
Abstract
We have previously reported that exogenous and endogenous estrogen can amplify residual cerebral blood flow during experimental cerebral ischemia. Because estrogen has been linked to nitric oxide and cyclic guanosine monophosphate (cGMP) signaling in noncerebral tissue, we tested the hypothesis that long-term 17β-estradiol treatment increases basal cGMP in brain homogenates and cerebral microvessels in female rabbits. We also determined whether there are important baseline gender-specific differences in regional cGMP. Adult female rabbits were implanted with 17β-estradiol pellets, 10 mg (F10, n = 10) or 50 mg (F50, n = 13), and compared with untreated females (F, n = 19) and males with negligible estrogen (M, n = 19) (plasma 17β-estradiol levels of 4 ± 4 pg/mL in M, 7 ± 5 pg/mL in F, 141 ± 74 pg/mL in F10, and 289 ± 110 pg/mL in F50). Cyclic GMP was determined by radioimmunoassay in cerebellum, hypothalamus, caudate nucleus, hippocampus, and cortex. Cerebral microvessels were harvested from additional cohorts of untreated males and females or estradiol-implanted females (n = 6 per group). Basal cGMP was higher in F versus M only in cerebellum. Estrogen-induced increases in regional cGMP were prominent in hippocampus at all doses (M = 43 ± 26, F = 43 ± 21, F10 = 84 ± 24, F50 = 117 ±55 fmol/mg protein) and in cortex at the high dose (M = 78 ± 55, F = 88 ± 51, F10 = 69 ± 34, F50 = 143 ± 52 fmol/mg protein). Similarly, microvascular cGMP increased only in females treated with the 50 mg dose (M = 77 ± 13, F = 86 ± 25, F10 = 106 ± 35, F50 = 192 ± 88 fmol/mg protein). Therefore, 17β-estradiol increases cGMP content in parenchymal regions that are known physiologic targets for reproductive steroids but are also areas of selective vulnerability to ischemic insult. Further, high doses of estrogenic steroids could amplify cGMP signaling within the cerebral microvasculature.
Women are at lower risk than men for cerebrovascular disease, including stroke and transient ischemic attack (Barret-Connor and Bush, 1991; Fisher et al., 1965; Ford et al., 1985; Sivenius et al., 1991; Wolf et al., 1987), potentially owing to estrogen's enhancement of vascular function or possibly by direct neuroprotection. Estrogen is a known vasodilator of many regional circulations, and acute administration of 17β-estradiol increases blood flow to diverse brain areas (Goldman et al., 1976). However, it is unclear whether estrogens are important in regulation of CBF or tonic vasodilatory mechanisms. Estrogen has been linked to nitric oxide and cyclic guanosine monophosphate (cGMP) signaling in uterine tissue (Rosenfeld et al., 1996; Weiner et al., 1994) and the coronary circulation (Wellman et al., 1996; White et al., 1995). Further, estrogenic steroids may employ cGMP-dependent signal transduction pathways that are unrelated to nitric oxide (NO). Recent data suggest that estradiol relaxes coronary vessels in vitro by an endothelium-independent mechanism involving cGMP-dependent phosphorylation of large conductance, calcium-activated K+ channels (White et al., 1995). The importance of cGMP in brain as a means of neuronal and vascular signaling is well known (Bredt and Snyder, 1989; Vallebuona and Raiteri, 1994), although the impact of estrogen on these signals is not known. Given the data that link estrogen to cGMP signaling in noncerebral tissue and our previous work implicating estrogen's vasoactivity in brain (Hurn et al., 1995; Alkayed et al., 1998), we tested the hypothesis that increasing plasma levels of 17β-estradiol in the female rabbit also increases basal cGMP in brain parenchyma and in cerebral microvascular tissue. In addition, age-matched females and males with equivalently low levels of endogenous estrogen were examined to determine the presence of inherent gender differences in regional cGMP.
METHODS
This study was approved by the Institutional Animal Care and Use Committee and is in compliance with the guidelines of the National Institutes of Health for care and handling of animals. Sexually mature female and male New Zealand rabbits (3 to 4 kg) were caged individually for a minimum period of 10 days under controlled light and temperature conditions. Four groups of animals were studied: (1) untreated females (F, n = 19 for tissue homogenates, n = 6 for cerebral microvessels), (2) females treated with 10 mg 17β-estradiol pellets (F10, n = 10 for homogenates, n = 6 for microvessels), (3) females treated with 50 mg 17β-estradiol pellets (F50, n = 13 for homogenates, n = 6 for microvessels), and (4) untreated, age-matched males (M, n = 19 for homogenates, n = 6 for microvessels). Untreated animals were sham-operated so that all groups received the same anesthetic exposure. In estrogen-treated animals, a 17β-estradiol pellet (Innovative Research, Toledo, OH, U.S.A.) was implanted subcutaneously on the dorsal neck surface (continuous release 2 µg/day, 21 days) 1 to 2 weeks before brain harvesting (Hurn et al., 1995). In rabbit, endogenous estrogen production is suppressed when exogenous estradiol is administered, allowing control of plasma estradiol levels and eliminating the need for oophorectomy (Dharmarajan et al., 1991). Plasma 17β-estradiol samples were obtained during tissue harvesting and measured by radioimmunoassay (Diagnostic Products Corp., Los Angeles, CA, U.S.A.) as previously described (Dharmarajan et al., 1991). All standards and samples were assayed in duplicate with interassay and intra-assay variabilities of 4% and 7%, respectively (Dharmarajan et al., 1991; Hurn et al., 1995).
For parenchymal cGMP measurements, the brain was harvested under deep barbiturate anesthesia with subsequent sub-dissection on ice into five areas: cerebellum, hypothalamus, caudate nucleus, hippocampus, and the anterior and posteriolateral cortex. The samples were frozen immediately in powdered dry ice and stored until assay at −80°C. For microvessel isolation (Gebremedhin et al., 1996), each animal was anesthetized with thiopental (100 to 150 mg) and mechanically ventilated while instrumented with a carotid artery catheter. After full-body perfusion through the left ventricle with cold HEPES-buffered physiologic salt solution (PSS), 100 mL of ice-cold iron oxide solution (10 mg/mL, particle size 0.15 µm, Aldrich Chemicals, Milwaukee, WI, U.S.A.) was injected through the carotid catheter. The brain was then harvested, surface pial vessels removed, and the cortex subdissected on ice. After mincing and passage through a 250-µm sieve (Cell Dissociation, Sieve—Fisher, Pittsburgh, PA, U.S.A.), iron-laden microvessels were separated from the parenchyma using a side-pull magnet (Bio Mag Separator; Advanced Magnetics, Cambridge, MA, U.S.A.). The retained tissue was resuspended in PSS with multiple repetitions of this magnetic separation. The crude microvessels were further separated by shearing through an 18-gauge needle, frozen in liquid nitrogen, and stored at −80°C for subsequent cGMP analysis. By light microscopy, the preparation appears as microvascular segments, predominantly vascular smooth muscle, with occasional adherent pericytes. To validate functional viability, we measured steady-state Na+-K+-ATPase activity as previously described (Fomitcheva and Kosk-Kosicka, 1996). Enzyme activity was preserved in the microvascular preparation at 4.4 µm Pi/mg protein per hour.
Each sample was subsequently homogenized in PSS and precipitated with 6% trichloroacetic acid. After centrifugation, the resulting pellet was resuspended in 1 mol/L NaOH, then 0.9 mol/L HCl, and quantified for protein with the Coomassie blue technique (Pierce, Rockford, IL, U.S.A.) as previously described (Pearce et al., 1991). The supernatants were washed with water-saturated ether, frozen in an ethanol/dry ice bath, then lyophilized for 12 to 72 hours. Cyclic GMP levels were measured in triplicate by commercial radioimmunoassay kit (Amersham Corp., Los Angeles, CA, U.S.A.) (Bredt and Snyder, 1989) and normalized relative to protein content (fmol/mg protein). Interassay and intra-assay variability were each approximately 7%. The data were analyzed with one-way analysis of variance with a posthoc Newman-Keuls test to distinguish between treatment effects. Statistical significance was set at P < 0.05. All data are presented as mean ± SD.
RESULTS
Plasma 17β-estradiol levels at the time of tissue or microvessel harvesting were 4 ± 4 pg/mL in M, 7 ±5 pg/mL in F, 141 ± 74 pg/mL in F10, and 289 ± 110 pg/mL in F50. Cyclic GMP was comparable in all brain regions and the microvascular fraction of males and untreated females, with the exception of cerebellum (M = 76 ± 39 versus F = 136 ± 46 fmol/mg protein, P = 0.032) (Fig. 1). Estrogen increased tissue cGMP in the F10 group as compared with F in hippocampus (P = 0.011) and further increased cGMP in the high-dose group (P = 0.002). In addition, cortical cGMP content was higher in homogenates obtained from estrogen-treated females as compared with F, but only at the high dose (F = 88 ± 51, F10 = 69 ± 34, F50 = 143 ± 52 fmol/mg protein, P = 0.042) (Fig. 2). Similarly, cGMP in the microvessels isolated from hemispheric tissue was higher only in the F50 group (P < 0.002) relative to untreated females (Fig. 3).
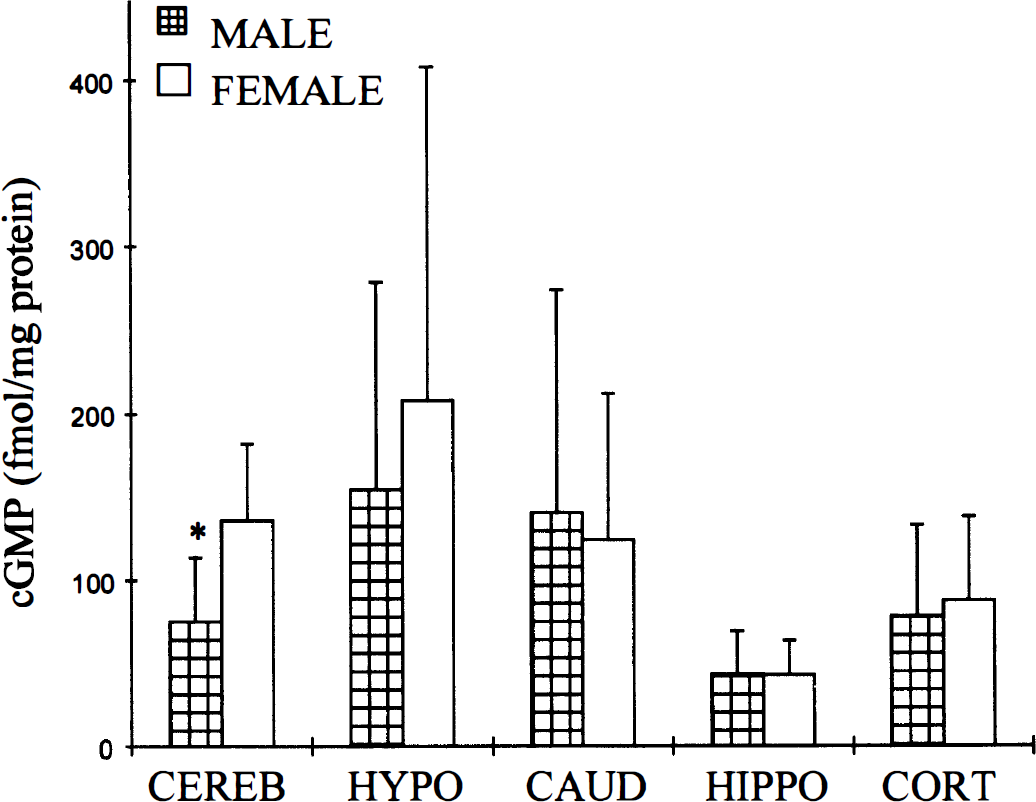
Basal brain cGMP content in male (n = 19) and female (n = 20) rabbits. All values are mean ± SD. * P < 0.05 from female group. Cereb, cerebellum; hypo, hypothalamus; caud, caudate nucleus; hippo, hippocampus; cortex, cortex.
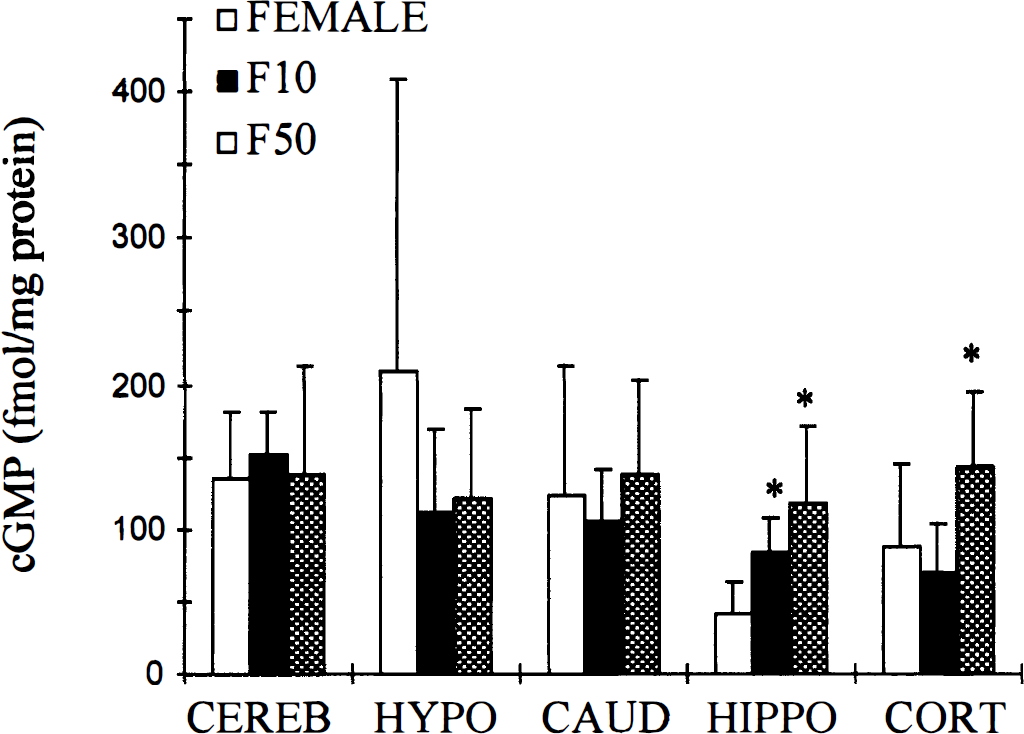
Basal brain cGMP content in untreated female rabbits (n = 20), females treated with 10 mg 17β-estradiol pellets (F10, n = 10), and females treated with 50 mg 17β-estradiol pellets (F50, n = 13). All values are mean ± SD. * P < 0.05 from female group. Cereb, cerebellum; hypo, hypothalamus; caud, caudate nucleus; hippo, hippocampus; cortex, cortex.
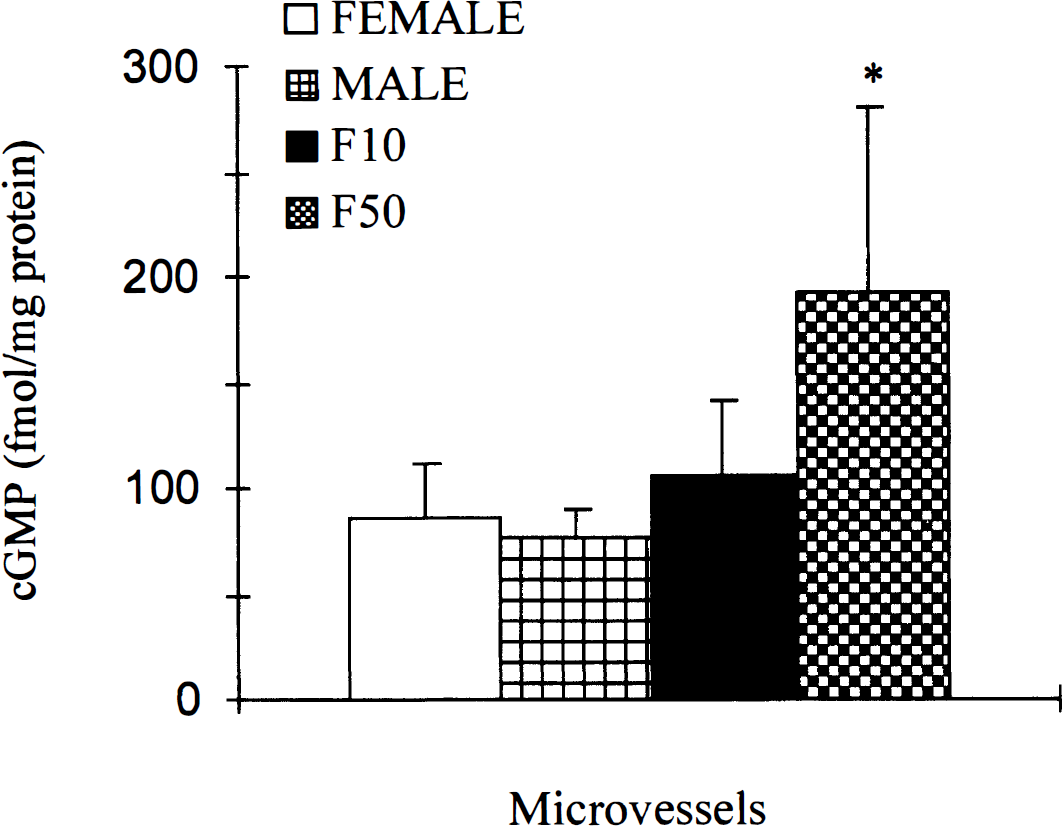
Effect of estrogen treatment on cGMP in isolated microvessels harvested from both cerebral hemispheres. Values represent means and SD from untreated female rabbits with endogenous estrogen, untreated males with near zero plasma estrogen, and females treated with 10 mg or 50 mg of exogenous 17β-estradiol (n = 6 per group). * P < 0.05 from female group.
DISCUSSION
The major findings in this study are that basal tissue cGMP levels in the female animal can be amplified in specific brain regions by long-term estrogen treatment and that the microvasculature is also responsive to exogenous steroid administration at high levels. Estrogen-induced increases in tissue cGMP are most striking in regions that are physiologic, neuronal targets for female sex steroids and in regions with known vulnerability to ischemic injury, e.g., hippocampus and cortex. These findings suggest that one mechanism by which estrogenic steroids act within brain is by amplifying regional cGMP signaling within the parenchyma and within small blood vessels. When brain cGMP content is compared in animals with low endogenous estrogen availability (unstimulated females and males), there are basal gender differences only in cerebellum.
We chose to quantify regional cGMP as one means of evaluating estrogen's potentially dual signaling targets in brain. There is evidence that estrogen can function as both a modulator of cerebral vasoreactivity (Hull et al., 1992; Ohkura et al., 1995) and of synaptic neurotransmission, particularly in interaction with excitatory neuronal activation (Woolley and McEwen, 1992; McEwen et al., 1997; Woolley et al., 1997). Estradiol is a known vasodilator steroid in many circulations, including the cerebrovascular bed, under normal (Mendelsohn and Karas, 1994) and most clearly under pathologic conditions (Hurn et al., 1995; Reis et al., 1994; Williams et al., 1994). Fairly consistent results indicate that women have a higher mean hemispheric CBF as compared to men (Rodriguez et al., 1988; Shaw et al., 1984) and a greater homogeneity in regional flow distribution (Rodriguez et al., 1988). In a recent positron-emission tomographic study, estrogen administration enhanced regional CBF responses to cognitive activation in young women (Berman et al., 1997). Further, our laboratory and others have shown that estrogen alters vasodilatory capacity during global cerebral ischemia (Hurn et al., 1995; Pelligrino et al., 1996).
Potential mechanisms by which estrogen could induce this vascular “plasticity” include hormonally enhanced release of NO and prostacyclin from vascular endothelium (Mendelsohn and Karas, 1994) or cGMP-dependent vascular smooth muscle hyperpolarization (White et al., 1995). Therefore, we determined whether long-term estrogen treatment increased basal brain cGMP content, and inferentially, the potential capacity to alter cGMP signal transduction in blood vessels or surrounding neural tissue. Cyclic GMP is an important mediator of vascular smooth muscle relaxation, as well as a critical element of many excitatory neurotransmitter systems in brain. Known cerebral vasodilators increase cGMP generation, including acetylcholine, substance P, and NO donors (Yu et al., 1995), and cell-permeable cGMP analogs produce dilation of cerebral arterioles in situ (Paterno et al., 1996). Our finding that cGMP content is elevated by 17β-estradiol is consistent with the observation that basal cGMP levels increase in ovine uterine artery during pregnancy (Magness et al., 1991). Both pregnancy and exogenous estradiol increase neuronal and endothelial NO synthase activity in uterine artery, heart, kidney, skeletal muscle, and cerebellum (Hayashi et al., 1994; Weiner et al., 1994). Further, estrogen enhances both basal and stimulated NO release in isolated rat aorta under isometric conditions (Rahimian et al., 1996). In light of the estrogen-responsive elements within the promoter region of the NOS gene (Venema et al., 1994), it may be that estrogen increases cGMP content subsequent to NO synthase upregulation. Nitric oxide-dependent effluxes of cGMP are well known in rat cerebellar cortex and hippocampus (Vallebuona and Raiteri, 1994; Luo et al., 1994; East and Garthwaite, 1991). However, cGMP is ubiquitous in the brain and increases may be linked to other NO-independent cellular processes. Recent data suggest that estradiol may act directly as a vasodilator by cGMP-dependent phosphorylation of large conductance, calcium-activated K+ channels (White et al., 1995). Our data do not distinguish among these potential mechanisms but indicate that estrogen elevates cGMP content in tissue likely to be potential targets for steroidal hormones.
Our measurements in rabbit indicate that microvascular cGMP is responsive only to high doses of exogenous estrogen. The high-dose pellet in the F50 group was chosen to be consistent with the doses known to produce vasodilation in the rabbit during low blood flow conditions (Hurn et al., 1995). The lower dose in the F10 group yielded physiologic levels similar to those present with chorionic gonadotropin (hCG)-stimulated pseudo-pregnancy in this species (Wu et al., 1977). This lower dose did not alter cGMP content compared with unstimulated females with estrogen levels at the hormonal nadir, which may indicate that physiologic estrogen fluctuation does not alter vascular cGMP signaling. These data do not exclude possible cGMP induction in larger, upstream conductive and resistance vessels that greatly influence cerebral blood flow regulation under normal conditions. Nevertheless, it must be emphasized that exogenous estrogen augmentation can more than double basal cGMP availability in vascular tissue.
Cortical cGMP levels likewise responded to estrogen but only at supraphysiologic doses. In contrast, hippocampal cGMP content increased stepwise with increasing estrogen doses, and the effect appeared to be regionally specific in that other noncortical regions such as caudate nucleus, hypothalamus, and cerebellum were unchanged. Estrogen has a widespread group of cellular targets within brain (Stumpf, 1968), and estrogen-receptor mRNA—containing cells are present in many brain areas not generally associated with reproductive function (Simerly et al., 1990). The location of the estrogen—cGMP interaction is consistent with previous work showing histologic and functional hippocampal sensitivity to estrogen over a range of concentrations (McEwen et al., 1997; Woolley et al., 1997). Estrogen can directly alter synaptic function in the CA1 region for hours, as evidenced by alterations in dendritic spine density during the estrous cycle and in response to pharmacologically amplified estradiol (Woolley and McEwen, 1992). In addition, although the regional and cellular estrogen receptor profile of the β-receptor Subtype is still unclear at present, immunocytochemical localization of classic estrogen receptors, likely α-subtype, on hippocampal interneurons has been documented (Weiland et al., 1996).
What remains to be shown is whether, and under what conditions, estrogen-mediated increases in cGMP content alter functional signaling in the cortex or hippocampus. Because these brain regions are known for their vulnerability to ischemic insult, one potential consequence may be to alter neuronal cell viability and loss. Endogenous estrogen provides substantial neuroprotection from carotid artery occlusion in the gerbil (Hall et al., 1991) and salvages cortical and striatal tissue after middle cerebral artery occlusion in rat, as does long-term estradiol treatment in ovariectomized female rodents (Alkayed et al., 1998; Rusa et al., 1998). The present data do not address the functional significance of estrogen's effect on cortical or hippocampal cGMP levels or establish a link to mechanisms of ischemic cell injury. Nevertheless, the regional specificity of estrogen's cGMP amplification is striking, particularly given the evidence that estrogen protects ischemic brain in several experimental stroke models.
In conclusion, estrogenic steroids likely employ multiple signaling mechanisms to alter neuronal and vascular physiology in brain. The present study demonstrates that long-term treatment with 17β-estradiol, the principal biologically active estrogen in mammals, increases cGMP content in a selective and dose-specific manner. Therefore, we hypothesize that long-term exposure to high estrogen results in augmented basal cGMP with a consequently greater sensitivity to cGMP-transduced vasodilatory and neural stimuli.