
Abstract
Previous studies have demonstrated that cortical spreading depression (CSD) induces neuronal tolerance to a subsequent episode of ischemia, The objective of the present investigation was to determine whether CSD alters levels of mRNA coding for putative neuroprotective proteins. Unilateral CSD was evoked in male Wistar rats by applying 2 mol/L KCl over the frontal cortex for 2 hours. After recovery for 0, 2, or 24 hours, levels of several mRNA coding for neuroprotective proteins were measured bilaterally in parietal cortex using Northern blot analysis. Levels of c-fos mRNA and brain-derived neurotrophic factor (BDNF) mRNA were markedly elevated at 0 and 2 hours, but not 24 hours after CSD. Tissue plasminogen activator (tPA) mRNA levels were also significantly increased at 0 and 2 hours, but not 24 hours after CSD. Levels of the 72-kDa heat-shock protein (hsp72) mRNA were not significantly increased by CSD, except for a small elevation (20%) at 2 hours recovery. Levels of the 73-kDa heat-shock cognate (hsc73) mRNA were slightly, but significantly, increased at 2 and 24 hours of recovery. Finally, levels of mRNA for protease nexin-1 and glutamine synthetase were not significantly altered by CSD at any time studied. The current results support the hypothesis that neuronal tolerance to ischemia after CSD may be mediated by increased expression of FOS, BDNF, or tPA, but not by increased expression of hsp72, hsc73, nexin-1, or glutamine synthetase.
Keywords
Preconditioning the brain with cortical spreading depression (CSD) has been shown to reduce neuronal injury resulting from a subsequent episode of ischemia (Kawahara et al., 1995; Kobayashi et al., 1995; Matsushima et al., 1996). This induction of neuronal tolerance to ischemia is similar to that reported previously using sublethal ischemia as the preconditioning stimulus (Kitagawa et al., 1991; Kirino et al., 1991; Glazier et al., 1994). Cortical spreading depression is characterized by transient perturbations of electrical activity, ion homeostasis, and metabolite levels that resemble the initial stages of ischemia (Hansen and Zeuthen, 1981; Lauritzen et al., 1990). Thus, the mechanisms by which CSD and sublethal ischemia induce neuronal tolerance may share common features. However, little is known about the molecular alterations that mediate the induction of neuronal tolerance after CSD or ischemia.
Previous studies have demonstrated that CSD induces rapid expression of immediate-early genes (IEG) such as c-fos (Herrera and Robertson, 1990; Herdegen et al., 1993). Many IEG encode transcription factors and, thus, may mediate expression of neuroprotective target genes. In situ hybridization studies have indicated that CSD also causes a rapid elevation of brain-derived neurotrophic factor (BDNF) mRNA (Kokaia et al., 1993). Brain-derived neurotrophic factor has been reported to protect neurons both in vitro and in vivo (Ghosh et al., 1994; Beck et al., 1994). Further, BDNF is induced by a variety of cerebral insults and may function to limit the extent of brain damage (Lindvall et al., 1994). Thus, BDNF is a prime candidate for mediating the induction of neuronal tolerance to ischemia after CSD. Accordingly, the first objective of the present study was to quantify the timecourse of expression of c-fos and BDNF mRNA after CSD using Northern blot analysis.
Cortical spreading depression has also been shown to induce delayed expression of glial fibrillary acid protein (GFAP), a marker of reactive astrocytes (Kraig et al., 1991). Reactive astrocytes may function to protect neurons from ischemia by releasing trophic factors or by scavenging glutamate. Protease nexin-1 is a serine protease inhibitor that was originally identified as a neurotrophic factor derived from glia (Monard et al., 1973). Increased expression of nexin-1 has been demonstrated in GFAP-positive astrocytes after ischemia (Hoffmann et al., 1992). Moreover, nexin-1 has been reported to protect neurons against glucose deprivation (Smith-Swintosky et al, 1995). However, the effect of CSD on nexin-1 expression has not been previously examined. Thus, a second objective of the present study was to determine whether CSD alters the level of nexin-1 mRNA. In addition, the effects of CSD on mRNA levels of tissue plasminogen activator (tPA), one of the serine proteases inhibited by nexin-1, were also measured. Neuronal depolarization has been reported to increase the expression and secretion of tPA (Qian et al., 1993; Gualandris et al., 1996). Furthermore, tPA has been implicated as a pathogenic factor in excitotoxic neuronal injury (Tsirka et al., 1995). Thus, alterations in the expression of nexin-1 and tPA after CSD may modulate the vulnerability of cortical neurons to ischemia.
Astrocytes also play an important role in scavenging and metabolizing glutamate. Glutamine synthetase is the rate-limiting enzyme in the metabolism of glutamate and is localized primarily in astrocytes (Martinez-Hernandez et al., 1977). Facilitation of glutamate metabolism would be expected to protect neurons from ischemia by diminishing their exposure to glutamate. Glutamine synthetase activity and immunoreactivity were reported to increase rapidly after cerebral ischemia (Petito et al., 1992). However, the influence of CSD on expression of glutamine synthetase has not been previously examined. Thus, an additional objective of the present study was to determine whether CSD alters the level of glutamine synthetase mRNA.
Finally, expression of the 72-kDa heat-shock protein (hsp72) has been associated with induction of neuronal tolerance in several experimental models of ischemic preconditioning (Kirino et al., 1991; Glazier et al., 1994). However, in situ hybridization studies have failed to detect increases in hsp72 mRNA after CSD (Nowak et al., 1991; Kobayashi et al., 1995). Thus, hsp72 is not likely to have an important role in the induction of neuronal tolerance after CSD. Recently, the 27-kDa heat-shock protein was reported to be strongly induced after CSD, suggesting that other stress proteins may contribute to neuronal tolerance after CSD (Plumier et al., 1997). The effect of CSD on expression of the 73-kDa heat-shock cognate (hsc73), a close relative of hsp72, has not been previously examined. Thus, the final objective of the present study was to determine whether CSD alters the level of hsc73 mRNA.
Preliminary results of this work have been reported previously (Karikó et al., 1997).
MATERIALS AND METHODS
Induction of cortical spreading depression
All procedures were performed in strict accordance with the NIH Guide for Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee. Cortical spreading depression was produced in 15 male Wistar rats using methods described previously (Kobayashi et al., 1995). In brief, anesthesia was induced with halothane, and the animals were intubated and ventilated mechanically with a mixture of 1% halothane, 25% O2, balance N2O. A cannula was placed in the tail artery for blood sampling, and core temperature was maintained at 37.5°C using a rectal probe connected to a heating lamp. The head of the animal was placed in a stereotaxic frame, and small burr holes (diameter, 2 mm) were made bilaterally over the frontal cortex, leaving the dura intact. Cortical spreading depression was induced by placing filter paper soaked with 2 mol/L KCl into one of the burr holes, chosen randomly in each animal. In the contralateral hemisphere, filter paper soaked with physiologic saline served as a control. The filter papers were replaced every 20 minutes for a period of 2 hours. In 5 animals, the blood flow response to CSD was measured using laser—Doppler flowmetry. In these animals, burr holes (diameter, 2 mm) were made bilaterally over the parietal cortex, and laser—Doppler probes (Model 433-4, Vasamedics, St. Paul, MN, U.S.A.) were placed on the intact dura. Cortical blood flow was measured bilaterally using two perfusion monitors (Model BPM2, Vasamedics).
After application of KCl for 2 hours, the animals were divided among three groups. In the first group (n = 5), the animals were killed immediately using an overdose of halothane. The remaining animals were permitted to recover for 2 hours (n = 5) or 24 hours (n = 5) before killing. Two animals served as unoperated on controls. One additional animal was used as a positive control for hsp72 mRNA. In this animal, forebrain ischemia was produced for 20 minutes, followed by reperfusion for 2 hours, using previously described methods (Kobayashi et al., 1995).
Northern blot analysis
The brain was dissected from the cranium and placed in a coronal rodent brain matrix slicer (Activational Systems, Warren, MI, U.S.A.) chilled to 4°C. The brain was sectioned in the coronal plane with a razor blade 5, 10, and 15 mm behind the frontal pole and neocortical samples (90 to 150 mg) were dissected bilaterally from the resulting frontal, parietal, and occipital sections. The site of KCl application was located within the frontal sample. Tissue samples were frozen in liquid N2 and pulverized using a mortar and pestle chilled in liquid N2. The powders were extracted for total RNA using Ultraspec RNA (Biotecx Laboratories, Houston, TX, U.S.A.). RNA concentrations were measured spectrophotometrically. RNA was denatured and separated (2 µg/lane) on 1.4% agarose, 0.22 mol/L formaldehyde gels. RNA bands were visualized with ethidium bromide and transferred to Nytran Plus filters (Schleicher and Schuell, Keene, NH, U.S.A.). After cross-linking, the filters were prehybridized at 42°C for 2 hours in 1× Northern hybridization buffer (5 Prime → 3 Prime, Boulder, CO, U.S.A.) containing 50% (vol/vol) deionized forrnamide (Clontech Laboratories, Palo Alto, CA, U.S.A.).
To probe the Northern blots, 50 ng DNA was labeled using Redivue [α-32P] dCTP (Amersham, Arlington Heights, IL, U.S.A.) and a random prime labeling kit (Boehringer Mannheim, Indianapolis, IN, U.S.A.). The filters were hybridized at 42°C for 12 to 16 hours with the prehybridization solution containing the labeled and denatured probe. The filters were washed and exposed to Kodak MS film using an MS intensifier screen at –70°C for 4 to 48 hours. Autoradiograms of the Northern blots were quantified by densitometry using a Vista-S6E scanner with transparency adapter and an image analysis program (Molecular Analyst, Bio-Rad Laboratories, Hercules, CA, U.S.A.). Densitometric values were normalized to those of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). For constitutively expressed mRNA, the normalized values of the ipsilateral (KC1) cortex were divided by those of the contralateral cortex to obtain the relative increase caused by CSD.
cDNA probes
Probes for c-fos, BDNF, nexin-1, and glutamine synthetase were rat-specific purified cDNA inserts generated by reverse transcription-polymerase chain reaction cloning using plasmid pCR II (Invitrogen, San Diego, CA, U.S.A.). Primers were designed using sequence information obtained from the Gen-Bank (R90.0), and the specificity of the probes was confirmed by sequencing (DNA Sequencing Facility, University of Pennsylvania).
c-fos. A 706-bp cDNA was generated from template RNA of rat cerebral cortex subjected to CSD. The specific 5′ primer (5′-CGCAGAGCATCGGCAGAA-3′) and 3′ primer (5′-GCGCAGCTAGGGAAGGAGTCA-3′) corresponding to nucleotides (nt) 493 through 510 and nt 1,198 through 1,178 of the coding sequence of rat c-fos cDNA were used (Accession: X06769). This probe detected a single 2.1-kb transcript in RNA samples from rat brain.
BDNF. A 777-bp cDNA was generated from template RNA of rat cerebral cortex subjected to CSD. The specific 5′ primer (5′-CTGCCTTGATGTTTACTT-3′) and 3′ primer (5′-CAGGAAGTGTCTATCCTTAT-3′) corresponding to nt 936 to 953 and nt 2,839 to 2,820 of the exons of rat gene for BDNF were used (Accession: X67107 and D10938, respectively). This probe detected transcripts in two major bands (1.6 to 1.8 kb and 4.2 to 4.4 kb), both of which were used for quantitation of BDNF mRNA.
Nexin-1. A 1017-bp cDNA was generated from template RNA of normal rat brain using 5′ primer (5′-CCAGGTTTTCAATCAGAT-3′) and 3′ primer (5′-GGCCTGTCTACTATAAAC-3′) corresponding to nt 108 to 125 and nt 1,124 to 1,107 of the coding sequence of rat nexin-1 cDNA (Accession: M 17784). This probe detected a single transcript of approximately 2.0 kb.
Glutamine synthetase. A 717-bp cDNA was generated from template RNA of rat cerebral cortex subjected to CSD. The specific 5′ primer (5′-CCCCTGGTTTGGAATGGA-3′) and 3′-primer (5′-TCGTCGCCAGTTTCGTTG-3′) corresponding to nt 515 to 532 and nt 1,231 to 1,214 of rat glutamine synthetase cDNA were used (Accession: M29579). This probe detected a single 2.8-kb transcript.
tPA. A purified 2.4-kb cDNA insert containing the full coding region of rat tPA was kindly provided by R. N. Pittman, Philadelphia, PA, U.S.A. This probe detected a single 2.5-kb transcript.
hsp72 and hsc73. A 2.2-kb cDNA containing the full coding region of rat inducible hsp72 was kindly provided by R. Mestril, San Diego, CA, U.S.A. This probe detected three transcripts: two inducible and a smaller, constitutive transcript. Both of the inducible transcripts were used to quantitate hsp72 mRNA and the constitutive transcript was used to quantitate hsc73 mRNA.
GAPDH. A linearized plasmid pHcGAP containing the full coding region of human GAPDH cDNA was obtained from American Type Culture Collection (ATTC, Rockville, MD, U.S.A.). This probe detected a single 1.2-kb transcript.
Statistical analysis
Differences in mean values of arterial variables between groups were tested for statistical significance using one-way analysis of variance. Differences in mean values of mRNA levels between hemispheres were tested for statistical significance using a paired t test.
RESULTS
Physiologic variables
There were no significant differences in arterial variables between recovery groups (Table 1). One of the 24-hour recovery animals exhibited arterial acidosis and hypotension; thus, the results from this animal were excluded from the statistical analysis. In animals monitored for cortical blood flow, CSD appeared as an abrupt increase in flow, rising to a peak several times higher than baseline and returning to control levels within 1 to 2 minutes (Fig. 1). Application of KCl for 2 hours triggered 11 ± 6 episodes of CSD (mean ± SD, n = 5) in the ipsilateral cortex. Episodes of CSD were not detected in the contralateral cortex.
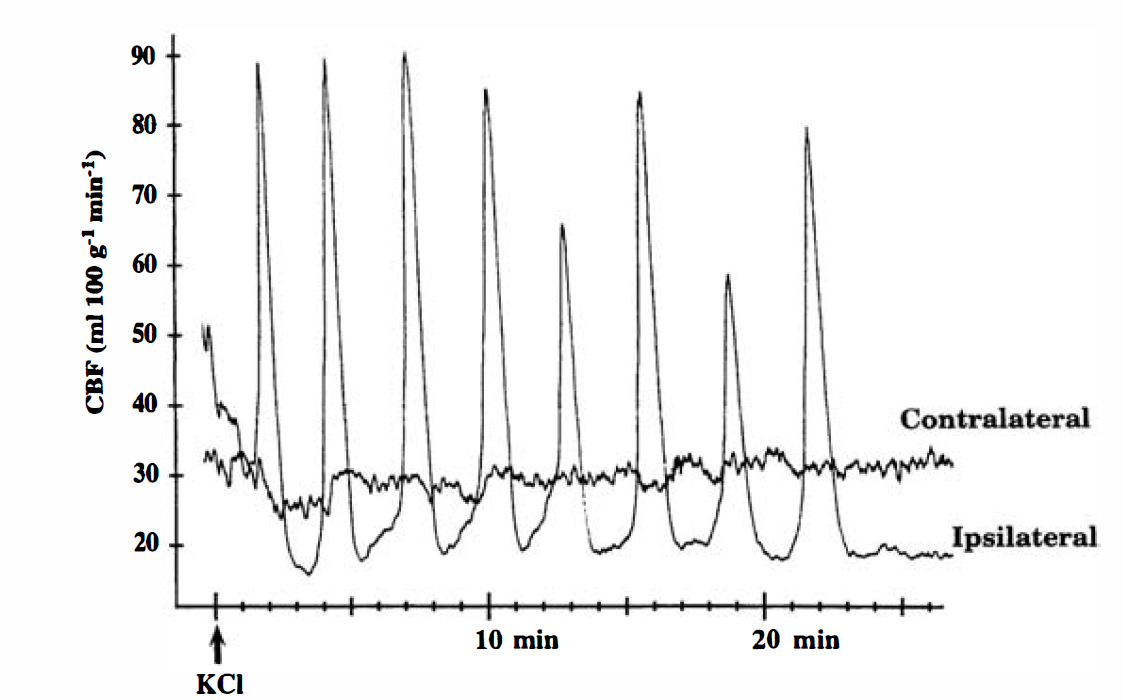
Changes in cortical blood flow during recurrent cortical spreading depression (CSD). KCl (2 mol/L) was applied to the intact dura over the frontal cortex of one hemisphere, and blood flow was monitored over the parietal cortex of the ipsilateral and contralateral hemispheres using laser—Doppler flowmetry. In the ipsilateral cortex, CSD appeared as a sharp increase in cortical blood flow. Episodes of CSD were not detected in the contralateral cortex.
Arterial variables
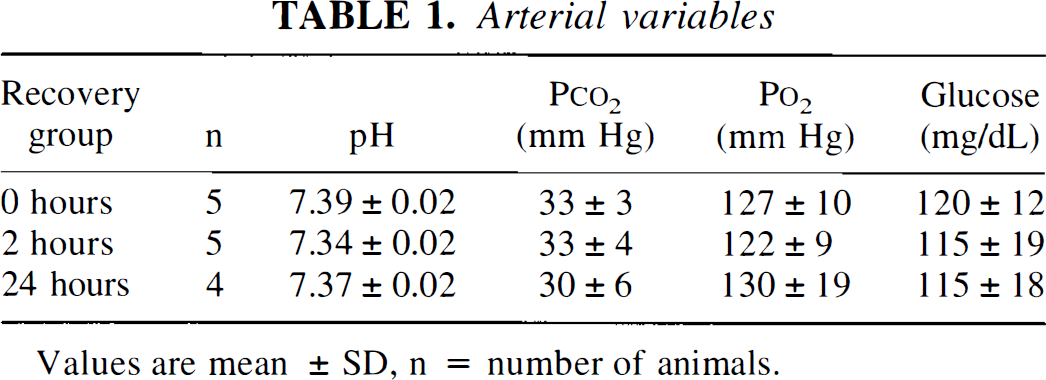
Values are mean ± SD, n = number of animals.
Levels of mRNA
Application of KCl markedly altered mRNA levels in several regions of the ipsilateral cortex (Fig. 2). In the frontal cortex, which includes the site of KCl application, ipsilateral levels of c-fos, BDNF, tPA, and hsp72 mRNA were higher than those in the contralateral cortex and those in the cortex of control animals (Fig. 2, region F). In the parietal cortex, ipsilateral levels of c-fos, BDNF, and tPA mRNA were also increased, whereas hsp72 mRNA was near the level of detection (Fig. 2, region P). In the occipital cortex, ipsilateral levels of c-fos, BDNF, and tPA mRNA remained higher than those in the contralateral cortex, but were lower than those in the frontal and parietal regions (Fig. 2, region O).
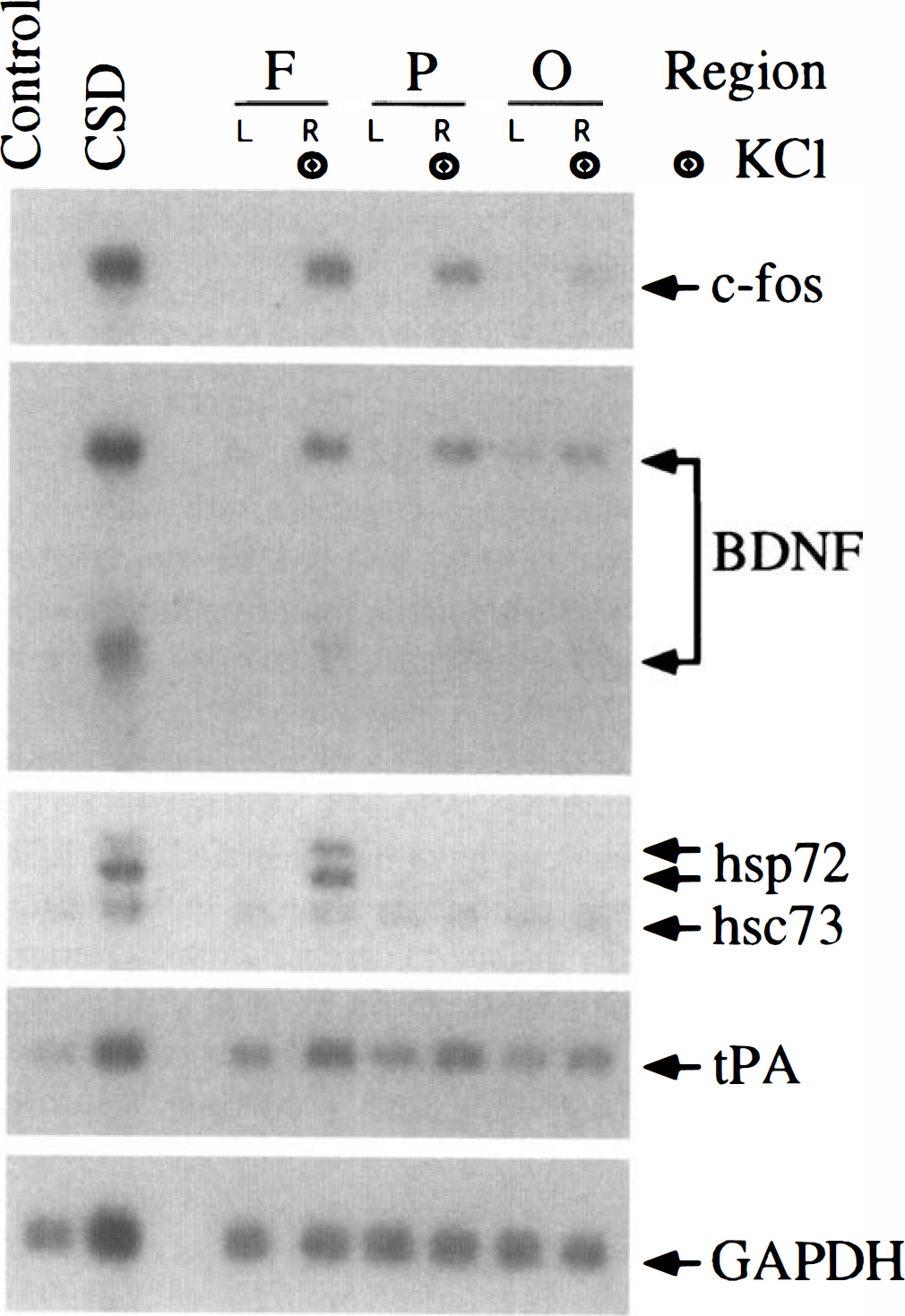
Regional changes in mRNA levels after CSD. Autoradiographic images of Northern blots are shown for c-fos, brain-derived neurotrophic factor (BDNF), 72-kDa heat-shock protein (hsp72), 73-kDa heat-shock cognate (hsc73), tissue plasminogen activator (tPA), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA. RNA samples were prepared from the cerebral cortex of an unoperated on animal (Control), from the frontal cortex (KCl site) of a CSD animal (CSD), and from the frontal (F), parietal (P), and occipital (O) cortex of both hemispheres in an animal at 0 hours of recovery after 2 hours of application of 2 mol/L KCl to the intact dura over the right frontal cortex. Lanes with samples ipsilateral to the KCl application are marked with the target symbol. See Fig. 1 for other abbreviations.
The timecourse of changes in mRNA levels after CSD was determined in parietal cortex to avoid the local effects of KCl in frontal cortex (Fig. 3). At the end of the period of KCl application (0 hours of recovery), ipsilateral levels of c-fos mRNA were markedly increased in all animals (Figs. 3 and 4). At 2 hours of recovery, c-fos mRNA in the ipsilateral cortex remained elevated, but the levels were lower than those at 0 hours of recovery. At 24 hours of recovery, c-fos mRNA in the ipsilateral cortex was below the level of detection in 3 of 5 animals, but was expressed at high levels in the remaining 2 animals. One of the latter animals was excluded from the study owing to arterial abnormalities (see above). This animal also exhibited bilateral elevation of c-fos and hsp72 mRNA. In the remaining 14 animals, mRNA levels in the contralateral cortex were similar to those in unoperated on controls (Fig. 3, controls).
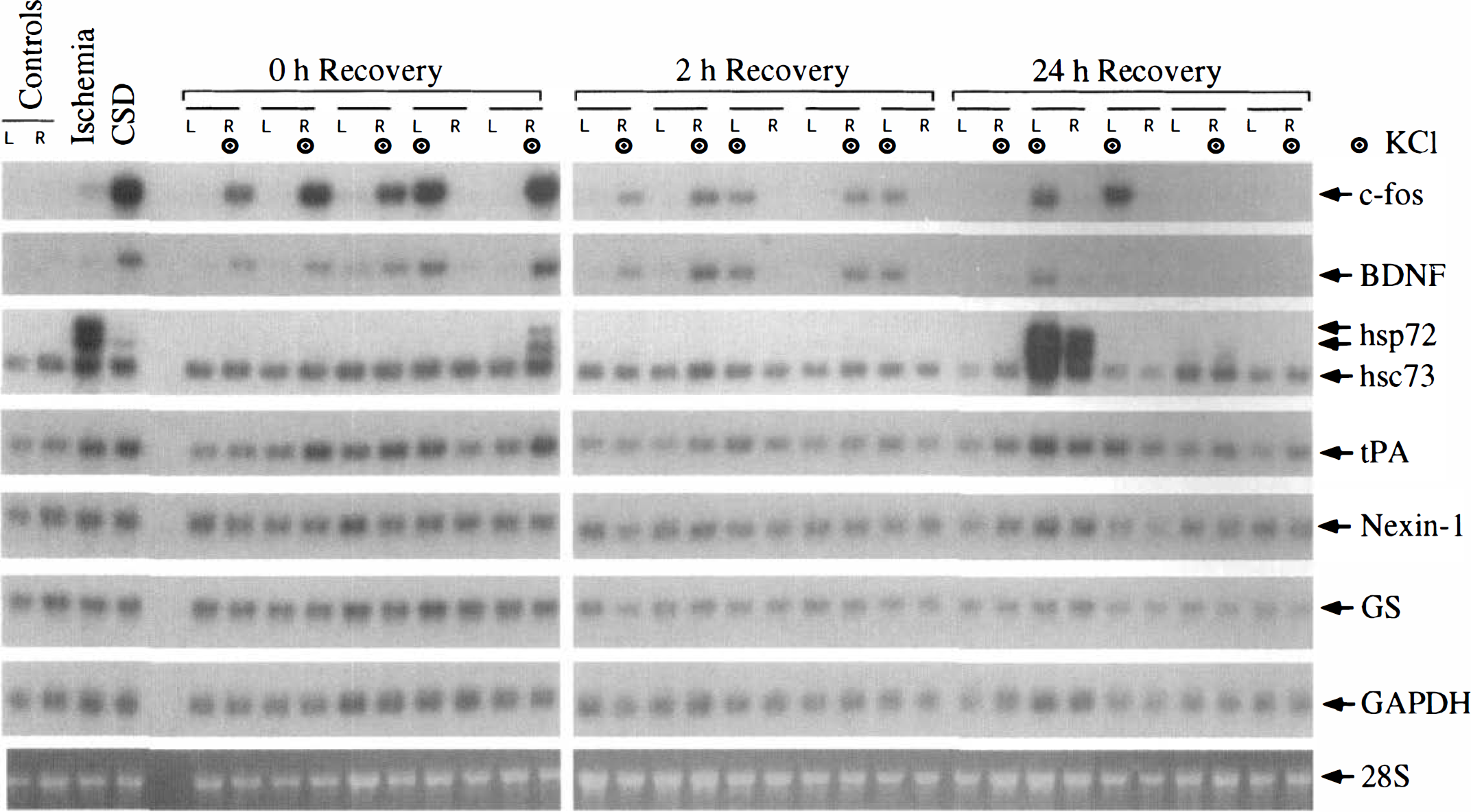
Effect of CSD on mRNA levels in parietal cortex. Autoradiographic images of Northern blots are shown for c-fos, BDNF, hsp72, hsc73, tPA, nexin-1, glutamine synthetase (GS), and GAPDH mRNA. Also shown is the fluorescent image of ethidium bromide—stained 28S rRNA. RNA samples were prepared from the parietal cortex of both hemispheres of an unoperated on animal (Control), from the cortex of an animal subjected to 20 minutes of forebrain ischemia and 2 hours of reperfusion (Ischemia), from the frontal cortex (KCl site) of a CSD animal (CSD), and from the parietal cortex of both hemispheres of 5 animals at 0 hours of recovery, 5 animals at 2 hours of recovery, and 5 animals at 24 hours of recovery after CSD. The side (L or R) of KCl application is marked with the target symbol. See Figs. 1 and 2 for other abbreviations.
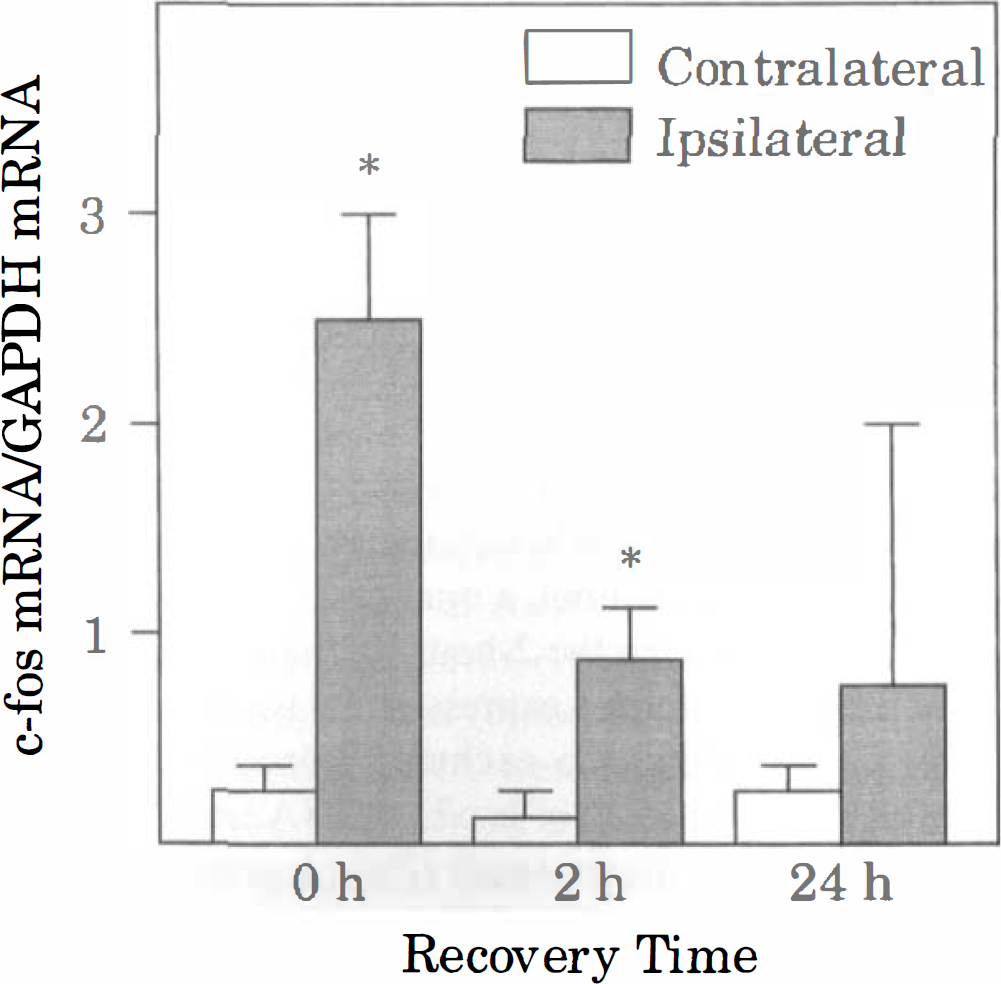
Effect of CSD on c-fos mRNA levels. Densitometric analyses were performed on the corresponding autoradiographic images in Fig. 3. For each sample lane, the optical density of the c-fos mRNA band was normalized with that of GAPDH mRNA. Bars represent mean values of 5 animals (4 animals at 24 hours), and brackets represent standard deviations. Asterisks denote significant differences from the value in the corresponding contralateral cortex, P < 0.05. See Figs. 1 and 2 for other abbreviations.
Cortical spreading depression also caused a robust increase in ipsilateral levels of BDNF mRNA at 0 and 2 hours of recovery (Figs. 3 and 5). However, by 24 hours of recovery, BDNF mRNA was near the level of detection in both hemispheres, with the exception of the excluded animal. Levels of hsp72 mRNA were barely detectable in the ipsilateral cortex in most of the animals at 0 and 2 hours of recovery (Fig. 3). However, in one of the animals at 0 hours of recovery, there was moderate induction of hsp72 mRNA in the ipsilateral cortex. Interestingly, in this animal, a total of 21 episodes of CSD were recorded during the 2-hour KCl application period. As noted above, robust expression of hsp72 mRNA occurred bilaterally in the excluded animal (24 hours of recovery). Quantitation of hsp72 mRNA levels indicated a statistically significant small (20%) increase at 2 hours of recovery, but not at 0 or 24 hours of recovery (Fig. 6). Similarly, levels of hsc73 mRNA were significantly higher (10% to 20%) in the ipsilateral cortex than in the contralateral cortex at 2 and 24 hours of recovery, but not at 0 hours of recovery (Fig. 7). Cortical spreading depression also caused a significant elevation of tPA mRNA in the ipsilateral cortex at 0 and 2, but not 24 hours of recovery (Figs. 3 and 7). Finally, there were no significant differences between hemispheres in the levels of nexin-1 and glutamine synthetase mRNA at any time studied (Figs. 3 and 7).
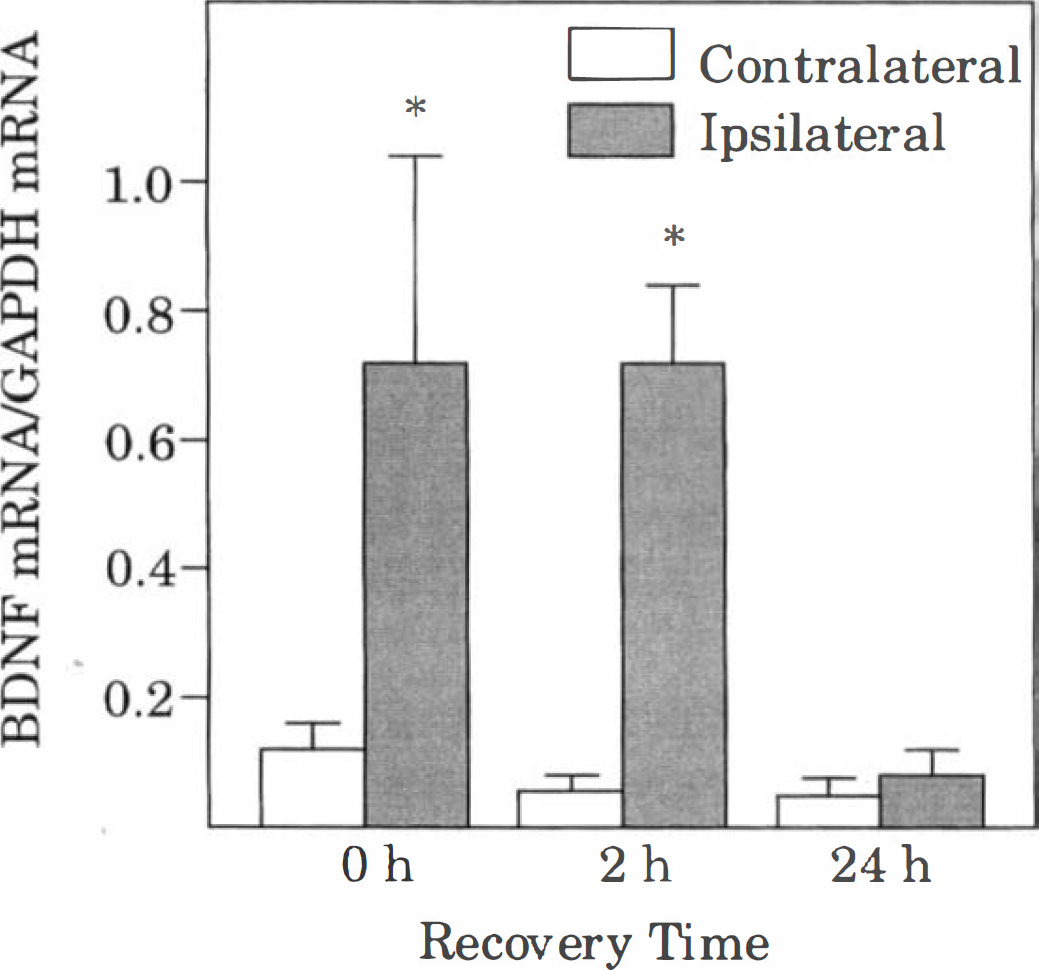
Effect of CSD on BDNF mRNA levels. For each sample lane, the optical densities of both BDNF mRNA bands (1.6 to 1.8 and 4.2 to 4.4 kb) were measured, and their sum was normalized with that of the GAPDH mRNA band. Bars represent mean values of 5 animals (4 animals at 2.4 hours), and brackets represent standard deviations. Asterisks denote significant differences from the value in the corresponding contralateral cortex, P < 0.05. See Figs. 1 and 2 for other abbreviations.
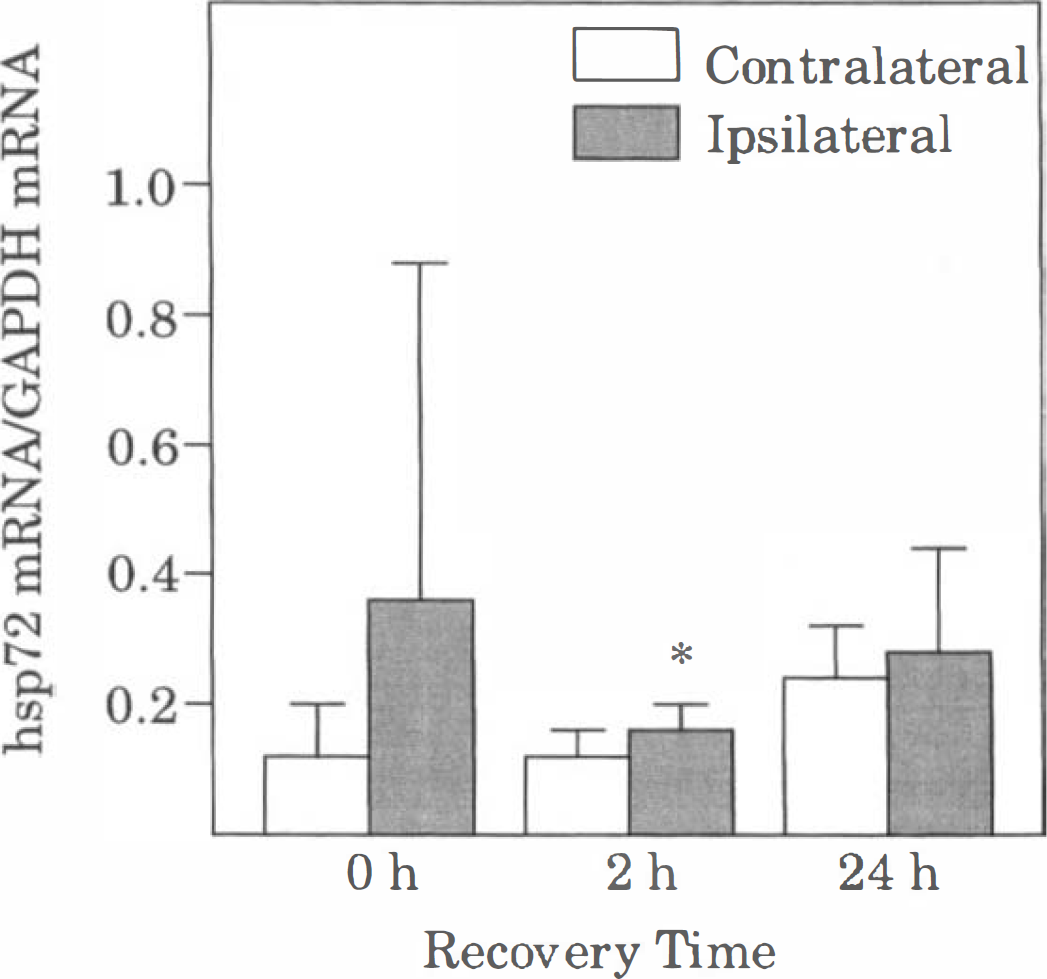
Effect of CSD on hsp72 mRNA levels. For each sample lane, the optical densities of both hsp72 mRNA bands were measured, and their sum was normalized with that of the GAPDH mRNA band. Bars represent mean values of 5 animals (4 animals at 2.4 hours), and brackets represent standard deviations. Asterisks denote significant differences from the value in the corresponding contralateral cortex, P < 0.05. See Figs. 1 and 2 for other abbreviations.
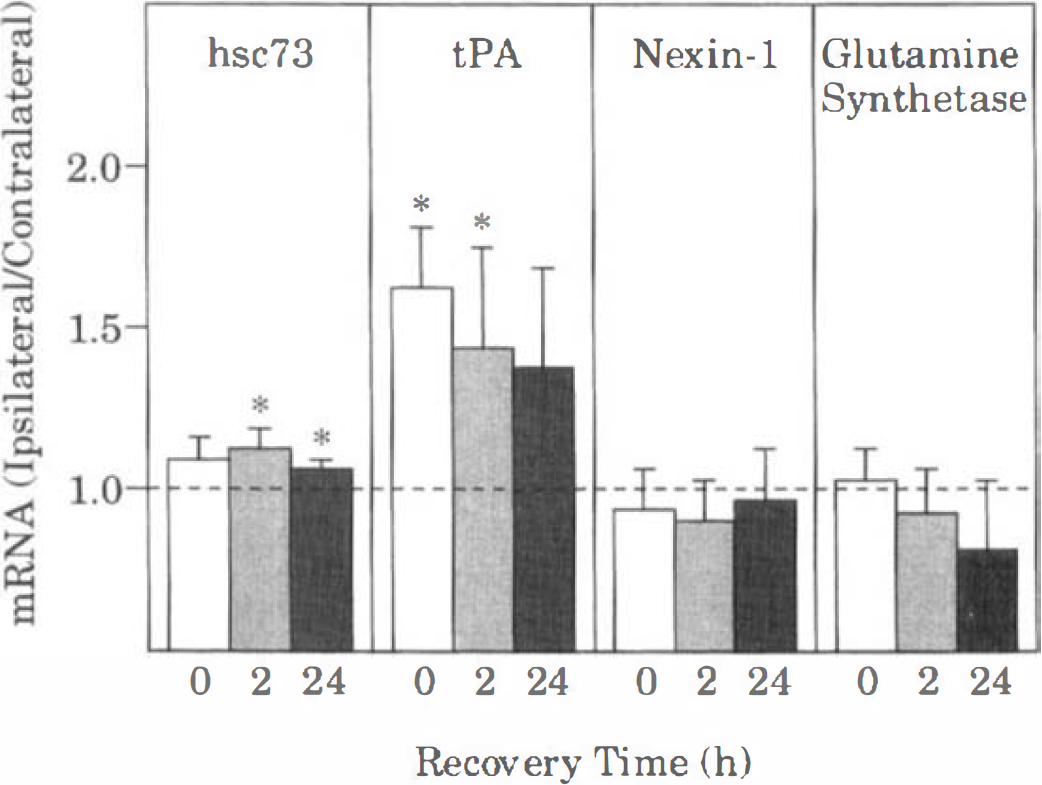
Effect of CSD on hsc73, tPA, nexin-1, and glutamine synthetase mRNA levels. Optical densities of the autoradiographic bands of the mRNA were first normalized to the density of GAPDH mRNA. In each animal, the normalized value from the ipsilateral hemisphere was divided by that in the contralateral hemisphere. Bars represent mean values of 5 animals (4 animals at 24 hours recovery), and brackets indicate standard deviations. Asterisks denote significant differences from the contralateral value (1.0), P < 0.05. See Figs. 1 and 2 for other abbreviations.
DISCUSSIONS
Preconditioning the brain with CSD induces a lasting tolerance of neurons to a subsequent episode of ischemia (Kawahara et al., 1995; Kobayashi et al., 1995; Matsushima et al., 1996). However, the long-term molecular alterations after CSD that contribute to the acquisition of neuronal tolerance remain poorly defined. The present results confirm and extend previous findings that CSD triggers transient induction of c-fos and BDNF mRNA (Kokaia et al., 1993; Kobayashi et al., 1995). In addition, the results demonstrate that CSD causes rapid elevation of tPA mRNA, but does not cause substantial changes in hsp72, hsc73, nexin-1, or glutamine synthetase mRNA at the times studied. Thus, the induction of neuronal tolerance after CSD may be mediated by changes in expression of FOS, BDNF, and tPA, but is not likely to involve altered expression of hsp72, hsc73, nexin-1, or glutamine synthetase.
Previous investigations have demonstrated that KCl-induced CSD triggers rapid expression of FOS-like immunoreactivity in the ipsilateral cerebral cortex (Herrera and Robertson, 1990; Herdegen et al., 1993). FOS-like immunoreactivity was detected as early as 1 hour after application of KCl, with maximal expression occurring at 3 hours. In our own prior work, in situ hybridization indicated that c-fos mRNA increased by the end of the KCl application period and at 2 hours of recovery, but returned to control by 24 hours (Kobayashi et al., 1995). To our knowledge, the present results are the first quantitative description of the timecourse of steady-state levels of c-fos mRNA after CSD. Consistent with the previous results, the present study clearly demonstrates that CSD triggers a robust elevation of c-fos mRNA by the end of the 2-hour KCl application and that levels of c-fos mRNA decline rapidly thereafter. Presumably, elevation of c-fos mRNA is mediated by alterations in intracellular Ca2+ and cyclic adenosine monophosphate (cAMP), alterations that are known to occur during CSD (Krivanek, 1977; Hansen and Zeuthen, 1981). Because FOS functions as part of a transcriptional activating system, its induction after CSD could potentially alter the expression of neuroprotective proteins. However, direct evidence linking FOS to expression of neuroprotective proteins after CSD is currently lacking.
The present results extend previous in situ hybridization studies demonstrating that BDNF mRNA is also elevated by CSD (Kokaia et al., 1993). The rapid elevation of BDNF mRNA after CSD is similar to that reported in rat hippocampus after focal trauma or electrical stimulation, occurring in both cases in the presence of inhibitors of protein synthesis (Hughes et al., 1993; Lauterborn et al., 1996). Thus, similar to c-fos mRNA, BDNF mRNA may be induced immediately by CSD without the need for synthesis of protein transcription factors. Indeed, examination of the promoter sequence of exon III reveals the presence of a cAMP-response element, which may account for the rapid increase in BDNF mRNA after CSD. However, BDNF mRNA levels did not decline in parallel with c-fos mRNA between 0 and 2 hours of recovery. The timecourse differences between BDNF and c-fos mRNA levels after CSD may be caused by differences in (1) the duration of active transcription or (2) the half-life of the two transcripts. However, the present observations cannot distinguish between these possible explanations.
More importantly, the influence of BDNF expression after CSD on the tolerance of neurons to ischemia is likely to be beneficial. Brain-derived neurotrophic factor has been shown to be neuroprotective in vitro (Ghosh et al., 1994). Brain-derived neurotrophic factor has also been reported to protect neurons in rodent models of cerebral ischemia (Beck et al., 1994). It should be noted, however, that the timecourse of expression of BDNF mRNA after CSD is brief, relative to the timecourse of induction of neuronal tolerance to ischemia (Kobayashi et al., 1995; Matsushima et al., 1996). Thus, neuronal tolerance has been demonstrated as early as 24 hours after CSD, a time at which levels of BDNF mRNA have returned to normal. Because the timecourse of induction of BDNF protein after CSD is not known, further work is needed to determine the role of BDNF in CSD-induced neuronal tolerance to ischemia.
Increased levels of tPA mRNA have been demonstrated previously after convulsive seizures, kindling, long-term potentiation, and motor learning in the rat (Qian et al., 1993; Seeds et al., 1995). Subsequent work has indicated that KCl-induced neuronal depolarization causes rapid secretion of tPA from PC12 cells and elevation of tPA activity in mouse hippocampus (Gualandris et al., 1996). These results suggest that depolarization of neurons by physiologic or pathologic stimuli increases the expression of tPA. The present findings showing increased levels of tPA mRNA after CSD are consistent with this suggestion. The rapid elevation of tPA mRNA after CSD is also consistent with the presence of a consensus cAMP-responsive element in the promoter of the rat tPA gene (Holmberg et al., 1995).
However, the consequences of tPA expression with respect to neuronal tolerance are controversial. Increased activity of tPA has been associated with neurite outgrowth, migration, and regeneration (Krystosek and Seeds, 1981). Thus, expression of tPA after CSD might facilitate synaptic remodeling, thereby promoting neuronal recovery after an episode of ischemia. However, recent evidence suggests that tPA activity may adversely affect neuronal injury after excitotoxic and ischemic insults (Tsirka et al., 1995). Mice lacking the tPA gene exhibited increased tolerance of hippocampal neurons to kainic acid, suggesting that tPA may be an important mediator of excitotoxic neuronal death. This apparent discrepancy may be explained, in part, by two significant differences in tPA expression between mouse brain and rat brain. First, the mouse tPA gene lacks a cAMP-response element and, thus, is not an IEG in the mouse (Holmberg et al., 1995). Second, in mouse brain, at least in hippocampus, tPA was found to be expressed primarily in microglia (Tsirka et al., 1995). In rat brain, tPA mRNA has been localized to ventricular ependyma, meninges, blood vessels, Purkinje cell layer of the cerebellum, scattered small cells throughout the brain, and a subpopulation of large cells (presumed neurons) in brainstem and cerebral cortex (Ware et al., 1995). Thus, the role of tPA in neuronal injury in rat brain may differ from that in mouse brain.
The present results indicate that CSD caused significant increases in the levels of hsp72 and hsc73 mRNA at 2 hours of recovery and of hsc73 nRNA at 24 hours of recovery. However, the biologic significance of the small increases (10% to 20%) is questionable. Nevertheless, overexpression of hsp70 by only 10% to 12% in Drosophila melanogaster during a heat stress was sufficient to improve subsequent survival at normal temperatures (Tatar et al., 1997). Thus, it is difficult to exclude the possibility of a contribution of hsp72 or hsc73 to the induction of neuronal tolerance after CSD. At present, however, there is no published evidence that CSD increases expression of hsp72 protein after CSD. In our own studies, hsp72 immunoreactivity has not been detected in cortical regions apart from the site of KCl application (unpublished observations). Thus, in our opinion, it is unlikely that CSD induces biologically significant amounts of hsp72 or hsc73 proteins.
Finally, the present results indicate that CSD failed to cause significant alterations in mRNA levels for two other putative neuroprotective proteins, nexin-1 and glutamine synthetase. Thus, these proteins are unlikely to have a role in the neuronal tolerance induced by CSD. It should be cautioned, however, that whereas expression of many proteins is regulated at the level of transcription, expression of other proteins is regulated at the level of translation. Because the present study examined levels of mRNA only, the possibility that CSD alters protein levels or enzymatic activity without changing mRNA levels cannot be excluded.