
Abstract
The rate of ischemic brain injury varies with the brain region, requiring only hours in striatum but days in hippocampus. Such maturation implies the existence of endogenous neuroprotective mechanisms. Adenosine is an endogenous neuroprotectant regulated by adenosine kinase (ADK). To investigate, whether adenosine might play a role in protecting the hippocampus after focal ischemia, we subjected transgenic mice, which overexpress ADK in hippocampal neurons (Adk-tg mice) to transient middle cerebral artery occlusion (MCAO). Although the hippocampus of wild-type (wt) mice was consistently spared from injury after 60 mins of MCAO, hippocampal injury became evident in Adk-tg mice after only 15 mins of MCAO. To determine, whether downregulation of hippocampal ADK might qualify as candidate mechanism mediating endogenous neuroprotection, we evaluated ADK expression in wt mice after several periods of reperfusion after 15 or 60 mins of MCAO. After 60 mins of MCAO, hippocampal ADK was significantly reduced in both hemispheres after 1, 3, and 24 h of reperfusion. Reduction of ADK-immunoreactivity corresponded to a 2.2-fold increase in hippocampal adenosine at 3 h of reperfusion. Remarkably, a significant reduction of ADK immunoreactivity was also found in the ipsilateral (stroked) hippocampus after 15 mins of MCAO and 3 h of reperfusion. Thus, transient downregulation of hippocampal ADK after stroke might be a protective mechanism during maturation hippocampal cell loss.
Introduction
Regional differences in brain cell responses to ischemia have been appreciated for decades. In particular, the rate of injury varies with the brain region, requiring only hours in striatum but days in hippocampus (Pulsinelli et al, 1982). The delayed injury of hippocampal neurons has not only been described in animal studies of stroke (Pulsinelli et al, 1982) but also in humans after cardiorespiratory arrest (Petito et al, 1987) and in cell culture models of ischemia (Xu et al, 2001). These findings imply that the delayed injury of hippocampal neurons after ischemia might be due to endogenous neuroprotective control. This might be of importance for the regenerative capabilities of the injured brain, for example, because the subgranular zone of the hippocampus, like the subventricular zone, constitutes one of the sources for stroke-induced neurogenesis (Arvidsson et al, 2002). Several endogenous adaptive responses of the brain have been described leading to decreased neuronal excitability, decreased synaptic glutamate accumulation, limited calcium mobilization in the postsynaptic neuron, protection against calcium-dependent degenerative effects, enhancement of neuronal energetics, and inhibition of apoptosis (Sapolsky, 2001).
In this study we addressed the question, whether adenosine, an endogenous neuroprotectant (Dragunow and Faull, 1988) and anticonvulsant (Boison, 2006) of the brain, might be involved in protecting the hippocampus after middle cerebral artery occlusion (MCAO). Adenosine inhibits neuronal activity through activation of adenosine receptors (A1; A2A, A2B, and A3) (Fredholm et al, 2005). Thus, by activation of A1 receptors, adenosine prevents the spread of seizure activity (Young and Dragunow, 1994) and of cell death after status epilepticus (Fedele et al, 2006) or traumatic brain injury (Kochanek et al, 2006). In the acute response to a harmful stimulus, activation of A1 receptors by adenosine exerts a potent inhibitory presynaptic feedback mechanism to reduce the release of injurious excitatory neurotransmitters, in particular glutamate. Simultaneously, adenosine hyperpolarizes the postsynaptic membrane, limits the activation of N-methyl-
Adenosine kinase is the key enzyme for the regulation of ambient levels of adenosine (Boison, 2006) removing adenosine by phosphorylation into AMP. A high flux rate in a substrate cycle between adenosine and AMP, involving a variety of different 5’ nucleotidases and adenosine kinase (ADK), allows for rapid changes in ambient adenosine levels as a response to subtle changes in ADK activity. Because equilibrative and concentrative nucleoside transporters rapidly exchange intra- and extracellular levels of adenosine, the extracellular levels of adenosine are largely dependent on the intracellular activity of ADK. We have previously shown that transgenic mice, which overexpress ADK in brain, but not in peripheral organs (Fedele et al, 2005), have reduced protective levels of endogenous adenosine in brain (Fedele et al, 2005) and therefore display aggravated cell death after ischemia (Pignataro et al, 2006). Because overexpression of ADK appears to be confined to brain, we do not expect a contribution of extracerebral physiologic effects to the observed brain phenotypes.
In contrast, inhibition of ADK is highly effective in increasing the concentration of extracellular adenosine (Boison, 2006). Thus, intracerebral implants of ADK-deficient stem cells provide powerful neuroprotection to the ischemic brain (Pignataro et al, 2006). These findings suggest that even subtle changes in ADK activity have a tremendous impact on the vulnerability of the injured brain. However, little is known how endogenous ADK responds to brain injury. To elucidate a potential endogenous mechanism of the hippocampal formation to delay injury after stroke, we investigated (i) the degree of stroke-induced hippocampal injury as a function of endogenous ADK levels, and (ii) expression levels of endogenous ADK as a function of stroke. We show (i) increased hippocampal vulnerability under high ADK levels and (ii) transient downregulation of hippocampal ADK as a consequence of focal ischemia, which might be a candidate neuroprotective mechanism of the hippocampus being activated by stroke.
Materials and methods
Animals
All animal procedures were conducted in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care in accordance with protocols approved by the Institutional Animal Care and Use Committee and the principles outlined by the NIH. Male C57BL/6 (wild type (wt); Charles River, Wilmington, MA, USA) and Adktm1−/−-Tg (UbiAdk) mice (Adk-tg) (Fedele et al, 2005) of the same genetic background weighing 25 to 30 g were housed under diurnal lighting conditions (12 h light).
Focal Ischemia
To investigate how different levels of ADK expression might contribute to protecting the hippocampal formation from cell loss after focal cerebral ischemia, we subjected mice to either 15 or 60 mins of focal ischemia. Focal cerebral ischemia was induced under anesthesia (1.5% isoflurane, 70% N2O, and 28.5% O2) by suture occlusion of the middle cerebral artery (MCAO), as described by Pignataro et al (2006). Achievement of ischemia was confirmed by monitoring regional cerebral blood flow in the area of the left middle cerebral artery. The ischemic brains were analyzed after 0, 0.25, 1, 3, and 24 h, or 3 to 7 days of reperfusion (n = 5 per condition).
Histologic Analysis
To evaluate the infarct volume, animals were killed with an overdose of isofluorane 24 h after ischemia. Brains were quickly removed, placed into a preformed mouse brain mold calibrated for removal of 1 mm slices with regular intervals, and sectioned coronally at 1 mm intervals using a razor blade, and stained by immersion in 2% (w/v) vital dye 2,3,5-triphenyltetrazolium hydrochloride (Pignataro et al, 2006).
For the immunohistochemical analysis, mice were transcardially perfused with 4% paraformaldehyde and 15% saturated picric acid solution in phosphate buffer (0.15 mol/L, pH 7.4). The brains were then postfixed in the same fixative at 4°C for 6 h, cryoprotected in 10% dimethylsulfoxide in phosphate-buffered saline (v/v), frozen and cut into 40 μm coronal sections using a sliding microtome. All sections were stored in antifreeze solution at −20°C before use for immunohistochemistry.
For the immunohistochemical detection of ADK or NeuN, brain sections were incubated overnight at 4°C with primary anti-ADK antiserum (Gouder et al, 2004) or a polyclonal antibody directed against NeuN (Chemicon International, Temecula, CA), diluted 1:5000, and 1:1000, respectively, in phosphate-buffered saline (pH 7.4), containing 2% normal goat serum and 0.2% Triton X-100. The sections were then washed 3 × 10 mins in Tris-buffered saline plus 0.05% Triton X-100 at pH 7.4, incubated for 30 mins with biotinylated goat anti-rabbit antibody diluted 1:300 in phosphate-buffered saline (pH 7.4), containing 2% normal goat serum, washed again three times in Tris-buffered saline, then incubated for 20–30 mins with avidin—biotin enzyme complex (Vectastain Elite Kit; Vector Laboratories, Burlingame, CA). After washing again three times with Tris-buffered saline, the tissue antigen was localized by incubation with hydrogen peroxide and diaminobenzidine hydrochloride (Sigma, St Louis, MO), which acts as a chromogen. The sections were then washed extensively, mounted on gelatin-coated slides, air-dried, dehydrated, and coverslipped.
To quantify ADK expression after stroke induction, immunoperoxidase-stained sections were analyzed with bright-field microscopy (Axoscope; Zeiss AG, Jena, Germany) and digitized with a high-resolution color camera (Zeiss AxioCam, Thornwood, NY, USA). ADK-expressing cells were identified by their dark nuclear staining. Numbers of ADK expressing cells were determined in the CA1 region of the hippocampal formation in fields of 3200 μm2. One field per mouse (n = 5) per condition was analyzed. Data analysis was performed by t-test and statistical significance was accepted at the 95% confidence level (P <0.05).
Determination of Adenosine in Isolated Hippocampi
A different set of mice was either sham treated or received 60 mins of MCAO followed by either 3 or 24 h of reperfusion (n = 4 each). At indicated time points, hippocampi ipsilateral to the stroked brain hemisphere were carefully dissected out from freshly killed animals. Each hippocampus was processed individually at 4°C by one burst of 5 secs of ultrasound homogenization in 200 μL of 50 mmol/L NaCl, 20 mmol/L Na2HPO4, 5 mmol/L EDTA (pH 6.5). The homogenates were heat inactivated (5 mins, 90°C), then centrifuged at 100,000 g for 10 mins. To obtain an internal standard for the samples, the protein contents of the supernatants were quantified with a commercial Bradford assay. To remove nucleotides from the supernatants 75 μl of each sample was applied to a Micro Bio-Spin 6 column (Bio-Rad, Hercules, CA, USA, #732-6221), which was equilibrated with 500 μL of the Adenosine 5′-Triphosphate Assay Mix Dilution Buffer (Sigma, St Louis, MO, USA). After centrifugation at 1000 g for 4 mins the flow-through was diluted with Adenosine 5′-Triphosphate Assay Mix Dilution Buffer to a final protein concentration of 1.0 mg/mL. Five microliters of each diluted sample was used for adenosine determinations with an enzyme-linked bioluminescent assay according to the following principle: adenosine was phosphorylated by an excess of recombinant ADK (own production) to AMP, which was then phosphorylated further to ATP by an excess of recombinant GTP:AMP phosphotransferase (own production) and pyruvate kinase (Sigma). The resulting ATP increase was then quantified with a luciferase assay (ATP Bioluminescent Assay Kit, Sigma). To run the reactions in a 96-well microtiter plate, 5 μL of each diluted sample containing an unknown amount of adenosine was added to 20 μL of a solution containing 5 mU/mL GTP: AMP phosphotransferase, 2 U/mL pyruvate kinase, 400 μmol/L GTP, 400 μmol/L phosphoenolpyruvate, and 10 μmol/L erythro-9-[3-(2-hydroxynonyl)]adenine, in 30% Adenosine 5′-phosphate Assay Mix (v/v) in Adenosine 5′-Triphosphate Assay Mix Dilution Buffer. Reactions were started by adding 15 mU/mL ADK and then followed for 15 mins at room temperature in a luminometer (Veritas Microplate Luminometer, Turner BioSystems, Sunnyvale, CA, USA). Adenosine levels were determined as an increase of relative light units per time and were normalized to total protein contents of the samples.
Results
Protection of the Hippocampus from Ischemic Injury by Endogenous Adenosine
To determine, whether ADK expression levels influence brain vulnerability after MCAO, we compared 2,3,5-triphenyltetrazolium hydrochloride staining performed after 24 h of reperfusion on brain sections from wt mice subjected to 60 mins of MCAO with those of Adk-tg mice subjected to 15 mins of MCAO (n = 5 animals, each). Adk-tg mice were subjected to 15 mins of MCAO, because 60 mins of MCAO is lethal in Adk-tg mice (Pignataro et al, 2006). Because overexpression of ADK and enhanced susceptibility to hypoxia-induced brain injury after 15 mins of MCAO Adk-tg mice displayed brain injury in cortex and caudate putamen (infarct volume: 19.8% ± 7.2% of occluded brain hemisphere), the extent of which was between wt animals treated with 15 mins of MCAO (6.3% ± 1.4%) and wt animals treated with 60 mins of MCAO (50.6% ± 2.4%) (Pignataro et al, 2006). However, at the level of the hippocampus, brain injury of wt animals treated with 60 mins of MCAO and Adk-tg animals treated with 15 mins of MCAO was comparable except for the fact that the hippocampus was consistently injured in the Adk-tg mice, whereas it was protected from cell loss in wt animals (Figures 1A and 1B, arrows). To document the integrity of the hippocampal formation in wt mice after transient focal ischemia, five additional mice were analyzed after 60 mins of MCAO and 24 h of reperfusion by NeuN immunohistochemistry. The expression of the neuronal marker NeuN (Figures 1C and 1D) revealed the integrity of the hippocampal formation (Figure 1A, arrow) after the stroke, although most of the ipsilateral brain hemisphere suffered intense damage (white in Figure 1A). In particular, the NeuN immunoreactivity revealed no regional differences in selective vulnerability between a sham-treated control hippocampus (Figure 1C) and the hippocampus of mice subjected to 60 mins of MCAO (Figure 1D). We hypothesize that the hippocampal formation is at least transiently protected from brain injury by an endogenous mechanism, involving expression levels of ADK.
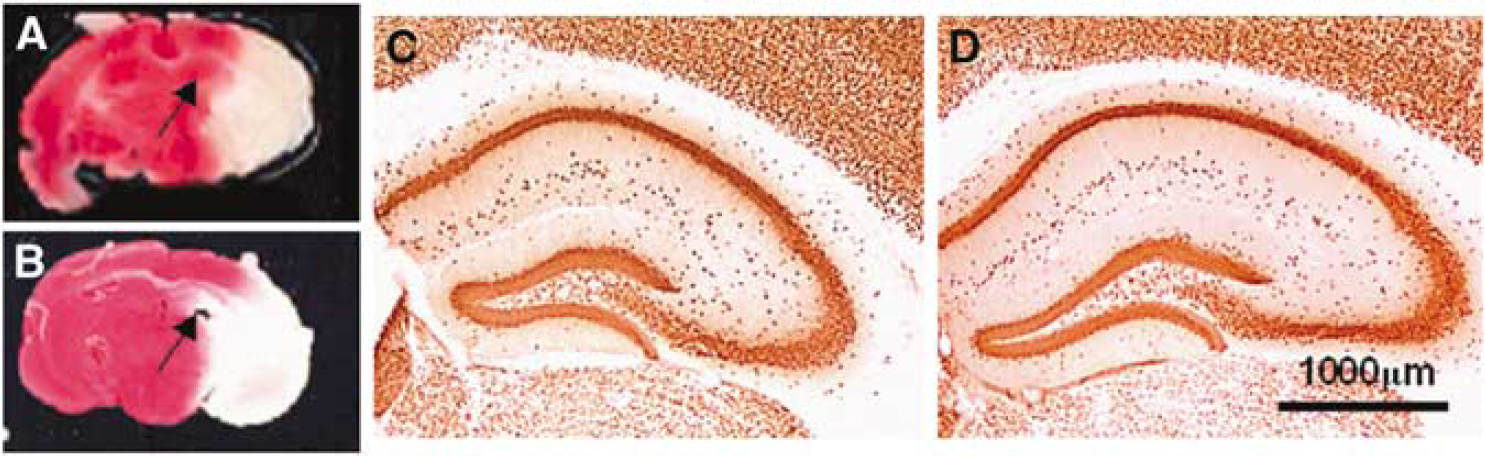
Resistance of the hippocampal formation to ischemic brain injury. (
Rapid Downregulation of Adenosine Kinase as a Consequence of Focal Ischemia
To determine, whether downregulation of endogenous hippocampal ADK might qualify as candidate mechanism mediating this endogenous neuroprotective mechanism, we evaluated ADK expression in wt mice immunohistochemically after several durations of reperfusion after either 15 or 60 mins of MCAO (n = 5 per condition). Because in adult brain ADK is expressed almost exclusively in astrocytes, ADK has emerged as a novel marker for astrocytes (Studer et al, 2006). Because astrocytic ADK immunoreactivity correlates with hippocampal enzyme activity and resultant levels of ambient adenosine (Fedele et al, 2005; Gouder et al, 2004; Huber et al, 2001), we quantified ADK expression by counting ADK-expressing cells in comparable subfields of the hippocampal CA1 region (Figure 2A). Although untreated control hippocampus contained 44.57 ± 4.27 ADK-positive cells per CA1 subregion, the number of ADK-positive cells was significantly (P <0.05) reduced in the same subregion after 60 mins of MCAO followed by 1 h (17.00 ± 8.76 cells), 3 h (6.75 ± 8.06 cells), 24 h (2.75 ± 2.22 cells), or 3 to 7 days (12.75 ± 5.5 cells) of reperfusion (Figure 2B). A significant reduction of ADK-positive cells was also found in comparable subfields of the contralateral CA1 region of the hippocampus with 27.00 ± 5.23 cells after 1 h of reperfusion, 5.75 ± 3.50 cells after 3 h of reperfusion, and 13.25 ± 12.23 cells after 24 h of reperfusion (Figure 2B). Thus, 3 h after 60 mins of MCAO hippocampal ADK expression was downregulated to approximately 13% to 15% of normal levels in hippocampi of both brain hemispheres.
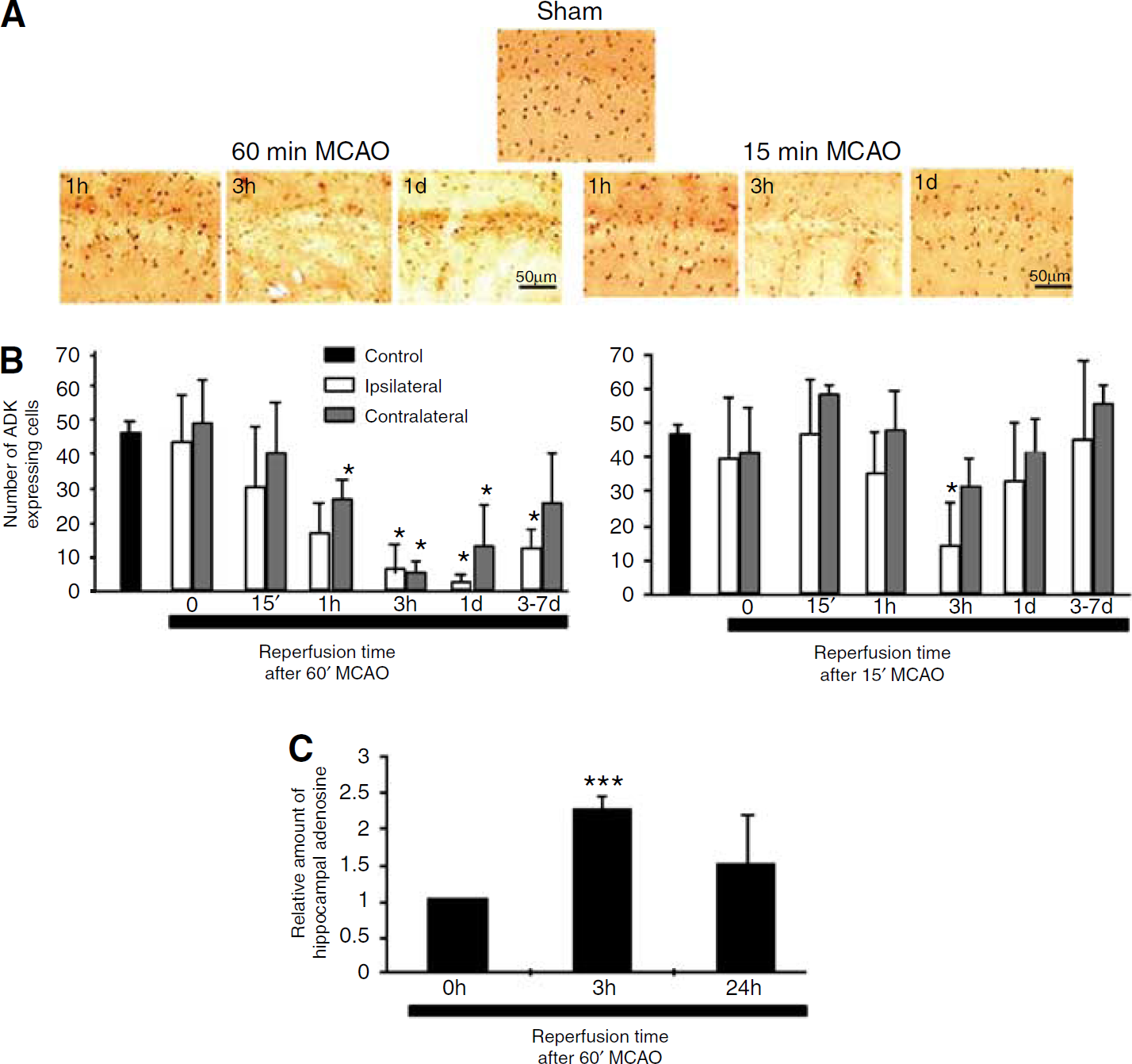
Downregulation of ADK and increase in adenosine after MCAO. (
To show that downregulation of hippocampal ADK leads to an increase in ambient hippocampal adenosine, we subjected a second set of mice to a sham treatment or to 60 mins of MCAO followed by either 3 or 24 h of reperfusion (n = 4, each). Hippocampi ipsilateral to the stroked brain hemisphere were rapidly dissected and relative adenosine levels, normalized to the sham control, determined. After 3 h of reperfusion relative adenosine values were elevated 2.2 ± 0.19-fold (P < 0.001, t-test) (Figure 2C) corresponding to the reduction in ADK immunoreactivity observed at this time point (Figures 2A and 2B). At 24 h of reperfusion, the increase in adenosine levels did not reach statistical significance.
To address the question, whether noninjurious ischemia (15 mins of MCAO), a stimulus known to induce ischemic tolerance (Stenzel-Poore et al, 2003), might influence the expression levels of ADK and thereby contribute to ischemic tolerance, we subjected a third group of mice (n = 5 animals per condition) to 15 mins of MCAO followed by 0, 0.25, 1, 3, and 24 h of reperfusion. Under these conditions, we found a significant reduction in the number of ADK immunopositive cells to 14.25 ± 13.00 cells per CA1 subfield only in the ipsilateral hippocampus after 3 h of reperfusion (Figure 2B).
Discussion
The delayed injury of hippocampal neurons after stroke (Pulsinelli et al, 1982) implies the existence of endogenous neuroprotective mechanisms. Here, we evaluate the adenosine/ADK system as a potential candidate mechanism regulating the endogenous neuroprotective potential of the hippocampus. We chose to study hippocampal ADK as an upstream effector of A1 receptors for the following reasons: (i) ADK is the key enzyme for the regulation of ambient levels of adenosine (Boison, 2006); (ii) hippocampal ADK expression is already known to be regulated during epileptogenesis (Gouder et al, 2004); (iii) hippocampal ADK is subjected to a striking spatio-temporal regulation during the early postnatal development of the mouse brain (Studer et al, 2006). These plastic and adaptive changes of ADK expression suggest an important role of the enzyme in controlling brain plasticity and vulnerability; (iv) previous in vivo and in vitro studies have clearly shown that ADK immunoreactivity correlates with enzyme activity and that reduced ADK protein levels led to increases in ambient adenosine (Fedele et al, 2005; Gouder et al, 2004; Güttinger et al, 2005; Huber et al, 2001).
The existence of endogenous adenosine-based neuroprotective mechanisms of the hippocampus is supported by our finding that transient focal ischemia with marginal consequences in wt mice (15 mins of MCAO) induces hippocampal cell death in mice overexpressing ADK in brain, whereas the hippocampus in wt animals is spared from injury after the induction of a sublethal stroke (60 mins of MCAO) (Figure 1). The critical role of ADK expression levels for the control of hippocampal vulnerability is further supported by our findings that transgenic overexpression of ADK in hippocampal neurons leads to increased electrographic activity due to reduced levels of adenosine (Fedele et al, 2005). Conversely, the focal ischemia-induced downregulation of ADK in wt mice we describe here (Figure 2) implies a transient increase in the inhibitory adenosinergic tone. Indeed, we find a transient 2.2-fold increase in ambient adenosine 3 h after 60 mins of MCAO (Figure 2C); the lack of statistically significant elevation of adenosine at 24 h of reperfusion may be due to compensatory metabolic mechanisms for the reduction of excessive adenosine (Boison et al, 2002). It has to be noted that the findings we describe here are based on relative adenosine determinations, which reflect changes in ambient adenosine compared with control tissue (sham-treated hippocampi). Absolute determinations of adenosine in intact living brain tissue are not reliable (e.g., downregulation of ADK by mechanical injury) and vary according to the methods used (Delaney and Geiger, 1996).
Similar to the findings we describe here, ADK was rapidly downregulated in the hippocampus within 2 h of status epilepticus (Gouder et al, 2004) or after oxygen-glucose deprivation in vitro (Lynch et al, 1998). Thus, rapid downregulation of ADK after stress (i.e., lack of oxygen in stroke or increased energy demand during seizures) might be a general mechanism to induce endogenous adenosine-mediated protective mechanisms of the brain. Although the mechanisms for stress-induced downregulation of ADK are not completely resolved, a recent study with Leishmania ADK suggests that rapid changes in enzyme activity become possible by an aggregation/disaggregation cycle with the inactive aggregated state of ADK stabilized by ADP (Sen et al, 2006). Thus, the oxygen-dependent energy charge of a cell (ATP/ADP ratio) may directly feed back into the regulation of ADK and ambient adenosine. Although highly attractive, this mechanism of enzyme regulation has not yet been shown in mammalian cells. In contrast, ADK does not appear to be regulated by phosphorylation (Sahin et al, 2004) or on the genomic level (Stenzel-Poore et al, 2003). The finding of downregulation of ADK in the contralateral nonhypoxic brain hemisphere implies additional, yet unknown, mechanisms to regulate ADK, and/or a cross-talk between brain regions. The spread of ADK downregulation to the contralateral side may thus be mediated by hippocampal projections that are involved in signal transmission after brain injury (Amaral and Witter, 1989; Butler et al, 2002). It is of interest to note that A1 receptor knockout mice display a spread of epilepsy-associated cell death to the contralateral brain hemisphere (Fedele et al, 2006). In conclusion, rapid downregulation of ADK might explain clinical observations of increased adenosine after cerebral ischemia, transient periods of hypoxia, or acute seizures.
The finding that adenosine A1 receptor knockout mice display a spread of epilepsy-associated cell death to the contralateral brain hemisphere (Fedele et al, 2006) indicates that the neuroprotective A1 receptors, acting as downstream effectors of the ADK/adenosine system, are involved in preventing the spread of brain injury to the contralateral brain hemisphere at least in epilepsy. Our finding that ADK immunoreactivity is reduced bilaterally as a consequence of 60 mins of unilateral MCAO, might thus be a mechanism to prevent the spread of brain injury to the contralateral brain hemisphere.
The hippocampal downregulation of ADK described here was not only observed after a sublethal stroke (60 mins of MCAO) but also, albeit to a lesser degree and only unilateral, after a mild stroke (15 mins of MCAO), which is commonly used as a preconditioning stimulus (Stenzel-Poore et al, 2003). Thus, even slight perturbations of oxygenation may result in transient downregulation of ADK and thus might provide a link to the established role of adenosine, A1 receptors, and ATP-sensitive K+ channels in cerebral ischemic preconditioning (Heurteaux et al, 1995).
In conclusion, we show that (i) stroke-induced hippocampal injury is dependent on expression levels of ADK and (ii) focal ischemia induces transient downregulation of ADK and increases in adenosine in the hippocampal formation within hours of the artery occlusion. These observations define stroke-induced downregulation of ADK as a candidate mechanism to delay or to prevent hippocampal injury after stroke and might be a mechanism in the delayed injury (maturation) of hippocampal neurons after ischemia.