
Abstract
We have previously demonstrated that the neuroprotective effect of the β2-adrenoceptor agonist clenbuterol in vitro and in vivo was most likely mediated by an increased nerve growth factor (NGF) expression. In the present study, we examined whether clenbuterol was capable of inhibiting apoptosis caused by ischemia. Transient forebrain ischemia was performed in male Wistar rats (300 to 350 g) by clamping both common carotid arteries and reducing the blood pressure to 40 mm Hg for 10 minutes. Clenbuterol(0.1, 0.5, and 1.0 mg/kg intraperitoneally) was administered 3 hours before ischemia or immediately after ischemia. The brains were removed for histologic evaluation 7 days after ischemia. The time course of DNA fragmentation was determined 1, 2, 3 and 4 days after ischemia. Staining with terminal deoxynucleotidyl transferase(TdT)-mediated dUTP nick end-labeling (TUNEL) was used for further analysis of DNA fragments in situ 3 days after ischemia. The NGF protein was assayed by enzyme-linked immunosorbent assay. Ten-minute forebrain ischemia damaged 80% to 90% of the neurons in the hippocampal CA1 region evaluated 7 days after ischemia. Pretreatment with clenbuterol (0.5 and 1.0 mg/kg) reduced the neuronal damage by 18.1% (P< 0.01) and 13.1% (P < 0.05), respectively. The neuroprotective effect also was found when clenbuterol (0.5 mg/kg) was administered immediately after ischemia (P < 0.05). The DNA laddering appeared in striatum 1 day and in hippocampus 2 days after ischemia and peaked on the third day in both regions. The DNA laddering was nearly abolished in the hippocampus and partially blocked in striatum and cortex by 0.5 mg/kg clenbuterol. These results were confirmed by TUNEL staining. Clenbuterol (0.5 mg/kg intraperitoneally) elevated the NGF protein level by 33% (P < 0.05) in the hippocampus and 41% (P < 0.05) in the cortex 6 hours after ischemia. Three days after ischemia, the NGF levels in these regions were no longer different between the clenbuterol-treated and control groups. This study clearly demonstrates that clenbuterol possesses a neuroprotective activity and a marked capacity to inhibit DNA degradation after global ischemia. The results suggest that clenbuterol increases NGF expression during the first hours after global ischemia and thereby protects neurons against apoptotic damage.
The term apoptosis describes a series of morphologic changes that are different from those observed in necrosis (Kerr et al., 1972). Apoptosis of neurons occurs naturally during the normal fetal and early postnatal development of vertebrates. There is increasing evidence that apoptosis also is involved in neurodegenerative disorders such as Alzheimer's disease, Huntington's disease, and Parkinson's disease (Dragunow et al., 1995; Mochizuki et al., 1997). It also has been reported that apoptosis can be induced by various insults (Linik et al., 1993; MacManus et al., 1993; Zhai et al., 1996). In particular, researchers have focused on the role of apoptotic cell death after cerebral ischemia (Kirino, 1982; Hill et al., 1995; Linik et al., 1995). The demonstration of apoptosis in neurodegenerative disease and cerebral ischemia suggests an extended window for therapeutic intervention (Linik, 1995). Therefore, it is important to characterize new pharmacologic strategies to interfere with apoptosis effectively. Nerve growth factor (NGF) has been found to prevent developmental apoptosis (Oppenheim, 1991; Johnson and Deckwerth, 1993). In vitro data have shown that deprivation of NGF results in neuronal death associated with apoptosis (Martin et al., 1992; Kew et al., 1996). In contrast, delivery of NGF prevents neuronal damage by apoptosis in vitro (Rabacchi et al., 1994; Kawamoto et al., 1995) as well as in vivo (Buchan et al., 1990; Frim et al., 1993). However, the clinical use of NGF may be limited by its inability to cross the blood-brain barrier. Catecholaminergic neurotransmitters and their analogues play a crucial role in the regulation of synthesis and secretion of NGF (Furukawa et al., 1986; Follesa and Mocchetti, 1993; Hayes et al., 1995). Our previous studies demonstrate that stimulation of β2-adrenergic receptor by clenbuterol, a lipophilic β2-adrenoceptor agonist, was able to elevate both NGF mRNA and NGF protein in vitro as well as in vivo. Moreover, clenbuterol showed neuroprotective effects in cultured hippocampal cells and in models of cerebral ischemia (Semkova et al., 1994; Semkova et al., 1996a,1996b). Thus, pharmacologic induction of NGF synthesis is a potential way to increase the NGF level in brain tissue.
In the present study, we further investigated the effect of clenbuterol on the delayed neuronal cell death, most likely apoptosis, in a rat model of global ischemia. Electrophoresis and staining with terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end-labeling (TUNEL) were used to measure DNA laddering and to detect DNA fragmentation in situ, respectively, to determine whether clenbuterol could prevent DNA degradation after global ischemia. The NGF protein in ischemic brain tissue was measured by enzyme-linked immunosorbent assay (ELISA) to demonstrate the association of NGF induction with the antiapoptotic effect of clenbuterol.
METHODS
Transient forebrain ischemia model
Forebrain ischemia was performed in male Wistar rats(300 to 350 g, Charles River, Sulzfeld, Germany) as described previously with some modifications (Semkova et al., 1996b). The rats were fasted overnight with free access to water and anesthetized with halothane (1.5%) in a mixture of N2O/O2 (70%/30%). Body and skull temperature were maintained at between 37.0° and 37.5°C by a temperature control feedback system (TSE, Bad Homburg, Germany). The skull temperature was monitored with a thermocouple placed subcutaneously over the midline of the skull, thereby supplying an approximate brain temperature (Corbett et al., 1992). Ischemia was induced for 10 minutes by clamping both common carotid arteries. The mean arterial blood pressure was lowered to 40 mm Hg by trimethaphan camphor sulphonate (5 mg/kg intravenously) and central venous exsanguination. After 10 minutes, the clips were removed and the blood that had been withdrawn was rapidly reinjected. Arterial pH, Pco2, Po2, blood pressure, and plasma glucose concentration were measured before and after ischemia. After the operation, the animals were kept at an environmental temperature of 30°C for 2 hours and then at 20°C in their home cages.
For evaluation of the time course of neuronal damage in the CA1 subfield of hippocampus, 1, 2, 3, 4, and 7 days after the onset of ischemia the rats were anesthetized with a lethal dosage of chloralhydrate and perfused transcardially with saline followed by 4% phosphate-buffered formalin. The brains then were removed and embedded in paraffin. The coronal sections(5 μM) were stained with 1% celestine blue and 1% acid fuchsin for histologic evaluation. Neuronal injury was evaluated as a percentage of damaged neurons in CA1 subfield of the hippocampus. Four rats were used for each recovery time point after ischemia. In a separate experiment, the animals (n = 8 to 10 per group) received clenbuterol (0.1, 0.5, and 1.0 mg/kg intraperitoneally) or saline 3 hours before ischemia. In another series of experiments, clenbuterol (0.5 mg/kg intraperitoneally) was administered immediately after ischemia. Seven days after ischemia, the brains were removed for histologic evaluation.
Detection of DNA fragmentation
The DNA in the hippocampus, striatum and occipital cortex was individually extracted using QIAamp Tissue Kit (QIA-GEN, Hilden, Germany) according to the manufacturer's protocol. The extracted DNA was labeled by Dig Oligonucleotide 3′-end Labeling Kit (Boehringer Mannheim, Germany). The DNA (5 μg) was incubated with the reaction solution containing 4 μL tailing buffer, 4 μL CoCl2 solution, 1 μL Dig-ddUTP solution, and 1 μL terminal transferase for 15 minutes at 37°C. The reaction was terminated by 2 μL glycogen ethylenediamine tetraacetic acid solution. The labeled oligonucleotide was precipitated at −70°C overnight using 2.5 μL of 4 mol/L LiCl and 75 μL prechilled (−20°C) 100% ethanol. After centrifugation (15000 rpm, −4°C, 15 minutes), the precipitates were washed with 50 μL of cold 70% ethanol and then dissolved in 50 μL of sterile water.
The labeled DNA from the striatum, hippocampus, and cortex (0.5, 1.0, and 2.0 μg per lane, respectively) underwent electrophoresis at 100 V in 2% agarose gel containing ethidium bromide for 3 hours. The DNA was transferred from the gel onto a nylon membrane before being detected by 1% CSPD (Disodium3-(4-methoxyspiro{1,2-dioxetane-3,2′-(5-chloro)tricyclo[3.3.1.1]decan}-4-yl)phenyl phosphate).
In this experiment, one series of rat brains was removed at 1, 2, 3, and 4 days after global ischemia (n = 3 per group) for measurement of the time courses of DNA fragmentation. Another series of animals (n = 3) was treated with clenbuterol (0.5 mg/kg intraperitoneally) 3 hours before ischemia. The brains were removed 3 days after ischemia to detect DNA fragmentation.
TUNEL staining
The rats were treated with either saline or clenbuterol (0.5 mg/kg intraperitoneally) 3 hours before ischemia, and the brains were removed 3 days after ischemia. Coronal paraffin sections (0.5 μM) were cut from the hippocampus, striatum, and occipital cortex (−3.6 mm, 1.00 mm, and −6.30 mm to bregma, respectively) for the evaluation of cellular DNA fragmentation in situ. The TUNEL staining was performed using ApopTag Kit (Oncor, U.S.A.). Briefly, after deparaffinizing, the sections were rinsed in three changes of phosphate-buffered saline and then incubated with proteinase K (20 μg/mL) for 20 minutes at room temperature. Two percent H2O2 was used to quench the endogenous peroxidase activity. Afterward, the slides were incubated with working-strength TdT enzyme for 1.5 hours at 37°C followed by incubation with working-strength stop/wash buffer. After incubation with two drops of antidigoxigenin-peroxidase for 30 minutes, the slides were equilibrated in 0.02% H2O2 for 5 minutes and then exposed to a freshly prepared staining solution containing 0.05% diaminobenzidine in 0.02% H2O2.
Measurement of nerve growth factor protein
The two-site NGF-ELISA was used for the measurement of NGF protein in the hippocampus, cortex, and striatum. One series of rats (n = 3 to 4 per group) was treated with clenbuterol (0.5 mg/kg intraperitoneally) 3 hours before ischemia, and the brains were removed 6 hours or 3 days after ischemia. A second series of animals (n = 3 to 4 per group) received clenbuterol (0.5 mg/kg intraperitoneally) 3 hours before ischemia and a daily injection for 3 days after ischemia. The brains were removed 3 days after ischemia. Both sides of the hippocampus, striatum, and occipital cortex were taken separately and frozen immediately on dry ice. The extraction buffer (0.1 mol/L Tris-HCl, 0.4 mol/L NaCl, 2% bovine serum albumin, 0.05% NaN3, 1 mmol/L phenylmethylsulfonylfluoride, 4 mmol/L ethylenediamine tetraacetic acid, 7 μg/mL trypsin inhibitor) was added to the tissue sample (19 mL/g wet tissue). The tissue was homogenized and then centrifuged (20000 rpm, 4°C, 30 minutes), and the supernatant was used for NGF protein assay.
The ELISA was performed as described previously with some modifications (Semkova et al., 1996b). The wells of microtiter plates (MaxiSorp, Nunc, Wiesbaden, Germany) were coated with buffer solution containing 50 mmol/L NaCO3/NaHCO3, 0.05% sodium azide, and 0.5 μg/mL anti-NGF monoclonal antibody (Boehringer Mannheim, Mannheim, Germany) for 2 hours at 20°C. Nonspecific binding sites were saturated with 1% bovine serum albumin, and the wells were washed three times with a wash buffer(50 mmol/L Tris-HCl, 200 mmol/L NaCl, 10 mmol/L CaCl2, 0.1% Triton X-100, and 0.1% sodium azide, pH 7.0). The extracted samples and standard solution were added, and the plates were incubated for 5 hours at 20°C followed by three washings with wash buffer. The β-galactosidase-conjugated anti-NGF antibody (Boehringer Mannheim) was added, and the plates were kept at 4°C overnight. After washing with wash buffer, the substrate solution containing 20 μg/mL 4-methylumbelliferyl-β-D-galactoside(Sigma, St. Louis, MO, U.S.A.) was added. The plate was incubated again for 5 hours at 20°C. The measurement of NGF was performed with the fluorospectrophotometer(BIO-TEK, Germany). The wavelengths for excitation and emission were set at 360 and 450 nm, respectively. Standard curves were prepared with 2.5 S NGF standard purified from mouse submaxillary gland by assaying parallel wells containing increasing amounts of NGF (0 to 500 pg/mL). The content of NGF in the samples was calculated from the standard curve.
RESULTS
Transient forebrain ischemia in rats
After 10 minutes of global ischemia, a progressive neuronal cell death was observed in the hippocampal CA1 subfield (Fig. 1). Twenty-four hours after reperfusion, no detectable neuronal damage was found in the hippocampus. Two days after ischemia, about 41% of the neurons in the CA1 region were injured. By the fourth day after ischemia, the percentage of damaged neurons in the hippocampus reached a plateau of more than 90% that was unchanged 7 days after ischemia.
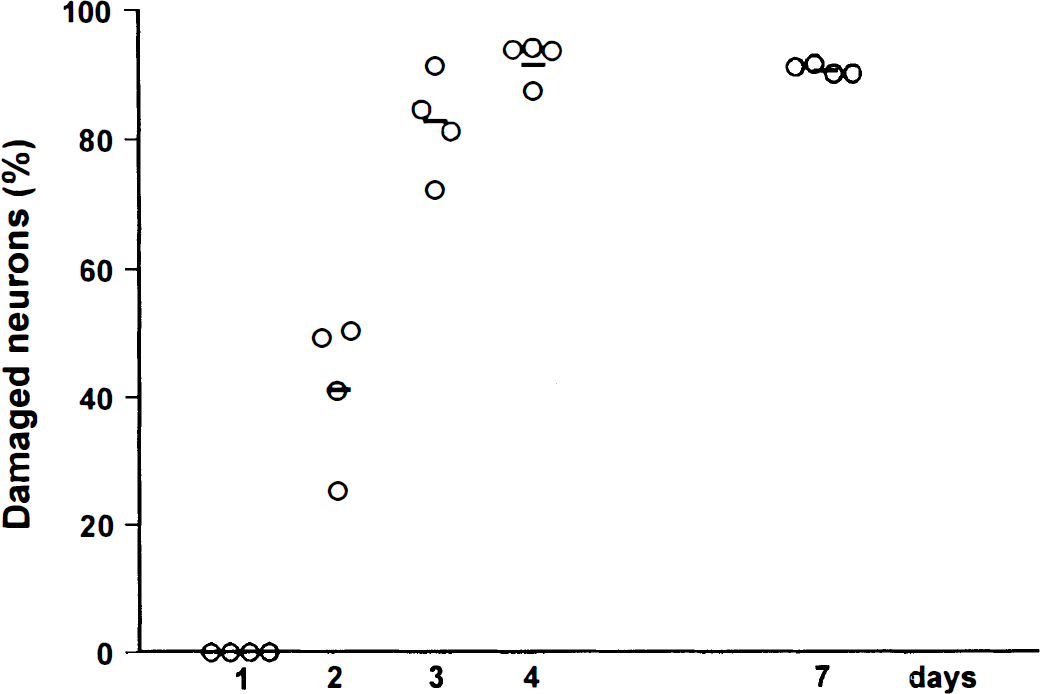
Time course of neuronal damage in CA1 subfield of rat hippocampus. The rat brains were removed 1, 2, 3, 4, and 7 days after 10-minute transient global ischemia. Neuronal damage was detected by celestine blue and acid fuchsin staining. n = 4 for each time point. Data are presented as scatter plot and mean values.
Intraperitoneal treatment with clenbuterol 3 hours before ischemia or immediately after ischemia reduced the CA1 neuronal damage measured 7 days after ischemia (Fig. 2). The neuronal damage in CA1 subfield was unaffected by the treatment with 0.1 mg/kg clenbuterol. Pretreatment with both 0.5 and 1.0 mg/kg clenbuterol resulted in a reduced neuronal loss. The former treatment reduced the number of damaged neurons in CA1 region by 23% (P < 0.01) and the latter by 15% (P < 0.05). The immediate injection of 0.5 mg/kg clenbuterol after ischemia (posttreatment) showed a similar neuroprotective effect(P < 0.05).
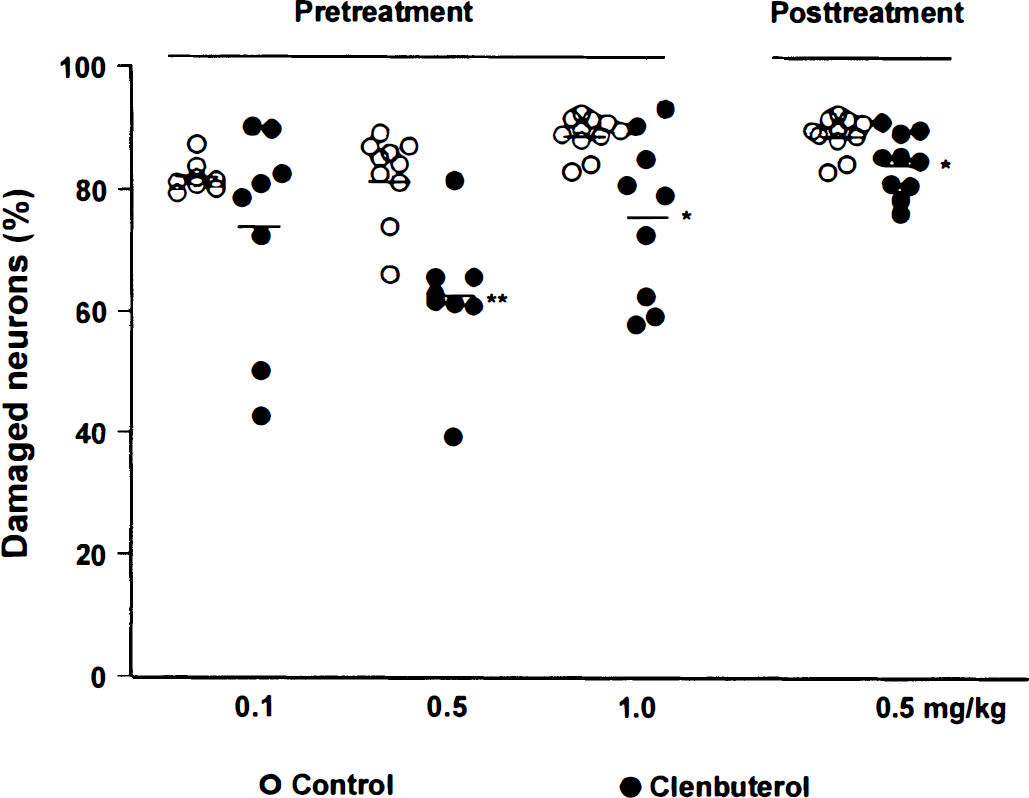
Clenbuterol protects neurons in CA1 subfield of rat hippocampus against ischemic damage. Neuronal damage was evaluated by celestine blue and acid fuchsin staining 7 days after ischemia. Pretreatment: Clenbuterol (0.1, 0.5, and 1.0 mg/kg) was administered intraperitoneally 3 hours before ischemia. Posttreatment: Clenbuterol (0.5 mg/kg intraperitoneally) was injected immediately after ischemia. The control group received saline only. n = 8 to 10 for each group. Data are presented as scatter plot and mean values. *P < 0.05, **P < 0.001 compared with the control (Student's t-test).
Compared with the control, animals treated with 0.5 and 1.0 mg/kg of clenbuterol showed a reduction of blood pressure to 85% (P< 0.05) and 70% (P < 0.001), respectively. The blood glucose level in the group treated with 1.0 mg/kg clenbuterol was significantly elevated by 71.2% (P < 0.001).
DNA fragmentation assay
The time courses of DNA fragmentation after global ischemia are presented in Fig. 3. The integrity of DNA from the hippocampus, striatum, and cortex of normal animals (Fig. 3A through C, lane 2) was preserved. On the first day after ischemia, DNA laddering was detectable only in the striatum (Fig. 3C, lane 3). No internucleosomal DNA cleavage was seen in hippocampus and cortex (Fig. 3A and B, lane 3). A typical DNA laddered pattern with oligonucleosomal fragments of 180 base pair and its multiples was observed in hippocampus and striatum on days 2 and 3 (Fig. 3A and C, lanes 4 and 5) and in the cortex on day 3 after ischemia (Fig. 3B, lane 4). The DNA fragmentation was reduced in all three regions at day 4 (Fig. 3A and C, lane 6; Fig. 3B, lane 5). The intensity of DNA laddering in the striatum was more pronounced than in the hippocampus and cortex.
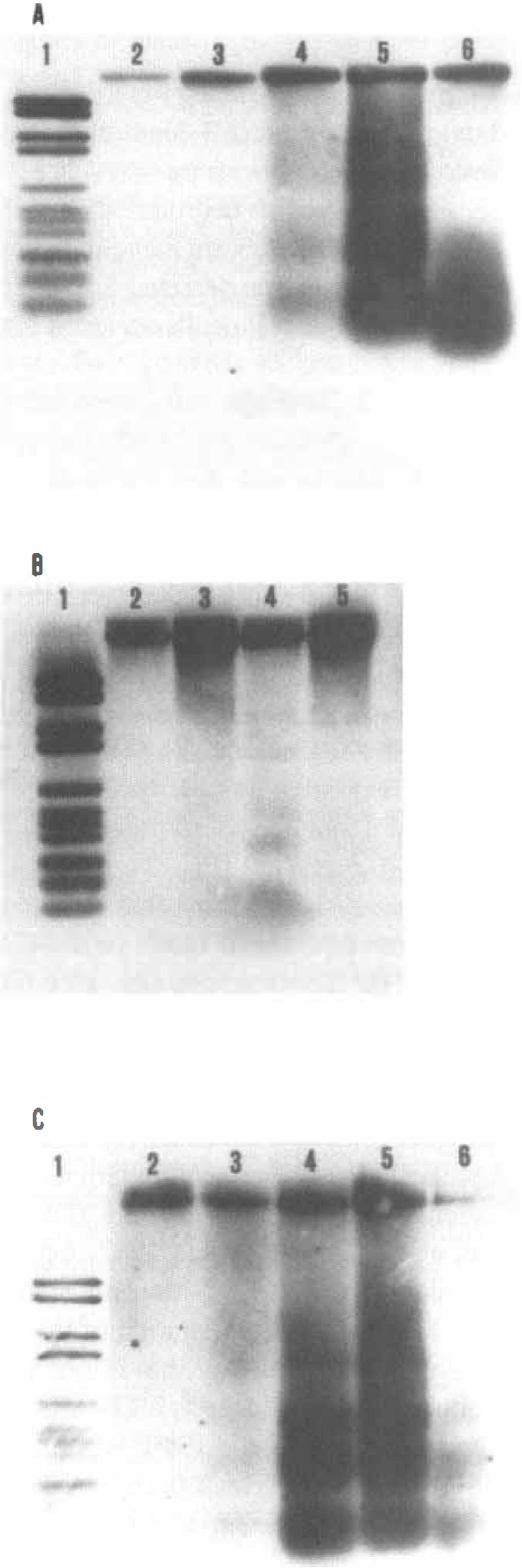
Time courses of DNA fragmentation extracted from hippocampus, striatum, and cortex after 10 minutes of transient global ischemia.
Based on these results, clenbuterol (0.5 mg/kg intraperitoneally) was administered 3 hours before ischemia. Three days after ischemia, the DNA from the hippocampus, striatum, and occipital cortex was prepared for DNA fragmentation assay. The effect of clenbuterol against DNA degradation is shown in Fig. 4. The typical DNA ladders of approximately 180 base pair and its multiples in the control group (Fig. 4, lanes 1, 3, and 5) confirm the results presented in the time course of DNA fragmentation (Fig. 3). In the hippocampus, the DNA fragmentation was nearly abolished after the treatment with clenbuterol (Fig. 4, lane 4). Clenbuterol partially blocked the DNA fragmentation in occipital cortex and striatum (Fig. 4, lanes 2 and 6). The protective effect of clenbuterol against DNA degradation in all of these regions investigated after global ischemia was confirmed by two repeated experiments.
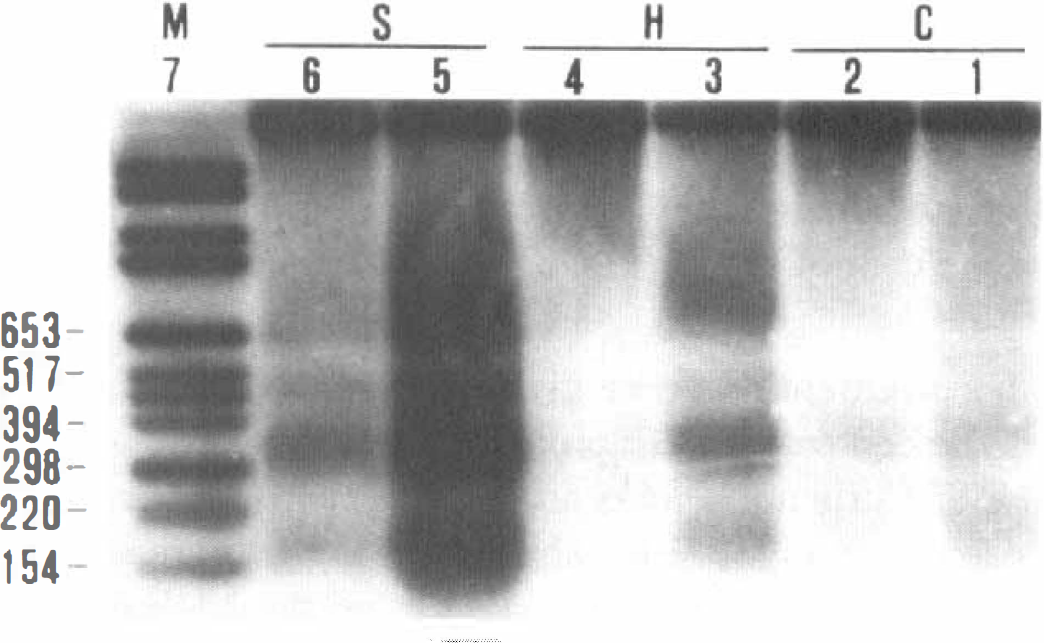
Clenbuterol prevents DNA fragmentation in hippocampus(H), striatum (S), and cortex (C) after 10-minute transient global ischemia. Clenbuterol(0.5 mg/kg) was intraperitoneally administered 3 hours before ischemia. Rat brains were removed 3 days after ischemia, and DNA was extracted from hippocampus, striatum, and cortex. Animals in the control group received saline. Lane 1: digoxigenin-labeled marker; lanes 2, 4, and 6: clenbuterol-treated ischemic animals; lanes 3, 5, and 7: untreated ischemic animals.
The result from TUNEL staining is shown in Fig. 5. In the control group (n = 3), which was subjected to 10-minute global ischemia followed by 3 days of recovery, extensively positively stained neurons were localized in the CA1 subfield of the hippocampus and in the striatum (Fig. 5A and C). In comparison, the number of positively stained neurons in the clenbuterol-treated group (n = 3) was smaller than in the corresponding region of the controls and the intensity of staining was much weaker than in the control group (Fig. 5B and D).

The TUNEL staining of hippocampal, striatal, and cortical tissue. Hippocampus
Measurement of nerve growth factor protein
The content of NGF protein in the hippocampus, cortex, and striatum 6 hours and 3 days after ischemic insult is shown in Fig. 6A. Clenbuterol (0.5 mg/kg) injected 3 hours before global ischemia elevated the NGF protein by 33.1% (P < 0.05) in the hippocampus and 44.4% (P< 0.05) in the cortex compared with the control group when measured 6 hours after ischemia. However, no significant increase of NGF was found in striatum (Fig. 6A). Three days after ischemia, the NGF levels in both clenbuterol-treated groups(0.5 mg/kg and 0.5 mg/kg × 4) were not different from controls (Fig. 6A and B).
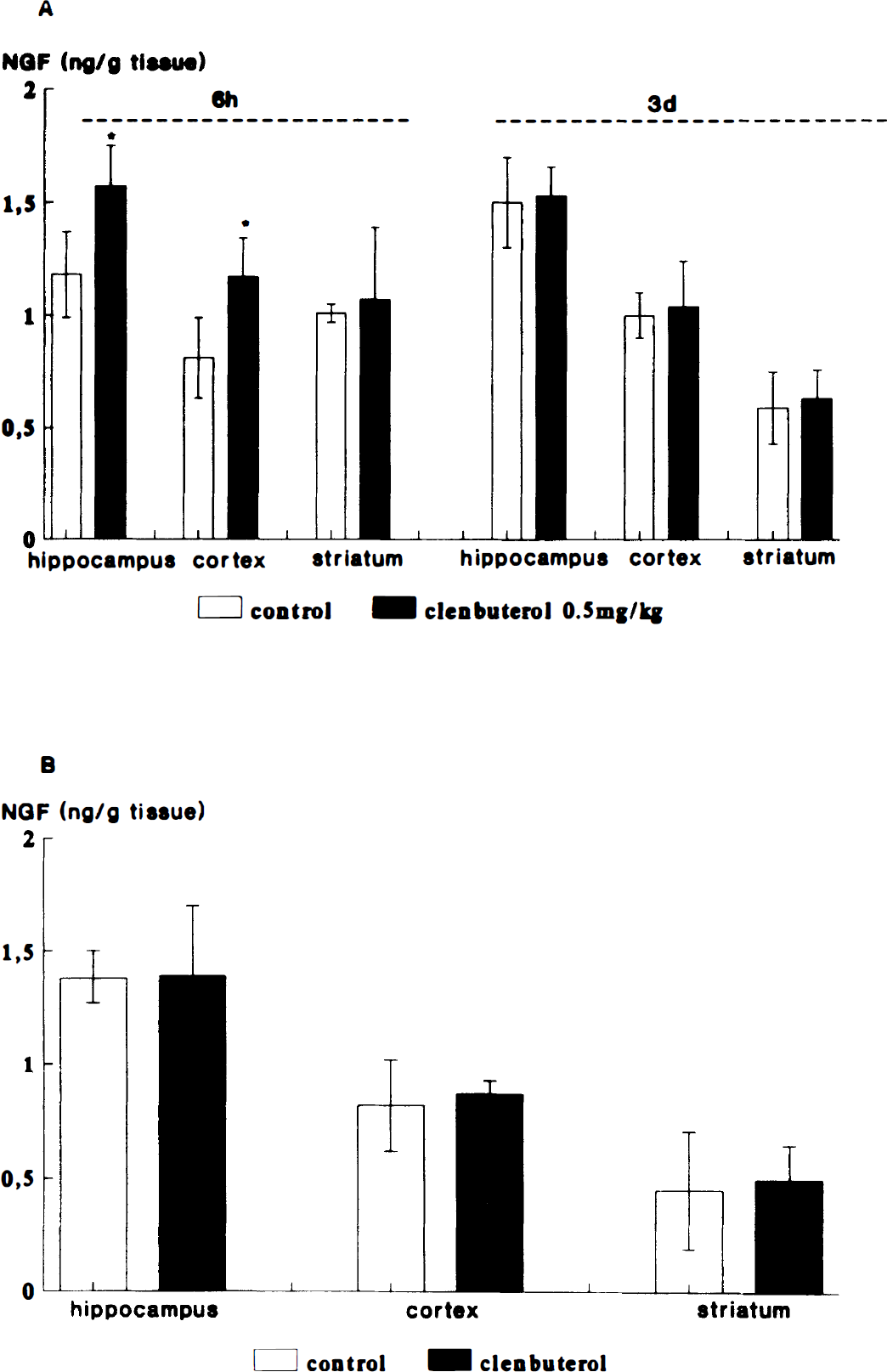
Effect of clenbuterol on nerve growth factor (NGF) protein in hippocampus, striatum, and cortex after the transient global ischemia.
DISCUSSION
In various models of transient global ischemia, a selective and delayed neuronal cell death in the vulnerable CA1 subfield of the hippocampus has been demonstrated. The time course of neuronal damage in CA1 subfield after 10-minute forebrain ischemia showed that the neuronal damage could be detected 2 days after ischemia, and the peak of the cell death occurred 3 to 4 days after ischemia (Fig. 1).
In the present study, a single intraperitoneal injection of clenbuterol was administered. Both doses of clenbuterol (0.5 mg/kg before and after treatment; 1.0 mg/kg pretreatment only) demonstrated neuroprotective activities. Hypotension and increased blood glucose concentration were found in the animals that were treated with 1.0 mg/kg clenbuterol. Increased blood glucose concentration has been suggested to accelerate tissue acidosis during ischemia and aggravate the neuronal damage (Smith et al., 1984; Warner et al., 1987; Smith et al., 1988). This could explain why treatment with 1.0 mg/kg clenbuterol caused a smaller neuroprotective effect than the lower dosage (0.5 mg/kg) (Fig. 2).
It has been previously reported that apoptosis is involved in the delayed neuronal death in the CA1 pyramidal cell layer of the hippocampus after global ischemia (Nitatori et al., 1995; Wiessner et al., 1996). Apoptosis can be described by the morphologic changes of dying cells, including cell shrinkage, nuclear and plasma membrane blebbing, chromatin condensation, and the formation of apoptotic bodies. The accumulation of intracellular free calcium after global ischemia (Dienel, 1984; Deshpande et al., 1987; Dux et al., 1987) has been suggested to activate the Ca++/Mg++-dependent endonucleases, resulting first in the fragmentation of the DNA into 180 to 300 kbp and subsequently into oligonucleosomal cleavage products, which appear as a DNA ladder in the electrophoresis (Arends et al., 1990; Tominaga et al., 1993; Siesjöet al., 1995). Thus, the DNA laddering is considered as the biochemical hallmark of apoptosis (Heron et al., 1993; MacManus et al., 1995). The time courses of DNA fragmentation in the hippocampus, striatum, and occipital cortex obtained in this study revealed that a typical DNA laddering pattern appeared first in the striatum 1 day after ischemia and occurred subsequently in the hippocampus on day 2 and in the occipital cortex on day 3 after ischemia. The peak of the DNA fragmentation in striatum, hippocampus, and cortex appeared 3 days after onset of ischemia. The DNA fragmentation occurred earlier in the striatum than in the hippocampus, and the intensity of DNA fragmentation in the striatum also was stronger than in either the hippocampus or the cortex. Results from TUNEL staining revealed extensively positive stained cells in the hippocampus and striatum 3 days after ischemia. By observing the morphologic features of the striatal neurons in paraffin sections stained with celestine blue, we observed that most of the striatal neurons kept their morphologic features intact 3 days after global ischemia (data not shown), suggesting DNA fragmentation could appear in the nuclei before the morphologic alterations observed in apoptotic cells. We can thus conclude that the delayed neuronal death or apoptosis after global ischemia is not only present in the CA1 subfield but also in the striatum. The latter seems to be especially susceptible to apoptosis.
In necrotic cells, the large degraded DNA fragments, which result from activation of hydrolytic enzymes in dead cells, usually show a smearing pattern (Afanas'ev et al., 1986; Bicknell and Cohen, 1995; MacManus et al., 1995). In our experiment, a typical DNA laddering with smearing was found in control animals (Figs. 3 and 4, lanes 1, 3, 5). These results suggest that both apoptosis and necrosis are present in ischemic brain tissue after global ischemia. It is difficult to estimate the percentages of apoptotic cells or necrotic cells in the total number of damaged cells induced by global ischemia. In situ end-labeling merely indicates the presence of DNA breaks that provide an available 3′-OH end. This may occur in both apoptotic and necrotic cells, so the TUNEL staining alone cannot determine the mechanism of cell death (Wijsman et al., 1993; Charriaut-Marlangue et al., 1995). Nevertheless, the DNA laddering is considered as a distinct characteristic of apoptosis.
In this study, we clearly demonstrate that 0.5 mg/kg of clenbuterol significantly blocked the DNA laddering in ischemic brain tissue (Fig. 4, lanes 2, 4, and 6). The TUNEL staining also showed that the number of positively stained cells in the clenbuterol-treated group is reduced and the intensity of the staining is much weaker than in the control group. These results prove that clenbuterol can efficiently antagonize apoptosis in all three brain regions studied here after global ischemia. On the other hand, a reduced DNA smearing pattern also was found in clenbuterol-treated animals. Combining the results obtained in this study with our previous findings that clenbuterol protects neuronal damage induced by excitotoxicity or focal ischemia (Semkova et al., 1994; Semkova et al., 1996a), we conclude that clenbuterol is able to attenuate necrosis as well.
Nerve growth factor is one of the best characterized neurotrophic factors that supports the growth, differentiation, and survival of neurons within both the peripheral and central nervous systems (Korsching et al., 1985; Rennert and Heinrich, 1986; Whittemore et al., 1986; Longo et al., 1993). Our previous studies showed that the stimulation of β2-adrenergic receptor by clenbuterol could attenuate the damage of cultured neurons by increasing synthesis of NGF. We further attempted to find out whether this drug could elevate the NGF protein content in the brain subjected to transient forebrain ischemia and whether induction of NGF synthesis by clenbuterol is associated with its antiapoptotic effect in the rat model of global ischemia.
The results showed that clenbuterol significantly elevated the NGF protein in the hippocampus and the occipital cortex, but not in the striatum 6 hours after the ischemic insult. Three days after ischemia, the NGF levels were no longer elevated in the clenbuterol-treated groups (0.5 mg/kg and 0.5 mg/kg × 4) when compared with the controls. We observed that after 3 days of recovery, the raised NGF level in controls was most likely induced by ischemia itself in the hippocampus and cortex. Furthermore, repeated treatment with clenbuterol may lead to a down-regulation of β-adrenergic receptors, which could limit NGF induction (Murugaiah and O'Donnell, 1994; Newmantancredi et al., 1996).
Our results suggest that the enhanced NGF synthesis by clenbuterol is detectable, especially at earlier time periods. Thus, we suggest that the NGF synthesis increased by clenbuterol in the initial first hours after ischemia might be important for the prevention of DNA degradation and the support of neuronal survival. The stabilization of the intracellular calcium homeostasis (Cheng and Mattson, 1991; Mattson et al., 1993; Mattson and Cheng, 1994) and the upregulation of the antiapoptotic protooncogene bcl-2 (Katoh et al., 1996; Lezoualc'h et al., 1996; Zhai et al., 1996) also may be involved in the neuroprotective activity of NGF.
In conclusion, this study clearly demonstrates that clenbuterol possesses a neuroprotective activity and a pronounced capacity to block the DNA degradation after global ischemia. We demonstrate for the first time that clenbuterol is able to antagonize apoptosis induced by transient global ischemia. Thus, the pharmacologic induction of endogenous NGF synthesis in brain tissue could become a new therapeutic strategy for neurodegenerative diseases and brain insults that are associated with apoptotic cell death.
Footnotes
Acknowledgements
The authors thank Sandra Engel and Renate Hartmannsgruber for their skillful technical assistance, and Dr. Jochen Prehn for his valuable advice.