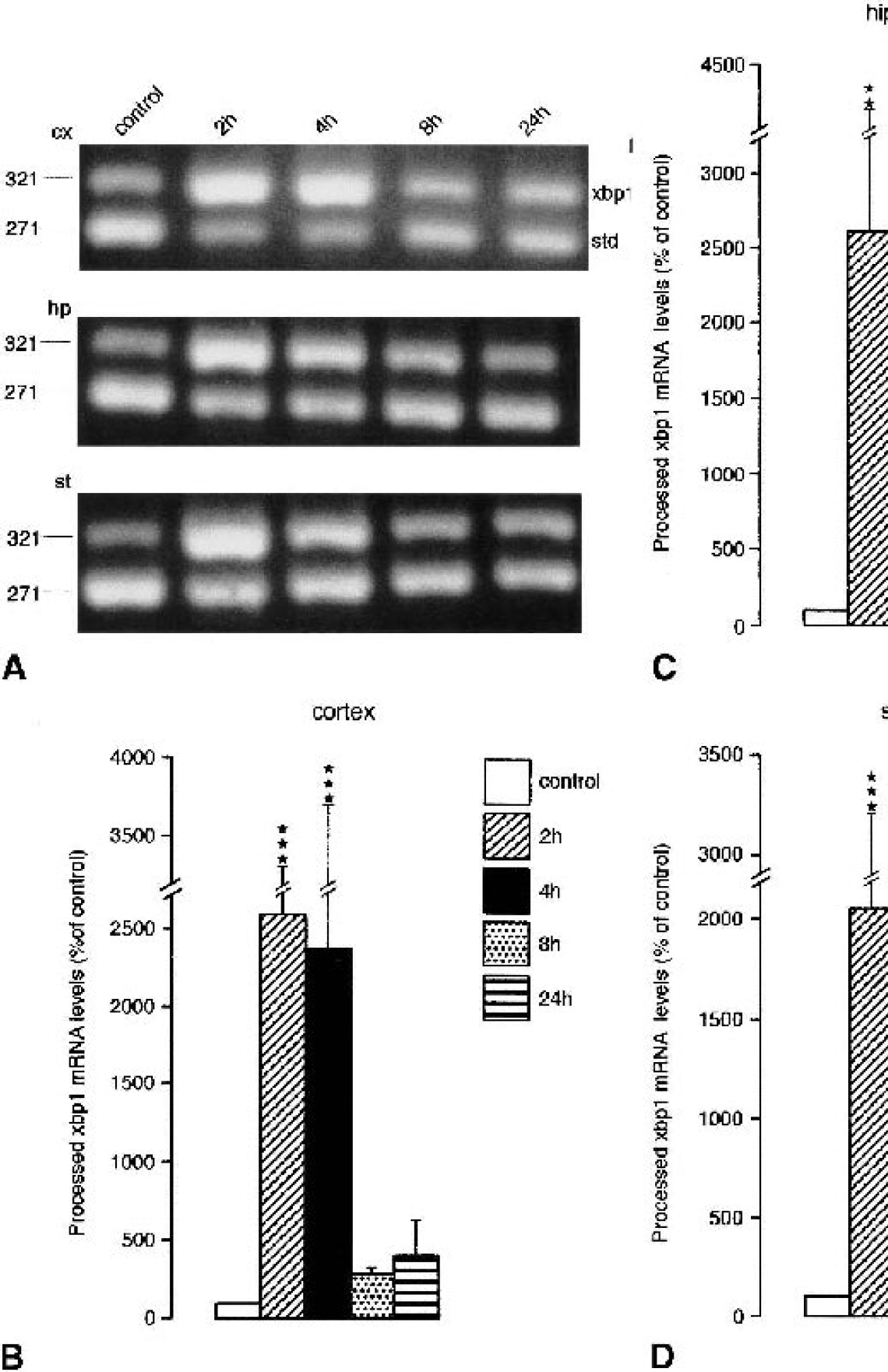
Changes in levels of processed xbp1 messenger RNA (mRNA) induced by transient global cerebral ischemia. Animals were subjected to transient forebrain ischemia for 30 minutes followed by 2 to 24 h of reperfusion. Tissue samples were taken from the cerebral cortex (cx), striatum (st), and hippocampus (hp), and were analyzed for changes in processed xbp1 mRNA levels. Changes in processed xbp1 mRNA levels were evaluated by quantitative polymerase chain reaction (PCR). PCR products were separated on a 2% agarose gel
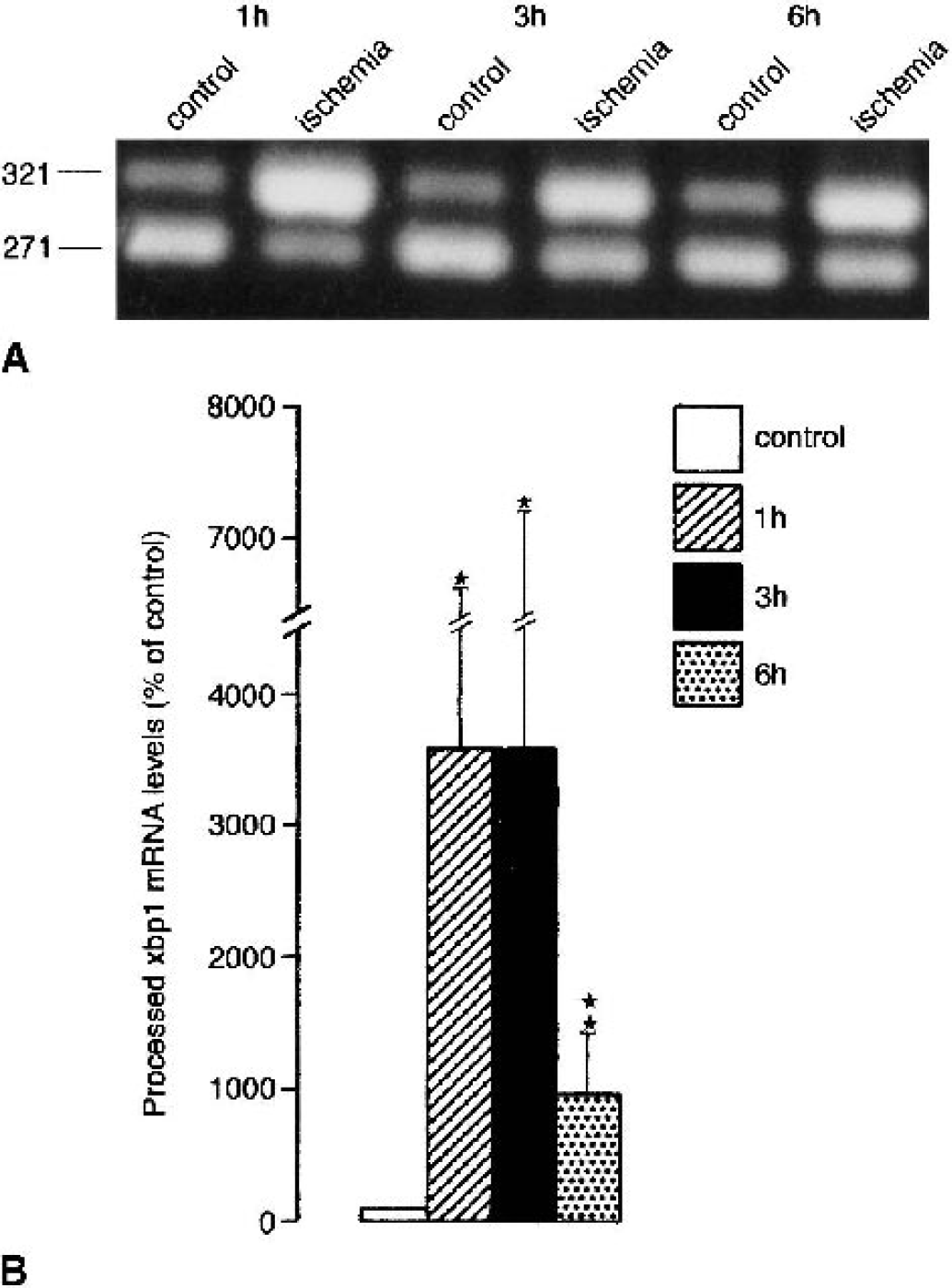
Changes in levels of processed xbp1 messenger RNA (mRNA) induced by transient focal cerebral ischemia. Animals were subjected to transient occlusion of the right middle cerebral artery (MCA) for 30 minutes followed by 1 to 6 h of reperfusion. Tissue samples were taken from the MCA territory of the left (nonischemic) and right (ischemic) hemisphere, and were analyzed for changes in processed xbp1 mRNA levels. Changes in processed xbp1 mRNA levels were evaluated by quantitative polymerase chain reaction (PCR). PCR products were separated on a 2% agarose gel
Changes in processed xbp1 mRNA levels induced by transient focal cerebral ischemia are summarized in Fig. 4B. Transient focal cerebral ischemia induced an about 35-fold increase in processed xbp1 mRNA levels after 1 h of reperfusion, and processed xbp1 mRNA levels were still significantly increased after 6 h of reperfusion. Individual processed xbp1 mRNA levels varied considerably (Fig. 4B, large standard deviation). To exclude a methodologic problem underlying this variability, we tested the reliability of our quantitative PCR assay. When the same sample was analyzed five times in parallel (including PCR amplification, gel electrophoresis, and image analysis), we calculated a value of 100 ± 10.9%, indicating that the quantitative PCR assay produced reproducible results. This suggests that the large scatter of processed xbp1 mRNA levels found in individual animals in the ipsilateral MCA territory after transient MCA occlusion resulted most probably from a variable response to vascular occlusion and not from variability introduced during sample processing.
We also investigated whether the rise in processed xbp1 mRNA levels observed after transient focal cerebral ischemia (Fig. 4A) was paralleled by a similar increase in processed XBP1 (XBP1proc) protein content. On Western blots run with brain and tissue culture samples, we could not find the 33-kd XBP1 protein, indicating that levels were too low to be detected. This is in line with the observation that the 33-kd XBP1 protein can be identified only in cells overexpressing xbp1 (Calfon et al., 2002). In control samples, we found only a faint band of the 54-kd processed XBP1 protein, suggesting low levels of XBP1proc protein in the physiologic state (Fig. 5A). After 1 and 3 h of reperfusion, we did not observe any major increase in XBP1proc protein levels. Only after 6 h of reperfusion was the optical density of the XBP1proc protein band increased, implying a rise in levels of newly synthesized XBP1proc protein. Thus, after transient focal cerebral ischemia the marked rise in processed xbp1 mRNA levels was not paralleled by a similar rise in levels of the XBP1proc protein during the first 3 h of reperfusion. In neuronal cell cultures exposed to the ER stressor thapsigargin, levels of XBP1proc protein rose already during exposure (Fig. 5B). The delayed increase in XBP1proc protein levels observed after transient focal cerebral ischemia, despite high levels of processed xbp1 mRNA, may have resulted from severe suppression of protein synthesis induced by transient focal cerebral ischemia (Fig. 6A). In this animal model of stroke, we observed a marked increase in eIF2α phosphorylation peaking at 1 h of recovery from ischemia (Fig. 6B). After 6 h of reperfusion, that is, at a time when levels of XBP1proc had markedly increased (Fig. 5A), eIF2α-P levels were clearly reduced relative to the peak level observed after 1 h of reperfusion (Fig. 6B).

Evaluation of ischemia-induced changes in levels of processed X-box protein 1 (XBP1) by Western blot analysis. Mice were subjected to transient occlusion of the right middle cerebral artery (MCA) for 30 minutes followed by 1 to 6 h of reperfusion. Tissue samples were taken from the MCA territory of the left (nonischemic) and right (ischemic) hemisphere, and analyzed for changes in processed XBP1 protein levels
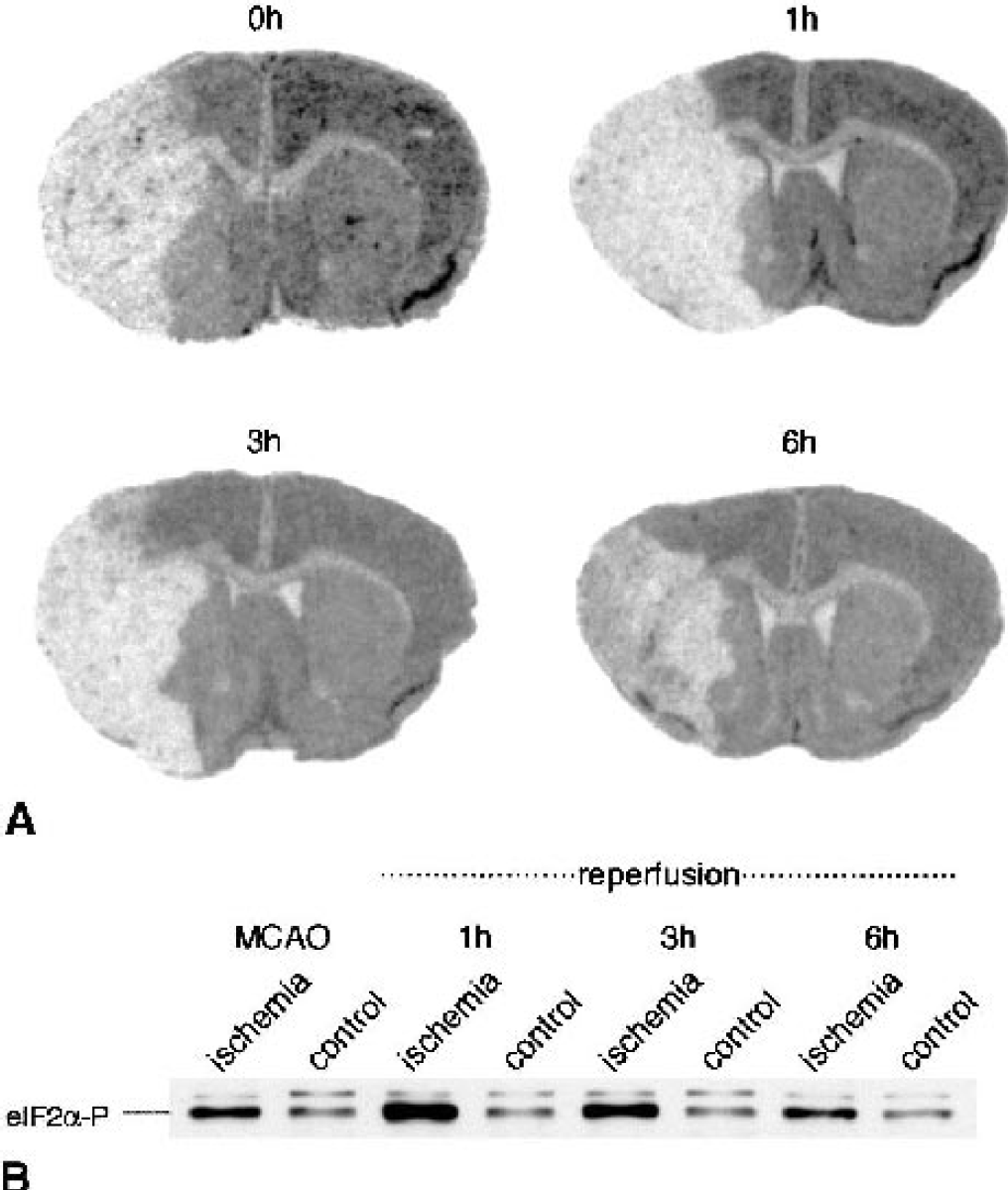
Ischemia-induced suppression of protein synthesis. Mice were subjected to transient occlusion of the right middle cerebral artery (MCA) for 30 minutes followed by 1 to 6 h of reperfusion. Protein synthesis was measured by autoradiography, as described in Materials and Methods
To investigate the consequence of ischemia-induced activation of xbp1 processing on the entire ER stress response, mRNA levels of genes coding for ER stress proteins were analyzed. XBP1proc protein is necessary as transcription factor for an activation of the expression of these genes. We evaluated changes in mRNA levels of grp78, grp94 and gadd153 induced by transient focal cerebral ischemia using the same tissue samples as those taken for quantification of processed xbp1 mRNA levels. Messenger RNA levels of ER stress genes were compared with mRNA levels of hsp70, an immediate-early gene, the expression of which is activated without the need for new protein synthesis (Massa et al., 1996). Results are illustrated in Fig. 7A and summarized in Figs. 7B to D. Transient focal cerebral ischemia induced a transient decrease of grp94 mRNA levels to 81% of control after 3 h of reperfusion, and a small increase in grp78 and gadd153 mRNA levels, peaking at 6 h of reperfusion, to 164% and 198% of control, respectively. Hsp70 mRNA levels, in contrast, rose sharply, with the highest values being found 3 and 6 h after ischemia (about 550-fold increase over control; Figs. 7A and 7E).
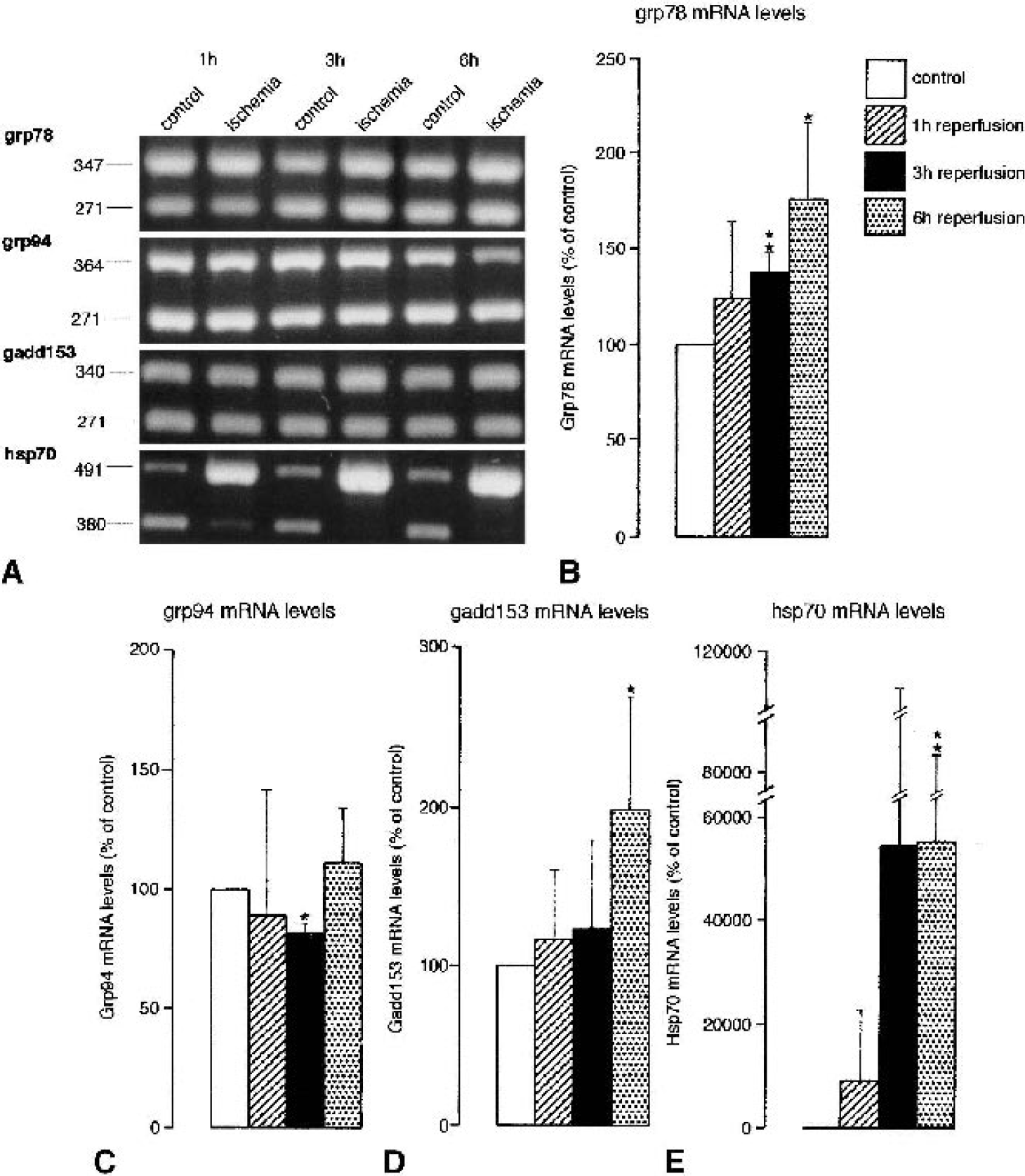
Ischemia-induced changes in the expression of stress genes. Mice were subjected to transient right middle cerebral artery (MCA) occlusion for 1 h, followed by 1 to 6 h of reperfusion. Tissue samples were taken from the MCA territory of the left (nonischemic) and right (ischemic) hemispheres. Ischemia-induced changes in the expression of genes activated specifically by endoplasmic reticulum stress (grp78, grp94, gadd153) or cytoplasmic stress (hsp70) were evaluated by quantitative polymerase chain reaction (PCR) as described in Materials and Methods. PCR products were separated on a 2% agarose gel
To investigate the possible mechanisms underlying ischemia-induced ER dysfunction and activation of the ER stress response, changes in xbp1 processing were evaluated in primary neuronal cell cultures exposed to the proteasome inhibitor MG-132 and proteasome inhibitor I (Figs. 8A and 8B). Transient cerebral ischemia has indeed been shown to induce disturbances of the ubiquitin/proteasomal pathway (Keller et al., 2000; Hu et al., 2001; Asai et al., 2002; Mengesdorf et al., 2002b), and evidence has been presented indicating that disturbances of the ubiquitin/proteasomal pathway may induce secondary ER dysfunction (Bush et al., 1997; Hampton, 2000; Zimmermann et al., 2000). Both proteasome inhibitors did indeed activate concentration-dependent xbp1 processing, as indicated by the pattern of restriction fragments after incubating xbp1 PCR product with Pst I, and by PCR with processed xbp1-specific primers (Figs. 8A and 8B).
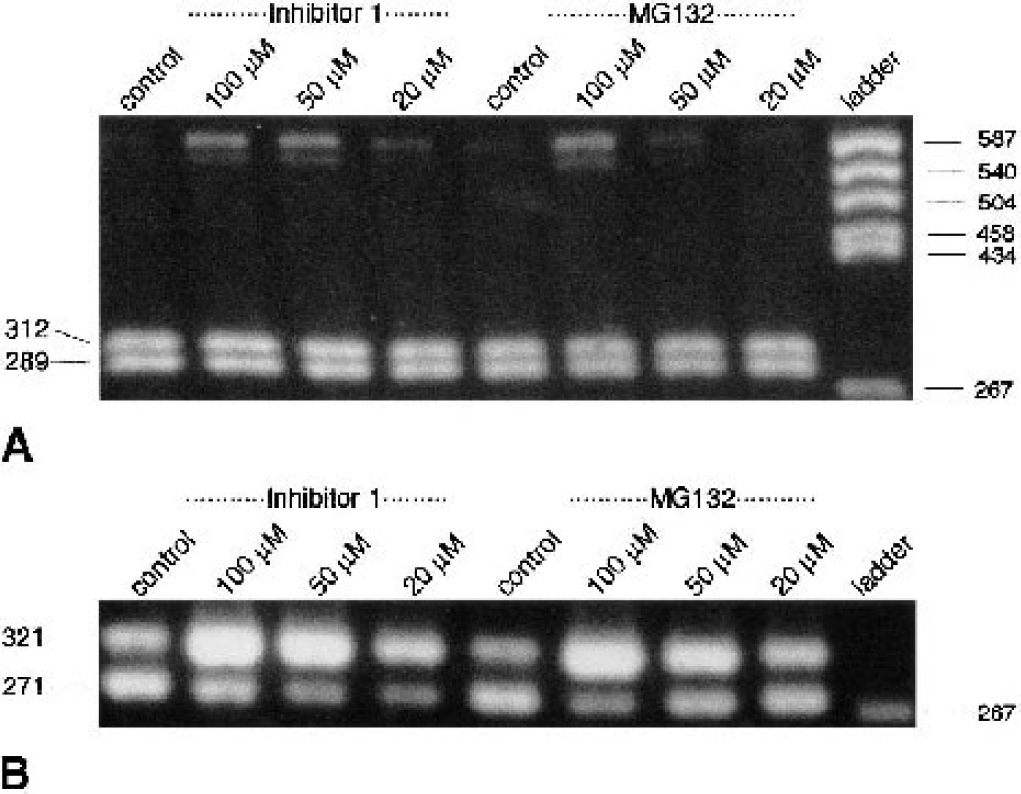
Activation of xbp1 processing in primary neuronal cell cultures exposed to proteasome inhibitors. Primary neuronal cell cultures were exposed to the proteasome inhibitors MG-132 and proteasome inhibitor I (20–100 μmol/L each) for 6 h. Changes in processed xbp1 messenger RNA (mRNA) levels were evaluated by xbp1-specific polymerase chain reaction (PCR) and restriction analysis of the PCR amplification product with Pst I
DISCUSSION
The ER is a subcellular compartment playing a central role in cells, because its lumen is the site of folding and processing of all membranes and secretory proteins (for review see Kaufman, 1999). For correct functioning of the folding and processing reactions, an oxidative environment and high ER intraluminal calcium activity is required. Under conditions associated with ER stress, unfolded or misfolded proteins accumulate in the ER lumen, a pathologic process resulting in the activation of the UPR. The first step induced by UPR is activation of the ER membrane resident enzymes PERK and IRE1. In the physiologic state, the ER chaperone GRP78 is bound to PERK and IRE1 and prevents these enzymes from being activated. When unfolded or misfolded newly synthesized polypeptides accumulate in the lumen of the ER, GRP78 is needed to refold these peptides. Dissociation of GRP78 protein from PERK and IRE1 causes oligomerization, autophosphorylation, and thus activation of these enzymes. The main goal of UPR is to hinder the further accumulation of newly synthesized misfolded polypeptides by blocking translation at the initiation step, and to activate specifically transcription and translation of grp78 and other ER stress genes to restore ER function. After being activated, PERK induces phosphorylation of eIF2α, resulting in blocking of translation at the initiation step, while IRE1 induces processing of xbp1 mRNA, resulting in new synthesis of processed XBP1 (XBP1proc) protein, which functions as an active transcription factor specific for ER stress genes. Cells in which UPR is activated in such a way that GRP78 protein and other ER stress proteins are synthesized to levels sufficient to restore ER functions are able to cope with this severe form of cellular stress, whereas cells in which the entire UPR signal transduction cascade fails to be sufficiently activated are destined to die.
This series of experiments was designed to investigate whether transient cerebral ischemia induces activation of one basic step of UPR downstream of activated IRE1, processing of xbp1 mRNA, synthesis of new XBP1proc protein, and transcription of ER stress genes such as grp78 and grp94. This is a key step in the ER stress response, which requires protein synthesis at a level sufficient to permit synthesis of new XBP1proc protein. With respect to activation of gene expression, UPR differs from the response of cells to a severe form of cytoplasmic stress (heat-shock response) because heat-shock genes are immediate-early genes that do not require new synthesis of a transcription factor before activation of transcription (Massa et al., 1996). Protein synthesis may thus become a factor limiting activation of UPR sufficiently to cope with ER stress, particularly under pathologic conditions of the brain associated with severe suppression of translation. We showed here that transient global and focal cerebral ischemia did indeed induce a marked increase in processed xbp1 mRNA levels, with most pronounced changes observed in the affected territory supplied by the MCA in mice subjected to 1-h MCA occlusion followed by 1 h of reperfusion. This is a reperfusion time when translation is almost completely suppressed. It is thus not surprising that we did not observe any rise in processed XBP1 protein levels during the first 3 h of recovery from MCA occlusion. After MCA occlusion in mice, the marked increase in processed xbp1 mRNA levels was not paralleled or followed by a similar rise in grp78/grp94 mRNA levels, suggesting that protein synthesis becomes a limiting factor in the activation of the synthesis of XBP1proc protein and grp78/grp94 transcription.
Using the quantitative PCR approach, we were able to quantify ischemia-induced changes in xbp1 processing. The 5′ primer spanning across the processed 26 bases was so designed that we produced the most pronounced difference in the amount of nonprocessed and processed xbp1-specific PCR product when cDNA from control or Tg-exposed cultures was used. We do not know whether primers used for processed xbp1-specific PCR also amplified small amounts of cDNA from samples exhibiting only nonprocessed xbp1. If so, the ischemia-induced increase in processed xbp1 mRNA levels would be even more pronounced than estimated in the present study.
We have found a large scatter of processed xbp1 mRNA levels in the ipsilateral MCA territory of animals subjected to transient focal cerebral ischemia (Fig. 4). For analysis of processed xbp1 mRNA levels, tissue samples were dissected from the MCA territory of the ipsilateral and contralateral hemisphere. We do not know at present whether the high levels of processed xbp1 mRNA found in individual animals after transient focal cerebral ischemia are representative for the entire ipsilateral MCA territory, or whether processed xbp1 mRNA levels are particularly high in certain subregions such as the border zone between the affected MCA territory and the surrounding tissue. If so, the rise in processed xbp1 mRNA levels would be even more pronounced than illustrated here. Experiments are under way to evaluate processed xbp1 mRNA levels in a more regional way.
The state of xbp1 processing has never been investigated before in rodent tissue samples. The results of the present study clearly indicate that transient global or focal cerebral ischemia induces activation of the processing of xbp1 mRNA. Endoplasmic reticulum stress is the only pathologic process identified as resulting in xbp1 processing, which implies that transient cerebral ischemia causes ER dysfunction. We suggested earlier that cerebral ischemia may disturb ER functions (Paschen, 1996; Paschen and Doutheil, 1999; Paschen and Frandsen, 2001), since the response of neurons to transient cerebral ischemia is in many respects similar to the response of primary neuronal cell cultures to ER dysfunction. Under both experimental conditions, the initiation step of translation is severely suppressed, as indicated by suppression of global protein synthesis, disaggregation of polyribosomes, and phosphorylation of eIF2α (Kleihues et al., 1975; Bodsch et al., 1985; Brostrom and Brostrom, 1990; Bonnekoh et al., 1992; Prostko et al., 1992, 1995; Burda et al., 1994; Srivastava et al., 1995; Paschen et al., 1996; Doutheil et al., 1997; Althausen et al., 2001), and the expression of ER stress genes is activated (Drummond et al., 1987; Wang et al., 1993; Lowenstein, 1994; Brostrom et al., 1995; Gissel et al., 1997a; Linden et al., 1998; Paschen et al., 1998a, b ; Jin et al., 2001; Tang et al., 2002). Our earlier observation that mRNA levels of the ER stress gene erp72 rise after transient global cerebral ischemia (Paschen et al., 1998a), and that this rise is more pronounced in the cortex than in the striatum or hippocampus, is in line with the observation of the present study that higher levels of processed xbp1 mRNA are found in the cortex than in other brain structures after a longer reperfusion period. Ischemia-induced activation of ER stress is also indicated by postischemic activation of PERK (Kumar et al., 2001), the only eIF2α kinase so far found to be activated after transient cerebral ischemia.
Despite clear indicators of ER stress as indicated by the marked rise in processed xbp1 mRNA levels (Figs. 3 and 4), we were surprised to find that grp78 mRNA levels rose only slightly after transient focal cerebral ischemia, whereas grp94 mRNA levels were transiently reduced. In cells in which ER function is disturbed by exposing cultures to Tg, we previously observed a several hundred–fold increase in grp78 and grp94 mRNA levels (Mengesdorf et al., 2001). The most plausible explanation for the absence of a clear rise in grp78/grp94 mRNA levels despite an approximately 35-fold increase in processed xbp1 mRNA levels is that this signal transduction pathway may be blocked after ischemia through severe suppression of translation. If this interpretation is valid, it can be assumed that in neurons subjected to a lethal period of transient ischemia, ER stress induced by ischemia causes suppression of protein synthesis to such a marked extent that affected cells cannot recover from ER dysfunction by upregulation of the expression of ER stress genes (Fig. 7).
At 6 h of reperfusion, levels of XBP1proc were increased in the ipsilateral MCA territory. This is a reperfusion time characterized by a transient moderate recovery of protein synthesis and dephosphorylation of eIF2α (Fig. 6; Althausen et al., 2001; Mengesdorf et al., 2002a). However, from 6 h of reperfusion onwards, the extent of protein synthesis suppression increased again (Mengesdorf et al., 2002a), in parallel with a secondary depletion of ATP levels (Paschen et al., 2000). At this stage, the pathologic process culminating in neuronal cell injury is most probably too much advanced in this animal model for a successful therapeutic intervention.
As pointed out previously, it is well established that, under conditions associated with ER stress, activation of the expression of glucose-regulated genes is triggered by the transcription factor XBP1proc, the formation of which is induced by activated IRE1-α in a splicing process on xbp1 mRNA (Shen et al., 2001; Yoshida et al., 2001; Calfon et al., 2002). The physiologic role of UPR induction downstream of activated IRE1 is indicated by the observation that cells transfected with a dominant-negative form of mouse IRE1-α are particularly sensitive to ER stressors (Miyoshi et al., 2000). Conversely, the first transcription factor recognized in higher mammals to activate UPR-responsive genes was the activating transcription factor (ATF)6 (Yoshida et al., 1998). ATF6 is a basic leucine zipper protein that activates expression of ER stress genes by binding to the ER stress response element (ERSE) located in the promoter regions of grp78, grp94, and calreticulin genes (Yoshida et al., 1998). ATF6 is constitutively expressed as a 90-kd protein. Under conditions associated with ER dysfunction, ATF6 is converted by Site-1 and Site-2 proteases into a 50-kd protein that translocates to the nucleus and binds to ERSE sequence of ER stress genes (Haze et al., 1999; Ye et al., 2000). Interactions between ATF6 and ERSE are also critical for transcriptional induction of xbp1 (Yoshida et al., 2000), and the slight increase in grp78 mRNA levels observed in the present study in animals subjected to 1-h MCA occlusion followed by 3 h of reperfusion may be caused by ATF6-activated grp78 expression, because we did not find increased levels of XBP1proc protein in the brains of these animals. In contrast, it has been proposed that XBP1proc may be a more effective transcriptional activator than ATF6 (Yoshida et al., 2001). However, this suggestion is based on transfection assays, and there is no information available regarding the relative effectiveness of endogenous XBP1proc or ATF6 in activating transcription of endogenous ER stress genes. Our observation that grp78/94 expression is almost not activated after transient focal cerebral ischemia despite clear evidence of ischemia-triggered induction of UPR [marked rise in processed xbp1 mRNA levels, Figs. 3 and 4; activation of PERK (Kumar et al., 2001)], implies that ATF6 is the less effective transcription factor in the brain: the only plausible explanation for the inability of the postischemic brain tissue to respond to high processed xbp1 mRNA levels with an activation of grp78/grp94 expression is by suppression of translation of the processed xbp1 mRNA owing to the block of protein synthesis. Endogenous ATF6, in contrast, needs only to be proteolytically cleaved into the active transcription factor, a process that would not be hindered in this pathologic situation.
The mechanisms underlying ischemia-induced ER dysfunction still have to be fully established. We have observed depletion of ER calcium stores in neuronal cultures exposed to a nitric oxide donor (Doutheil et al., 2000), implying that excessive activation of neuronal nitric oxide synthase plays a role in this pathologic process. This notion is corroborated by the observation that in vulnerable neurons of the hippocampal CA1 sector, neuronal ER calcium stores were depleted immediately after ischemia, and restoration of ER calcium stores was found only in animals pretreated with a neuroprotective nitric oxide synthase inhibitor (Kohno et al., 1997). Furthermore, increased production of oxygen-free radicals, which is known to occur during reperfusion after ischemia (Siesjö et al., 1995), may cause ER dysfunction because the ER calcium pump is particularly sensitive to radicals (Dreher et al., 1995; Racay et al., 1995; Viner et al., 1996). Finally, ischemia-induced dysfunction of the ubiquitin–proteasomal pathway (Keller et al., 2000; Hu et al., 2001; Asai et al., 2002; Mengesdorf et al., 2002b) may contribute to ER dysfunction because blocking proteasome function causes ER stress (Fig. 8) (Bush et al., 1997; Hampton, 2000; Zimmermann et al., 2000).
It is well established that a severe form of ER dysfunction is sufficient to induce cell death (for reviews see Kaufman, 1999; Paschen and Doutheil, 1999). Cells exposed to ER stress conditions can react in two different ways (see scheme given in Fig. 9): cells unable to cope with this pathologic process are destined to die, whereas cells in which UPR is sufficiently activated to restore GRP78 protein to levels high enough to bind and block IRE1 and PERK will survive ER stress conditions. After transient cerebral ischemia, ER stress-induced suppression of protein synthesis is so pronounced that it critically limits new synthesis of XBP1proc protein and consequently grp78 mRNA and thus GRP78 protein. Conversely, suppression of global protein synthesis triggered by transient cerebral ischemia does not necessarily exclude cap-independent translation of messages such as grp78 exhibiting an internal ribosome entry site (IRES). Messages containing IRES sequence (such as ornithine decarboxylase) may indeed be preferentially translated after ischemia. A marked increase in ornithine decarboxylase immunoreactivity has been observed in the CA1 subfield of gerbils after transient global cerebral ischemia (Müller et al., 1991), that is, in a brain region in which protein synthesis is almost completely suppressed during reperfusion (Bodsch et al., 1985; Thilmann et al., 1986). It has also been shown that GRP78 immunoreactivity is increased in injured neurons after transient focal cerebral ischemia (Ito et al., 2001). For this study however, an antibody was taken directed against the ER retrieval motif KDEL, which is not specific for the GRP78 protein, implying that increased immunoreactivity is not necessarily an unequivocal proof of a rise in GRP78 protein levels. Furthermore, ischemia-induced activation of grp78 expression would clearly be more pronounced if the xbp1 pathway would not be blocked by severe suppression of translation, as indicated in the present study. This suggests that ER stress-induced block of translation may indeed contribute to the pathologic process culminating in neuronal cell injury after transient cerebral ischemia.
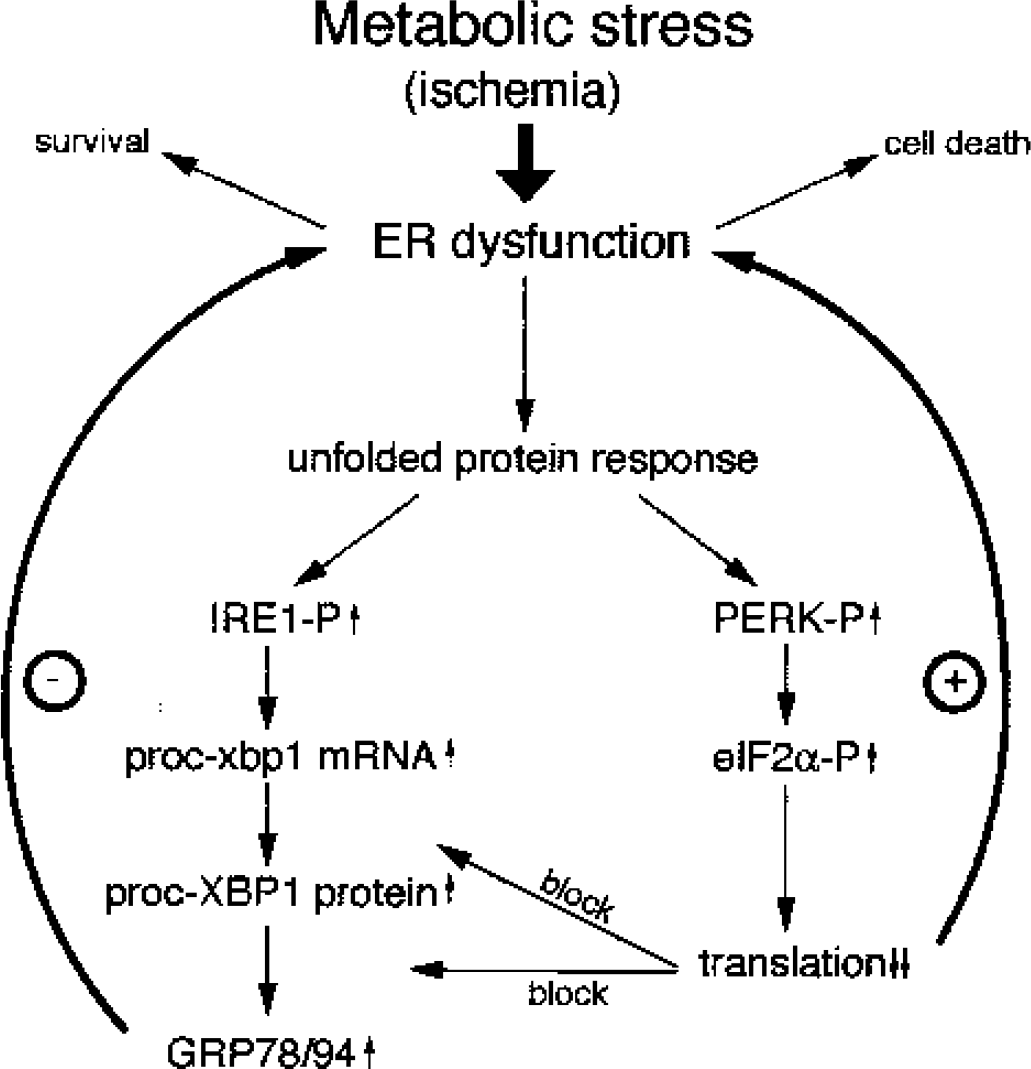
Scheme of the possible mechanism connecting endoplasmic reticulum (ER) dysfunction with the occurrence of ischemia-induced neuronal cell death. Ischemia induces ER stress, resulting in the activation of the unfolded protein response. If ER stress–induced suppression of protein synthesis is so severe that it blocks synthesis of new XBP1 protein, which is needed to activate expression of ER stress genes, cells are notable to recover from ER stress (+) and will consequently die. Cells in which translation recovers to such an extent that new XBP1 and GRP78 protein are synthesized in a way sufficient to restore ER function will recover from ischemia-induced ER dysfunction. eIF2α, alpha subunit of the eukaryotic initiation factor 2; PERK, PKR-like endoplasmic reticulum kinase; mRNA, messenger RNA.
Footnotes
Acknowledgments:
The excellent technical assistance of Änne Pribliszki, Cordula Strecker, and Ursula Gerster is gratefully acknowledged.