
Abstract
Ischemic and excitotoxic insults to the brain induce rapid production of tumor necrosis factor-α (TNF), but the role of TNF in neuronal responses to brain injury are unclear. Two different TNF receptors (p55 and p75) are expressed in neurons and glia, To understand the role of TNF in brain injury, we generated mice that lack p55, p75, or both receptors, We report that neuronal damage after focal cerebral ischemia—reperfusion is significantly increased in mice lacking p55 receptors (85 ± 7 mm3 infarct volume; mean ± SD) compared with wild-type mice (70 ± 8 mm3) and mice lacking p75 receptors (72 ± 6 mm3). Moreover, mice lacking p55 receptors exhibited increased degeneration of CA3 hippocampal neurons after administration of the excitotoxin kainic acid compared with wild-type mice and mice lacking p75 receptors. When taken together with recent data showing that TNF can prevent apoptosis of cultured neurons exposed to oxidative and metabolic insults, our findings suggest that TNF plays a neuroprotective role after acute brain insults.
The dramatic increase in tumor necrosis factor (TNF) production by glial cells and neurons after ischemic and excitotoxic brain injury (Tchelingerian et al., 1993; Liu et al., 1994; Bruce et al., 1996; Buttini et al., 1996; Shohami et al., 1994; Botchkina et al., 1997; Zhai et al., 1997) suggests an important role for this cytokine in modifying the neurodegenerative process. Exposure of cultured hippocampal and cortical cell cultures to TNF was reported to either promote neuronal degeneration (Chao and Hu, 1994) or protect neurons against excitotoxic, metabolic, and oxidative injury (Cheng et al., 1994; Barger et al., 1995; Mattson et al., 1997). Tumor necrosis factor is a potent activator of microglia, and might thereby induce production of neurotoxic substances such as nitric oxide and excitotoxins (McGeer et al., 1993; Rothwell and Hopkins, 1995). On the other hand, TNF can act directly on neurons to increase their resistance to oxidative stress (Barger et al., 1995; Mattson et al., 1997) and stabilize calcium homeostasis (Cheng et al., 1994). In vivo studies have yielded conflicting data concerning the role of TNF in ischemic brain injury. Intracerebroventricular administration of TNF to spontaneously hypertensive rats before permanent or transient middle cerebral artery occlusion significantly increased infarct volume and worsened behavioral outcome (Barone et al., 1997). Intravenous (Lavine et al., 1998) and intracortical (Meistrell et al., 1997) administration of a TNF-neutralizing antibody significantly reduced cortical infarct volume after permanent or transient middle cerebral artery occlusion. Intracortical administration of a TNF-binding protein reduced cortical infarct volume after permanent middle cerebral artery occlusion in mice (Nawashiro et al., 1997a). On the other hand, mice rendered genetically unresponsive to TNF exhibit increased infarct size 24 hours after middle cerebral artery occlusion (Bruce et al., 1996), and intraventricular administration of TNF to adult mice before middle cerebral artery occlusion significantly reduces cortical infarct size (Nawashiro et al., 1997b).
Tumor necrosis factor binds two different receptors, p55 and p75, both of which are expressed in neurons throughout the brain (Kinouchi et al., 1991; Cheng et al., 1994). Activation of the p55 receptor results in recruitment of TNF receptor-associated proteins and a phosphorylation cascade that results in activation of the transcription factor NF-κB (Kruppa et al., 1992; Smith et al., 1994); the signaling pathways of p75 receptor are not established. Studies of cultured primary rat hippocampal neurons (Barger et al., 1995; Mattson et al., 1997) and tumor cell lines (Beg and Baltimore, 1996; Van Antwerp et al., 1996; Wang et al., 1996) have clearly shown that activation of NF-κB prevents apoptosis, possibly by inducing the expression of the antioxidant enzyme manganese superoxide dismutase (Mn-SOD) (Mattson et al., 1997). Because the roles of p55 and p75 receptors in mediating cellular responses to brain injury are unknown, we have generated mice lacking either p55 or p75 receptors, or both p55 and p75 receptors (Zheng et al., 1995; Bruce et al., 1996). In the present study we used these mice to directly address the role of endogenous TNF in modifying neuronal injury after focal cerebral ischemia and excitotoxin administration. The data suggest a critical role for activation of p55 receptors in enhancing neuronal resistance to ischemic and excitotoxic injury.
MATERIALS AND METHODS
Generation and maintenance of TNF receptor knockout mice
The gene targeting strategy used to generate lines of mice lacking either the p55 TNF receptor (p55–/–) or the p75 receptor (p75–/–) has been described previously (Zheng et al., 1995). Mice lacking both p55 and p75 receptors (p55/p75–/–) were generated by cross-breeding of p55–/– mice with p75–/–mice. Wild-type mice were C57BL/6 × 129 F(1); all knockout lines were maintained on a random C57BL/6 × 129 background. Experiments were performed in 3-month-old male mice (25 to 30 g body weight); mice were fed ad libitum and maintained on a 12 hours light/12 hours dark cycle. Mice lacking either p55 or both TNF receptors show no overt phenotypes and reproduce normally, but do exhibit altered responses of lymphocytes to a variety of infectious agents (Pfeffer et al., 1993; Zheng et al., 1995). Previous analyses revealed no overt alterations in brain structure or in performance on behavioral tests in p55/p75–/– mice compared with their wild-type counterparts (Bruce et al., 1996).
Focal cerebral ischemia and administration of kainic acid
The focal cerebral ischemia—perfusion model used was similar to that of Yang et al. (1994) and has been described in our previous studies (Bruce et al., 1996; Keller et al., 1998). The method involves occluding the middle cerebral artery for 1 hour with a nylon thread and then removing the thread to allow reperfusion. Thermistor probes were inserted into the rectum and temporalis muscles to monitor body and brain temperature, which were maintained at 36°C to 37°C by external warming. Blood samples were taken at baseline (10 minutes before occlusion), during ischemia (30 to 35 minutes after thread placement), and 10 minutes after reperfusion for measurements of blood gases and pH. In addition, cerebral blood flow and blood pressure were measured before, during, and after ischemia using methods described previously (Endres et al., 1998). Twenty-four hours after ischemia mice were anesthetized with chloral hydrate and killed. Brains were cut into 2-mm coronal sections, and the sections were stained with triphenyltetrazolium chloride for 30 minutes at 37°C. Images of the stained brain slices were captured using a digital camera, and the infarct volume was quantified as described previously (Smith-Swintosky et al., 1996).
Methods for stereotaxic administration of kainic acid into the hippocampus are detailed in our previous studies (Bruce et al., 1996; Smith-Swintosky et al., 1996). Briefly, kainic acid (0.3 µg in a volume of 0.5 µL) was injected unilaterally into dorsal hippocampus (dorsoventral −2.0, mediolateral +2.4, anteroposterior –1.8 from bregma) of anesthetized mice. Mice were monitored during a 4-hour period after kainate administration; all mice exhibited wet dog shakes and tonic-clonic seizures during this period with no overt differences among genotypes. Mice were killed 24 hours later and perfused transcardially with 4% paraformaldehyde. Coronal brain sections (30 µm) were cut on a freezing microtome and were stained with cresyl violet. Nissl-positive undamaged neurons were counted in hippocampal region CA3 (three 45× fields per section and three sections per brain). Cell counts were performed without knowledge of the genotype or treatment history of the mice.
RESULTS AND DISCUSSIONS
Mice of the four different genotypes (wild-type, p55–/–, p75–/–, and p55/p75–/–) were subjected to 1 hour of middle cerebral artery occlusion and were killed 24 hours after the onset of reperfusion. Infarct sizes were not significantly different in wild-type and p75–/– mice (Fig. 1). In contrast, infarct sizes were significantly greater in p55–/– and p55/p75–/– mice compared with wild-type and p75–/– mice (Fig. 1). There were no significant differences in physiologic parameters among the four groups of mice (Table 1), suggesting that the increased lesion size in mice lacking p55 TNF receptors was not caused by vascular alterations. Additional groups of mice of the four different genotypes were administered either saline or kainic acid into the dorsal hippocampus and were killed 24 hours later. In wild-type and p75–/– mice kainic acid caused degeneration of approximately 15% to 20% of CA3 hippocampal neurons (Fig. 2). The extent of CA3 neuron degeneration was significantly increased to 40% to 50% in mice lacking either p55 or both TNF receptors (Fig. 2). The increased damage to CA3 neurons in mice lacking p55 receptors might result from a direct neuroprotective effect of endogenous TNF acting on this receptor (Cheng et al., 1994). However, although all mice exhibited seizures after kainate administration, we cannot rule out the possibility of subtle differences in intensity of seizure activity among the different genotypes.
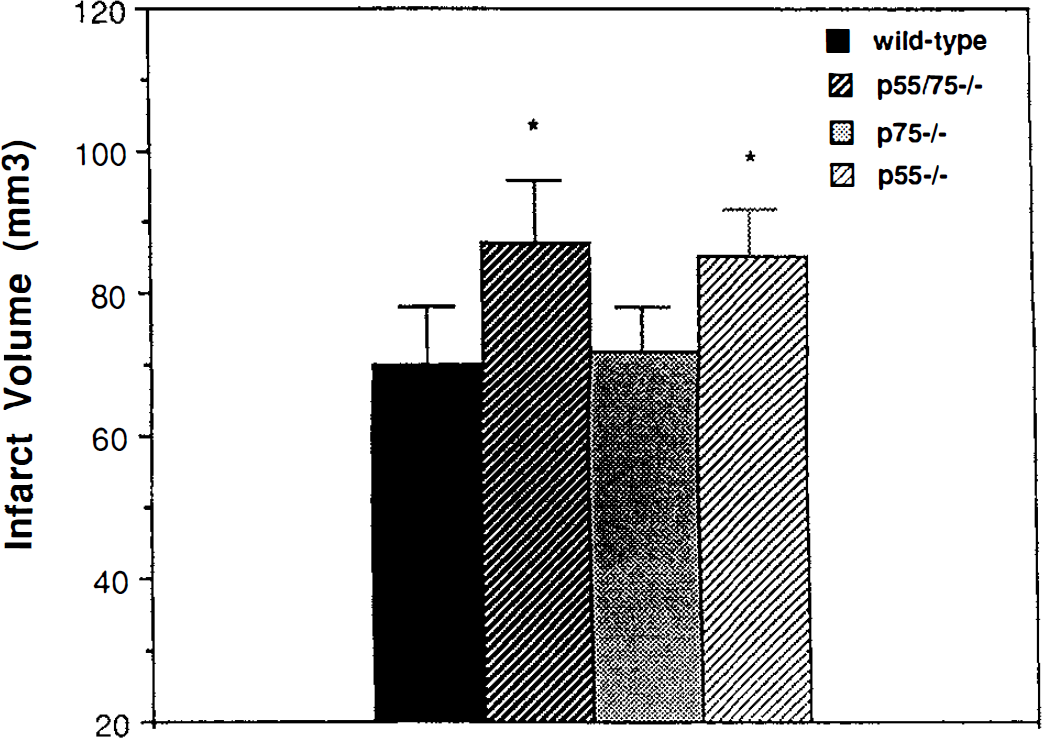
Infarct size after focal ischemia—reperfusion is increased in mice lacking the p55 tumor necrosis factor (TNF) receptor. Middle cerebral artery occlusion was performed on wild-type (n = 10), p55/p75–/– (n = 11), p75–/– (n = 9), and p55–/– (n = 12) mice for 1 hour. Twenty-four hours after reperfusion mice were killed and infarct volume was quantified (see Methods). Values are the mean and SD. (*, P < 0.05 compared with values for wild-type and p75–/– mice by analysis of variance with Scheffe's post hoc tests.)
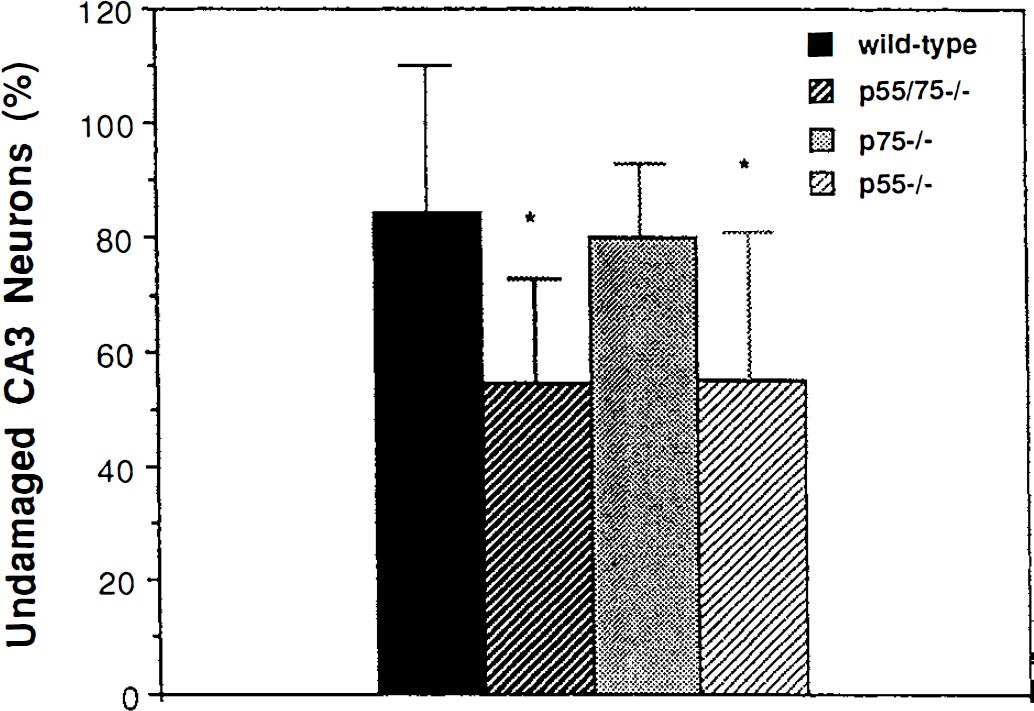
Seizure-induced damage to hippocampal CA3 neurons is increased in mice lacking the p55 TNF receptor. Kainic acid was administered into the dorsal hippocampus of wild-type (n = 20), p55/p75–/– (n = 16), p75–/– (n = 12), and p55–/– (n = 13) mice. Twenty-four hours later mice were killed and damage to CA3 neurons was quantified (see Methods). Values are the mean and SD. (*, P < 0.01 compared with values for wild-type and p75–/– mice by analysis of variance with Fisher's post hoc tests.)
Physiologic parameters before, during, and 30 minutes after middle cerebral artery occlusion in wild-type mice, and in mice lacking one or both TNF receptors
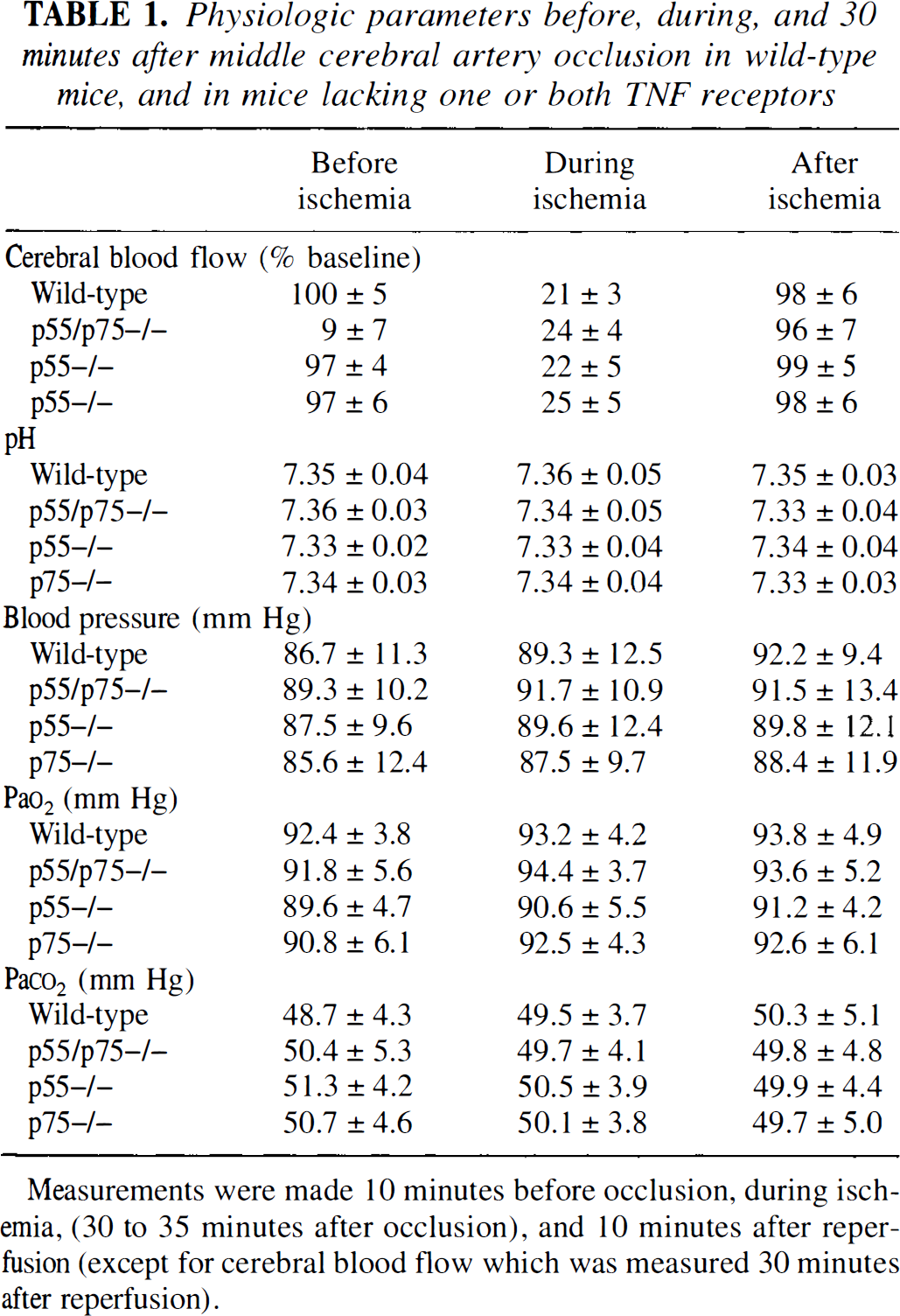
Measurements were made 10 minutes before occlusion, during ischemia, (30 to 35 minutes after occlusion), and 10 minutes after reperfusion (except for cerebral blood flow which was measured 30 minutes after reperfusion).
In previous studies exogenous TNF (Barone et al., 1997) or TNF-blocking antibodies or peptides (Barone et al., 1997; Meistrell et al., 1997; Nawashiro et al., 1997a) were used to assess the role of TNF in ischemic brain injury. The latter studies did not establish the distribution of TNF or TNF-blocking agents within the brain, and the cellular sites of action responsible for their effects on injury extent are therefore unclear. In contrast to the prior studies, we used a gene targeting approach in which mice were generated that lack TNF receptors and are therefore incapable of responding to injury-induced increases in TNF. Our data suggest that activation of the p55 TNF receptor by endogenous TNF plays an important role in transducing an antideath signal in neurons after ischemic and excitotoxic brain injuries. The antideath signal may involve activation of the transcription factor NF-κB, which is known to occur after activation of p55 receptors (Kruppa et al., 1992; Barger et al., 1995) and after cerebral ischemia (Salminen et al., 1995; Clemens et al. 1997), and this transcription factor is known to be critical for prevention of apoptosis by TNF and nerve growth factor in cell culture studies (Barger et al., 1995; Beg and Baltimore, 1996; Mattson et al., 1997; Taglialatela et al., 1997). Indeed, we have found that NF-κB activation is delayed for at least 6 hours after traumatic brain injury in mice lacking both TNF receptors (P. Sullivan, S. Scheff, and M. P. Mattson, manuscript in preparation). Recent studies have shown that TNF can induce the expression two different genes, the antioxidant enzyme Mn-SOD (Mattson et al., 1997) and the calcium-binding protein calbindin D28k (Cheng et al., 1994; Mattson et al., 1995), that may contribute to neuroprotection. Interestingly, levels of Mn-SOD increase after ischemic brain injury (Liu et al., 1993), and TNF may mediate such increases as kainic acid-induced increases in Mn-SOD levels in hippocampal neurons occur in wild-type mice, but not in mice lacking both TNF receptors (Bruce et al., 1996). Another potential mechanism whereby TNF might modify ischemic and excitotoxic neuronal injury is by altering the expression or activity of glutamate receptors and voltage-dependent calcium channels (Furukawa and Mattson, 1998).
There is increasing agreement that pretreatment with TNF (in cell culture or in vivo) can protect neurons against excitotoxic, oxidative, and ischemic injuries (Cheng et al., 1994; Barger et al., 1995; Nawashiro et al., 1997b). However, the role of endogenous TNF after brain injury is less clear, and likely quite complex. Because both the p55 and p75 TNF receptors are widely expressed by neurons, astrocytes, microglia, and vascular cells (macrophages and endothelial cells), both direct and indirect actions of TNF on neurons must be considered. Activation of microglia and circulating macrophages by TNF may adversely affect neurons because activated microglia and macrophages generate toxic oxygen radicals (e.g., superoxide and nitric oxide) and excitotoxins (McGeer et al., 1993). However, microglia and macrophages are unlikely to contribute to the initial neuronal injury that occurs after cerebral ischemia or severe epileptic seizures as there is a time lag of many hours to days that is required for their full activation and movement to the site of injury. Tumor necrosis factor is also well-known to have effects on vascular endothelial cells that might be expected to impact on delayed neuronal damage after brain injury (Lefer and Ma, 1993). Although we found no differences in cerebral blood flow, mean arterial blood pressure, and blood gas and pH levels among wild-type mice and mice lacking either or both TNF receptors, we cannot completely rule out effects of TNF on other vascular parameters (e.g., blood-brain barrier functions). Clearly, a better understanding of the actions of TNF on the various cell types involved in ischemic brain injury will be required to develop effective interventions that are based on cell type-specific manipulations of TNF signaling pathways.
Footnotes
Acknowledgments
The authors thank W. Fu, R. McFall, L. Yang, and J. Yu for technical assistance.