
Abstract
Epidermal growth factor (EGF) has been considered to be a candidate for neurotrophic factors on the basis of the results of several in vitro studies. However, the in vivo effect of EGF on ischemic neurons as well as its mechanism of action have not been fully understood. In the present in vivo study using a gerbil ischemia model, we examined the effects of EGF on ischemia-induced learning disability and hippocampal CA1 neuron damage. Cerebroventricular infusion of EGF (24 or 120 ng/d) for 7 days to gerbils starting 2 hours before or immediately after transient forebrain ischemia caused a significant prolongation of response latency time in a passive avoidance task in comparison with the response latency of vehicle-treated ischemic animals. Subsequent histologic examinations showed that EGF effectively prevented delayed neuronal death of CA1 neurons in the stratum pyramidale and preserved synapses intact within the strata moleculare, radiatum, and oriens of the hippocampal CA1 region. In situ detection of DNA fragmentation (TUNEL staining) revealed that ischemic animals infused with EGF contained fewer TUNEL-positive neurons in the hippocampal CA1 field than those infused with vehicle alone at the seventh day after ischemia. In primary hippocampal cultures, EGF (0.048 to 6.0 ng/mL) extended the survival of cultured neurons, facilitated neurite outgrowth, and prevented neuronal damage caused by the hydroxyl radical-producing agent FeSO4 and by the peroxynitrite-producing agent 3-morpholinosydnonimine in a dose-dependent manner. Moreover, EGF significantly attenuated FeSO4-induced lipid peroxidation of cultured neurons. These findings suggest that EGF has a neuroprotective effect on ischemic hippocampal neurons in vivo possibly through inhibition of free radical neurotoxicity and lipid peroxidation.
Keywords
Epidermal growth factor (EGF), a single-chain polypeptide composed of 53 amino acids, has been originally isolated from the submaxillary glands of male mouse. EGF is a well-known potent mitogen for a variety of cell types (Cohen, 1962; Carpenter and Cohen, 1979; Carpenter and Wahl, 1990). The mitogenic activity is mediated by the binding of EGF to a 170-kd cell surface receptor with intrinsic tyrosine kinase activity (Cohen et al., 1980; Plowman et al., 1990). Immunohistochemical studies have shown that EGF and its receptor are distributed in certain brain regions including the hippocampus (Fallon et al., 1984; Gomez-Pinilla et al., 1988; Araujo et al., 1989; Birecree et al., 1991). EGF has been shown to modulate short-term and long-term potentiation of evoked potentials in the hippocampus (Abe et al., 1991; Abe and Saito, 1992), suggesting its physiologic role for synaptic plasticity. Furthermore, EGF facilitates the survival and the process outgrowth of cultured telencephalic neurons from the neonatal rat brain (Morrison et al., 1987). Not only under normal culture conditions but also under pathologic conditions in vitro, EGF has been shown to protect cortical neurons against anoxia-induced damage (Kinoshita et al., 1990) and to attenuate the neurotoxicity of potassium cyanide or N-methyl-D-aspartate on cultured neurons (Pauwels et al., 1989). Recently, Maiese et al. (1993) have shown that pretreatment of cultured hippocampal neurons with EGF prevents nitric oxide-induced neuronal damage. These in vitro findings raise the possibility that EGF subserves an in vivo neuroprotective function in the ischemic brain, but the experimental support of this speculation has not been shown.
In the first set of the present study, we used a gerbil model with transient forebrain ischemia and investigated whether EGF infusion into the cerebral ventricles prevented the ischemia-induced reduction of response latency time in a passive avoidance task and the ischemic neuronal death in the hippocampal CA1 region. The second set of experiments was designed to estimate with in situ detection of DNA fragmentation how many CA1 neurons were in the course of degeneration after the 7-day infusion of EGF or vehicle. The third set of experiments was to see whether EGF, similar to other well-known peptide growth factors (Mattson et al., 1993; Zhang et al., 1993; Mattson and Scheff, 1994), could protect cultured neurons against oxidative injuries caused by the hydroxyl radical-producing agent FeSO4 and by the peroxynitrite-producing agent 3-morpholinosydnonimine (SIN-1) (Braughler et al., 1986; Floyd, 1990; Stadtman and Oliver 1991) because hydroxyl radicals (Braughler et al. 1986; Mattson et al., 1993) and peroxynitrite (Bolanos et al., 1997; Iadecola et al., 1997; Samdani et al., 1997) induced lethal damage to neurons during brain ischemia and reperfusion.
MATERIALS AND METHODS
In vivo ischemic experiments
Animals
Male Mongolian gerbils weighing 70 to 80 g (about 12 weeks of age) were housed communally at constant temperature (22 ± 1°C) with a 12:12 hour light-dark cycle, and bred with food and water ad libitum. They were handled once a week for cage cleaning. The following experiments were conducted in accordance with the Guide for Animal Experimentation at Ehime University School of Medicine.
Osmotic minipump implantation
The animals were anesthetized with 1.5% halothane in a 4:3 mixture of nitrous oxide and oxygen and placed in a stereotaxic apparatus. An osmotic minipump (Alza Corp., Palo Alto, CA) was implanted subcutaneously into the back of each animal, and a needle from the minipump was placed in the left lateral ventricle at the point 1.5 mm anterior, 1.0 mm lateral, and 2.7 mm ventral to bregma as shown in the atlas of Thiessen and Yahr (1977).
Pre-ischemic infusion of EGF
EGF was purchased from Chemicon International Inc. (Temecula, CA) and dissolved in 0.05 mol/L phosphate-buffered saline (PBS) containing 0.1% bovine serum albumin. EGF (24 or 120 ng/d) or vehicle was infused for 7 days into the left lateral ventricles of normothermic gerbils exposed to 3-minute forebrain ischemia (n = 8 in each group). The molar concentrations of infused EGF were similar to those of the other growth factors so far examined in the same gerbil ischemia model (Wen et al., 1995a,b; Matsuda et al., 1996). Six animals received insulin infusion in a dose of 120 ng/d to exclude the possibility of nonspecific effects of protein. Although insulin with a molecular weight similar to that of EGF has been shown to exhibit neurotrophic action at high concentrations (Zhu and Auer, 1993; Hamilton et al., 1995), we did not detect any neuroprotective action of insulin in vitro when the same molar concentrations of insulin as those of EGF were added to neuronal cultures (data not shown). Sham-operated animals received vehicle infusion (n = 8). The infusion was started at 2 hours before an ischemic insult as described elsewhere (Sano et al., 1994; Wen et al., 1995a,b; Kotani et al., 1996; Matsuda et al., 1996).
Post-ischemic infusion of EGF
To investigate the effect of post-ischemic treatment with EGF on delayed neuronal death, 2 ng or 10 ng EGF in 2 μL of vehicle was injected into the left lateral ventricle through a Hamilton syringe (MS-25, Ito, Japan) immediately after 3-minute forebrain ischemia, and then EGF in a dose of 24 ng/d or 120 ng/d was continuously infused for 7 days into the cerebral ventricles as described above (n = 8 in each group). In control experiments, ischemic animals and sham-operated animals received vehicle infusion (n = 8 in each group).
Occlusion of the common carotid arteries
Occlusion of the common carotid arteries was performed as previously described (Wen et al., 1996). Briefly, both common carotid arteries were exposed through a ventral midline incision and separated carefully from the adjacent veins and nerves under inhalation anesthesia. Immediately after the termination of inhalation anesthesia, the common carotid arteries were clamped for 3 minutes with aneurysm clips.
During forebrain ischemia, brain temperature has been shown to fall differently in individual animals, thereby affecting the number of viable CA1 neurons after ischemia (Mitani et al., 1991). To avoid the effect of unstable brain temperature on ischemic neuronal loss, we kept brain and rectal temperatures at 37.0 ± 0.2°C while clamping the common carotid arteries (Mitani et al., 1991; Sano et al., 1994; Wen et al., 1995a,b; Kotani et al., 1996; Matsuda et al., 1996; Wen et al., 1996). This enabled us to induce an invariable neuronal damage in the hippocampal CA1 field even after such a brief ischemic insult (Sano et al., 1994; Wen et al., 1995a,b; Kotani et al., 1996; Matsuda et al., 1996) and to evaluate accurately the in vivo effect of EGF on delayed neuronal death.
Passive avoidance task
Seven days after forebrain ischemia, the gerbils were trained in a conventional step-down passive avoidance apparatus which was divided into a safe platform and a chamber with a stainless-steel grid floor (Araki et al., 1986; Sano et al., 1994; Wen et al., 1995a, b; Kotani et al., 1996; Matsuda et al., 1996; Wen et al., 1996). Each animal was placed initially on the safe platform. When the gerbil stepped down onto the grid floor, it received a foot shock. Although the gerbil went repeatedly up and down between the platform and the grid, it eventually remained on the platform. This training session lasted 300 seconds. Twenty-four hours later, the gerbil was again placed on the safe platform while the shock generator was turned off, and the response latency, i.e., the time until it stepped down onto the grid floor, was measured. This test session also lasted 300 seconds. Each animal received only one training session and only one test session.
Histopathologic study of hippocampal CA1 region
At 1 hour after the passive avoidance experiments, each animal was anesthetized with pentobarbital, and the osmotic minipump was disconnected from the needle placed in the lateral ventricle. Bromophenol blue was injected through the needle to ascertain the infusion of EGF, vehicle, or insulin into the cerebral ventricles. Then the animals were perfused transcardially with 4% paraformaldehyde-2.5% glutaraldehyde in 0.1-mol/L phosphate buffer (pH 7.4) for light and electron microscopy (Wen et al., 1996). Gerbils (approximately 1 in 16 animals) not showing diffusion of bromophenol blue were excluded from subsequent experiments. A brain region including the dorsal hippocampus from 0.5 to 1.5 mm posterior to bregma was removed and kept in the same fixative overnight at 4°C. Four serial coronal sections with 50 μm in thickness at the level 1.0 to 1.2 mm posterior to bregma were cut with a microslicer (Dosaka EM, Japan) for electron microscopy. The remaining dorsal hippocampus was embedded in paraffin, and 5 μm serial frontal sections were stained with 0.1% cresyl violet. All neurons with intact morphologic appearance along 1 mm linear length of the CA1 field in six serial coronal sections (1.20 to 1.23 mm posterior to bregma) were counted. The mean number of neurons per section was calculated for each animal. For electron microscopy, the specimens were postfixed with 1% osmium tetroxide for 30 minutes, dehydrated with a graded series of ethanol, transferred to propylene oxide and embedded in epoxy resin. The strata moleculare, radiatum, pyramidale, and oriens of the CA1 region were identified on semithin sections stained with 1% toluidine blue, and 70-nm ultrathin sections were made with a Reichert-Nissei ultramicrotome (Optische Werke, Austria) and mounted on single-slot (2 × 0.5 mm) grids that were coated with Formbar film. They were subjected to dual staining with uranyl acetate and lead citrate and observed in a transmission electron microscope.
In situ detection of DNA fragmentation (TUNEL staining)
To analyze quantitatively the changes in the nuclear chromatin of the hippocampal CA1 neurons as revealed by electron microscopy, we conducted TUNEL staining. Two additional paraffin sections from each animal in the EGF- and vehicle-infused groups were processed for in situ detection of DNA fragmentation (TUNEL staining) to estimate the number of CA1 neurons degenerating after the termination of EGF or vehicle infusion. Briefly, the sections were (1) deparaffinized in xylene and in a graded series of ethanol; (2) washed two times in 0.05 mol/L PBS for 10 minutes and then incubated with 20 μg/mL of proteinase K (Sigma) for 15 minutes; (3) rinsed with distilled water four times for 2 minutes each and quenched in 2.0% hydrogen peroxide for 10 minutes; (4) rinsed with PBS twice for 5 minutes each and incubated with equilibration buffer (Oncor, Inc.), and then incubated with a mixture of terminal deoxynucleotidyl transferase and reaction buffer containing digoxigenin-2′-deoxyuridine 5′ triphosphate (Oncor, Inc.) in a humidified chamber for 1 hour at 37°C; (5) immersed in wash buffer (Oncor, Inc.) for 10 minutes and washed with PBS three times for 5 minutes each; (6) incubated with anti-digoxigenin-peroxidase (Oncor, Inc.) for 1 hour at room temperature; and (7) rinsed with PBS three times for 5 minutes each and exposed to 0.05% diaminobenzidine and 0.02% hydrogen peroxide. All TUNEL-positive neurons along 1 mm linear length of the CA1 field in the two serial coronal sections were counted and the mean number of positive neurons was calculated in each animal.
In vitro culture experiments
Hippocampal neuron culture
The hippocampi of 17-day-old rat embryos were aseptically dissected and hippocampal neurons were dissociated from the tissues as described elsewhere (Kira et al., 1995). The dissociated cells were seeded on 24-well plastic plates (Corning, NY) coated with poly-L-lysine (Sigma) at a density of 3 to 5 × 104 cells/cm2. The cells were cultured at 37°C in Dulbecco's modified Eagle's medium (0.1% glucose; Nipro, Japan) supplemented with 10% fetal calf serum (Equitech-Bio, TX) under a humidified atmosphere with 5% CO2. On the second day of culture, the culture medium was replaced with serum-free Dulbecco's modified Eagle's medium containing 0 to 1 nmol/L (or 0 to 6.0 ng/mL) EGF, 20 mmol/L N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES), 0.45% glucose, 5 μg/mL pig insulin (Boehringer-Mannheim), 5 μg/mL human transferrin (Boehringer-Mannheim), 5 ng/mL sodium selenite (Boehringer-Mannheim), 25 nmol/L progesterone (Sigma) and 0.1% bovine serum albumin (Sigma). Then the cells were cultured for 3 days. On the third day of culture, freshly prepared 90 μmol/L FeSO4 or 20 μmol/L SIN-1 solution was added to the medium to introduce oxidative damage. The neurons exposed to FeSO4 were maintained for 24 hours at 37°C and those exposed to SIN-1 for 6 hours at 37°C (Braughler et al., 1986; Floyd, 1990; Stadtman and Oliver 1991; Zhang et al., 1993; Cheng and Mattson, 1995). Neurons not exposed to FeSO4 or SIN-1 were also treated with the different concentrations of EGF. Then, the cells were fixed with the PHEM buffer (60 mmol/L 1,4-piperazinediethanesulfonic acid, 25 nmol/L HEPES, 2 mmol/L MgCl2 and 1 mmol/L ethylenedioxybis-N,N,N′,N′-tetraacetic acid, pH 6.9) containing 4% paraformaldelhyde and 0.2% glutaraldelhyde for morphologic examination (Tanaka and Maeda, 1996).
Immunoblot analysis
For immunoblot analysis, the cultured hippocampal neurons from each well were solubilized in a sample solution containing 2% sodium dodecyl sulfate. The final volume was adjusted to 100 μL, and elecrophorsis was used on 15 μL of the sample and placed into each lane by using 6% polyacrylamide gel in the Laemmli's buffer system (Laemmli, 1970). The electrophoretic bands were transferred to nitrocellulose sheets (Towbin et al. 1979) and immunoblotted with a monoclonal antibody (Sternberger Monoclonals Inc., Baltimore) against microtubule-associated protein 2 (MAP2), which is known to be a specific neuronal marker. Anti-mouse immunoglobulin G coupled with alkaline phosphatase (Promega, Madison, WI) was used for the second immunoreaction. The immunoreactive bands were visualized as described elsewhere (Kira et al., 1995). Prestained molecular weight markers were purchased from Bio Rad (Richmond, CA). For quantitative evaluation, the immunoreactive bands of MAP2 were subjected to densitometric analysis (Tanaka and Sobue, 1994).
Lipid peroxidation assays
Relative levels of lipid peroxidation in cultured neurons were quantified by using 2-thiobarbituric acid fixation and subsequent measurement of light absorbance at 535 nm (Buege and Aust, 1987). Because this experiment needed a large number of neurons, we chose the cerebral cortex for material. The cortical neurons of 17-day-old rat embryos were cultured at a density of 8.0 × 105 cells/cm2. Freshly prepared 10 μmol/L FeSO4 solution was added to the medium, which was pretreated with 0 to 6.0 ng/mL EGF on the third day of culture, and the neurons were maintained for 2 hours at 37°C. Then the culture medium was removed and 150 μL of 1% sodium dodecyl sulfate was added to each culture plate. The neurons were scraped off and collected into a 1-mL tube, and 450 μL of 0.375% thiobarbituric acid solution containing 0.015% dimethyl sulfoxide, 15% trichloroacetic acid, and 0.25 mol/L of hydrochloric acid was added to the tube. After heating in a boiling water bath for 15 minutes, the homogenate was centrifuged at 10,000 × g for 5 minutes, and light absorbance at 535 nm was measured with a UV-120-02 spectrophotometer (Shimadzu, Japan).
Statistics
All in vivo experiments were performed blindly with respect to experimental group. The effects of EGF on response latency, CA1 neuron density, and TUNEL-positive neuron number were evaluated by the two-tailed Mann-Whitney U test, which enabled us to compare the EGF-treated groups with the vehicle- or insulin-treated ischemic group. Densitometric data of the immunoreactive bands and lipid peroxidation assays were evaluated by the one-way analysis of variance followed by post hoc test (Fisher's protected least significant difference). All data were represented as mean ± SD.
RESULTS
In vivo ischemic experiments
The mean response latency and CA1 neuronal density of sham-operated animals with vehicle infusion were 240 ± 42 seconds and 241 ± 15 cells/mm, respectively, whereas those of 3-minute ischemic gerbils infused with vehicle alone were 123 ± 30 seconds and 133 ± 35 cells/mm, respectively. There were significant differences in response latency (U = 0, P < .01) and CA1 neuronal density (U = 0, P < .01) between the two groups (Fig. 1A, B). In histologic sections, the CA1 region of 3-minute ischemic gerbils exhibited a marked decrease in viable neurons when compared with the CA1 field of sham-operated animals (Fig. 2A, B).
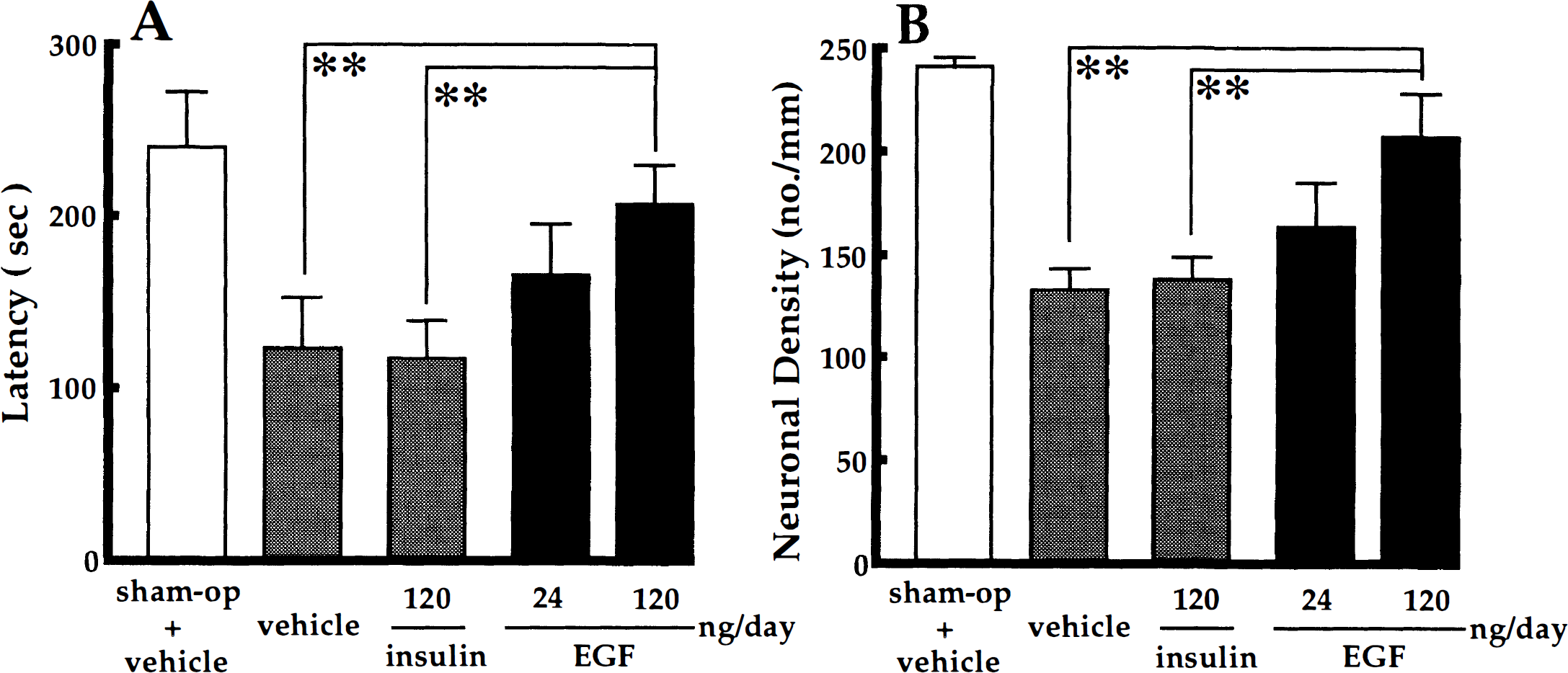
Effects of preischemic epidermal growth factor (EGF) infusion on the response latency (
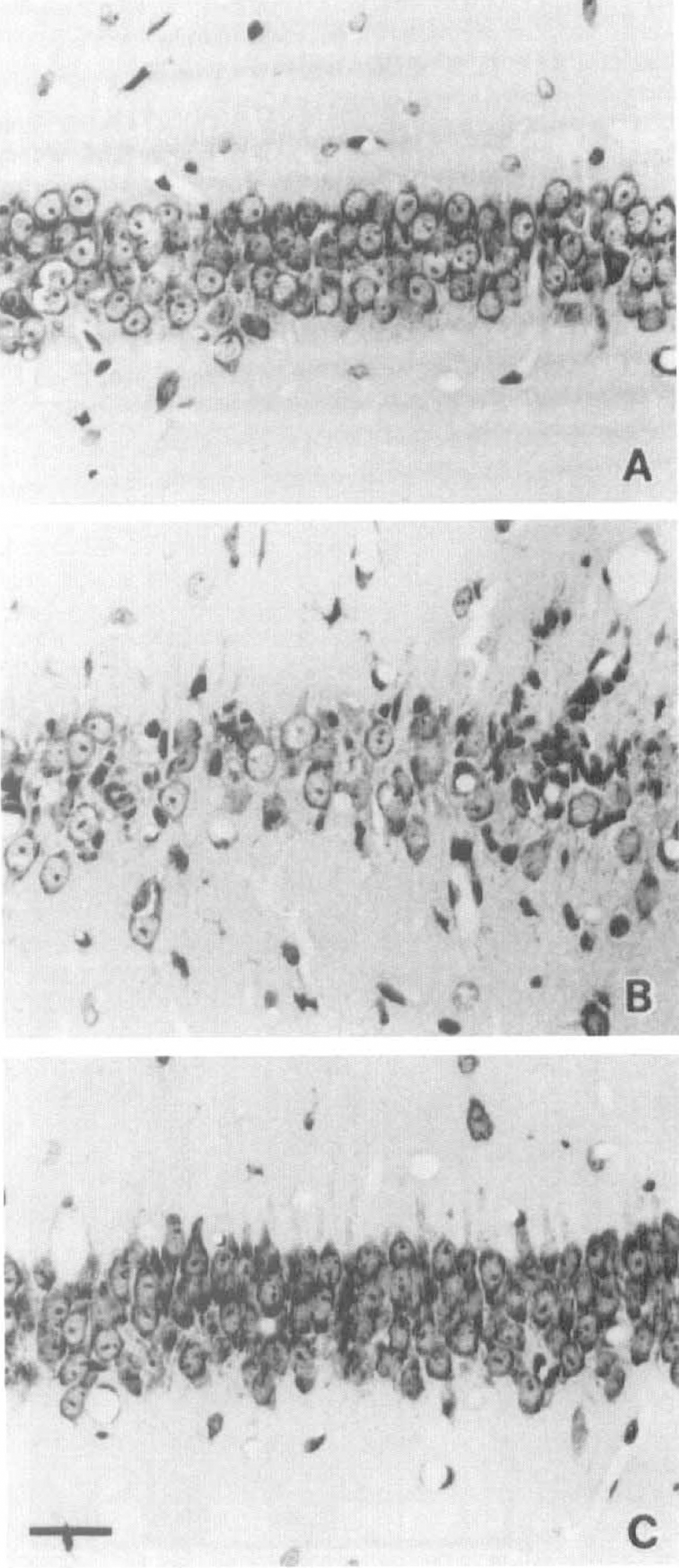
Photomicrographs of the hippocampal CA1 field of sham-operated and ischemic gerbils. (
The infusion of EGF in a dose of 120 ng/d into the lateral ventricle for 7 days, starting 2 hours before 3-minute forebrain ischemia, caused a significant prolongation in response latency time in the step-down passive avoidance task (EGF versus vehicle in ischemic gerbils: U = 12, P < .01; EGF versus insulin in ischemic gerbils: U = 9, P < .01) (Fig. 1A). Histologic examinations revealed that the treatment with 120 ng/d of EGF rescued many ischemic neurons that were destined to degenerate without EGF infusion (EGF versus vehicle in ischemic gerbils: U = 10, P < .01; EGF versus insulin in ischemic gerbils: U = 13, P < .01) (Figs. 1B, 2C). The lower dose of EGF (24 ng/d) was ineffective in the rescue of the ischemic neurons, although the mean response latency and CA1 neuronal density were higher in EGF-infused ischemic animals than in vehicle/insulin-infused ischemic animals (Fig. 1A, B).
EGF infusion in a dose of 120 ng/d for 7 days, starting just after forebrain ischemia, also caused a significant prolongation in response latency time in comparison with the response latency of vehicle-treated ischemic gerbils (EGF versus vehicle in ischemic animals: U = 6, P < .01) and rescued a significant number of ischemic CA1 neurons (EGF versus vehicle in ischemic animals: U = 7, P < .01) (Fig. 3). The lower dose of EGF (24 ng/d) was ineffective in the rescue of the ischemic neurons (Fig. 3).
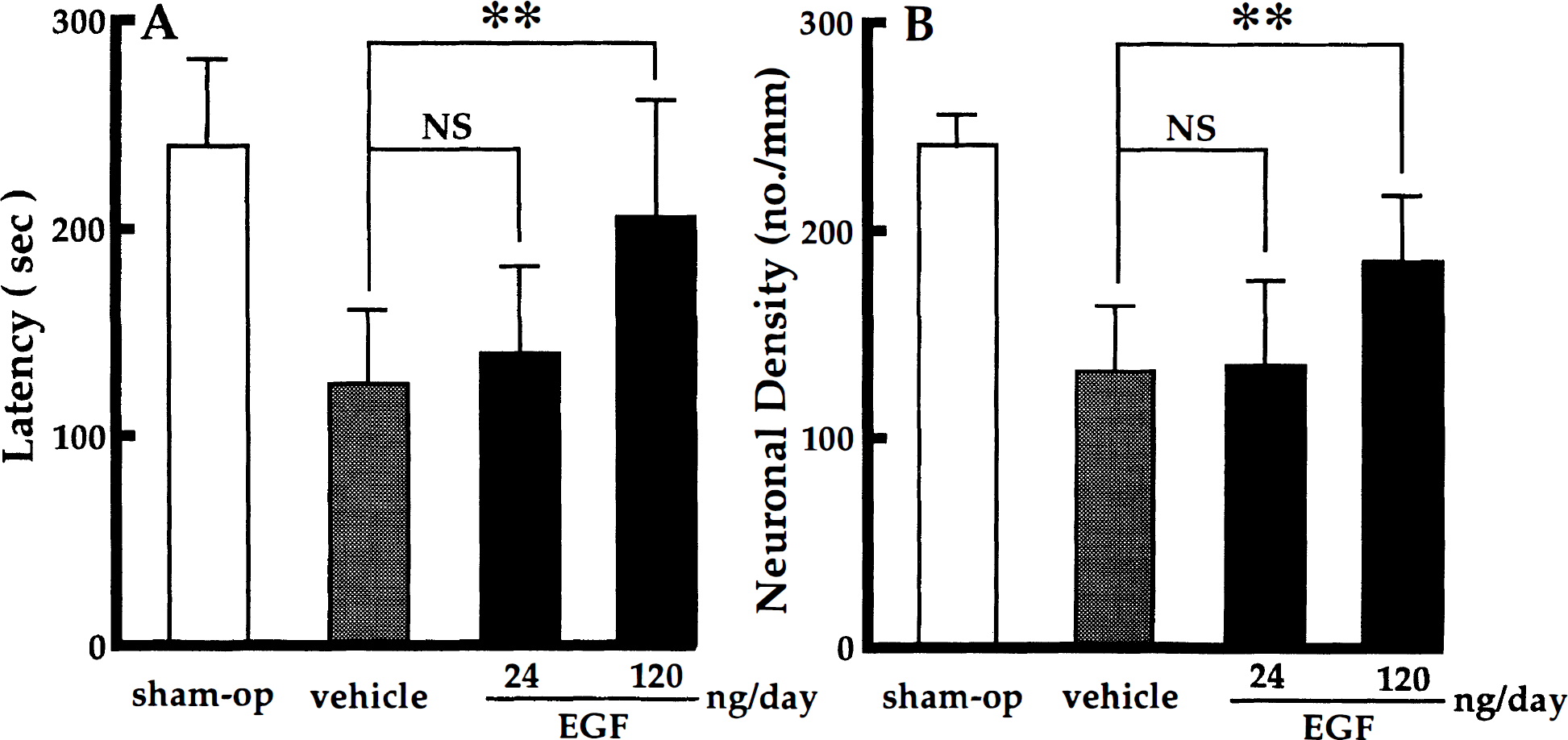
Effects of postischemic EGF infusion on the response latency (
In line with the results of the passive avoidance task and light microscopic observations, electron microscopy showed that the number of intact synapses within the stratum moleculare, stratum radiatum, and stratum oriens of the hippocampal CA1 region was more in EGF-treated than in vehicle-treated ischemic gerbils (Fig. 4). Under light microscopy, vehicle-treated hippocampal CA1 neurons surviving at the seventh day after transient forebrain ischemia seemed to be intact. However, a careful observation with an electron microscope revealed that a significant number of neurons was possibly in the course of nuclear chromatin fragmentation and/or condensation to different degrees (Fig. 5A, B); the nuclei of vehicle-treated ischemic neurons at early stages of degeneration had an irregular euchromatin with low electron density as compared with intact nuclear euchromatin (Fig. 5A). On the other hand, most of surviving pyramidal neurons in the CA1 field of EGF (120 ng/d)-treated gerbils retained normal morphologic features even at the seventh day after forebrain ischemia (Fig. 5C).
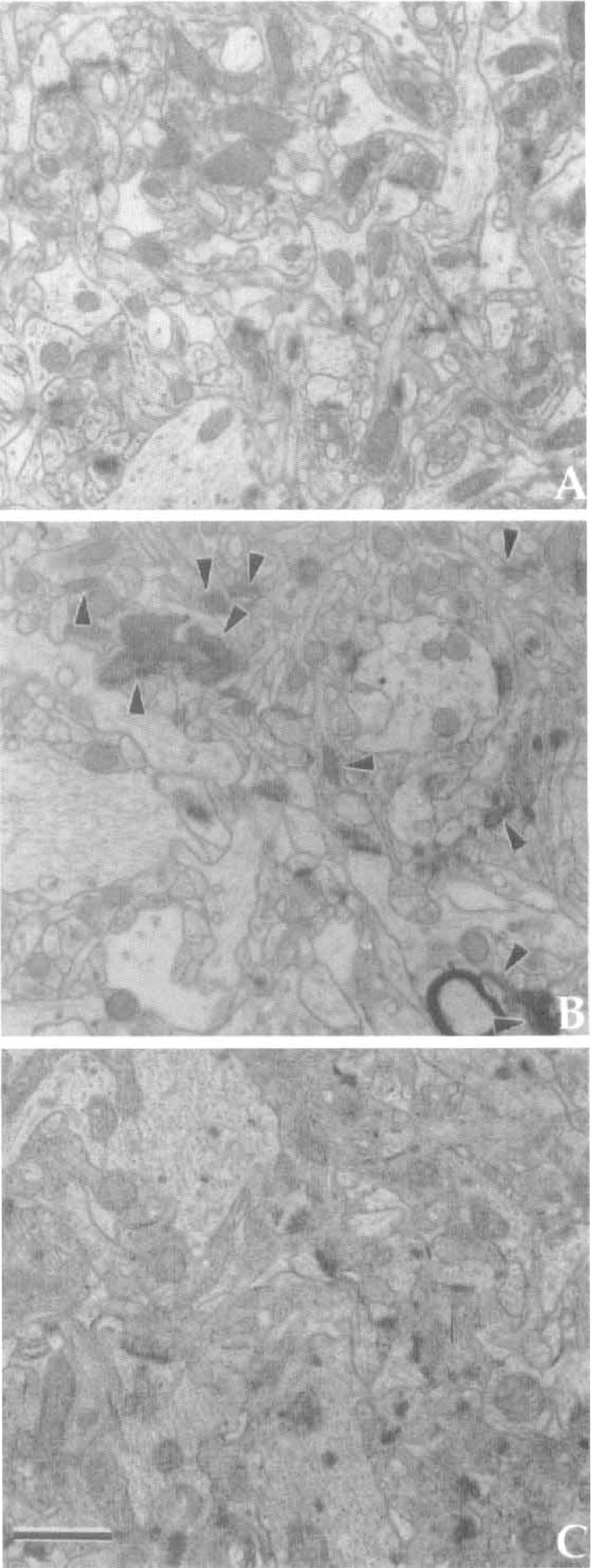
Electron micrographs of the stratum radiatum in the hippocampal CA1 field of sham-operated and ischemic gerbils. (
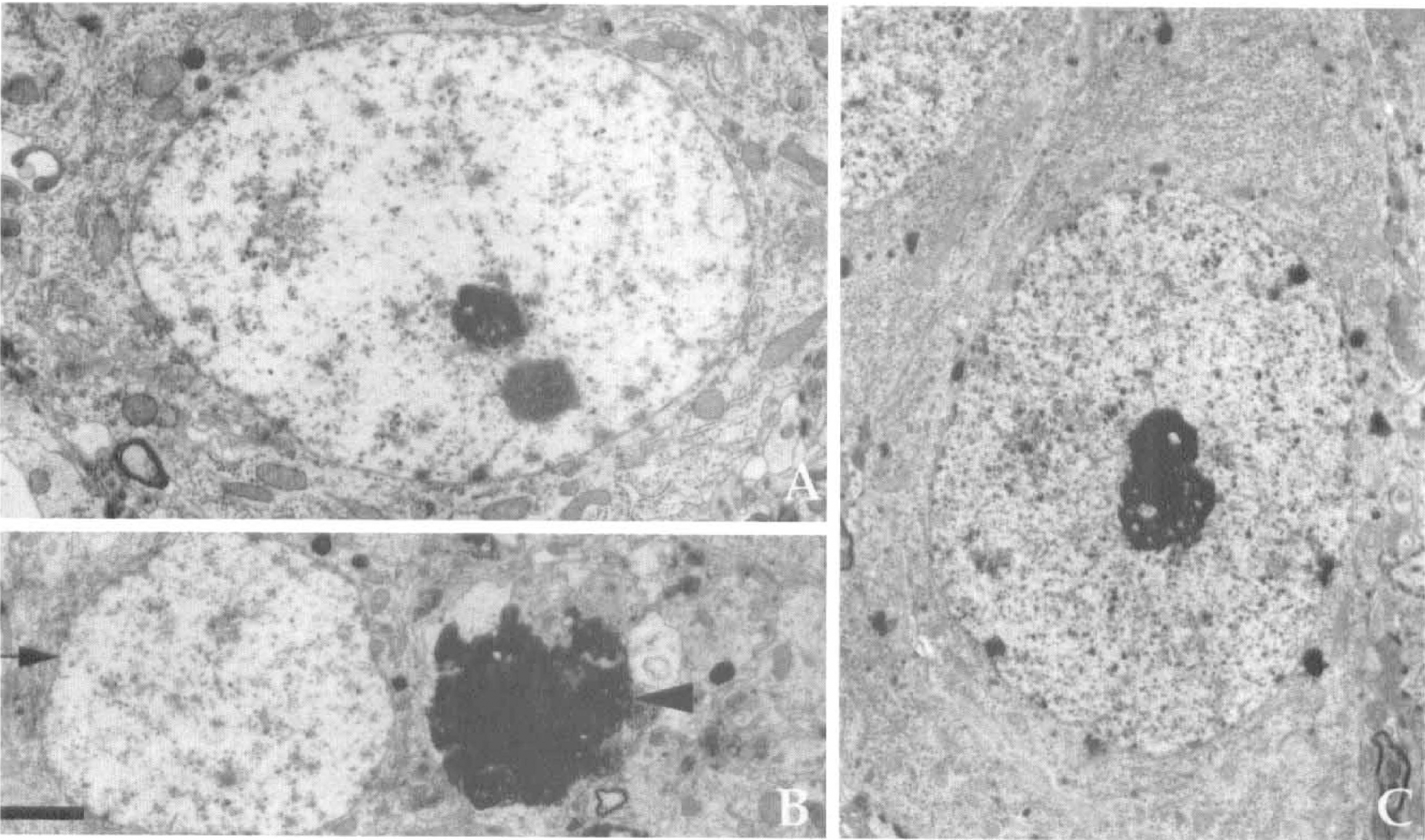
Electron micrographs of hippocampal CA1 pyramidal neurons treated with vehicle (A, B) or 120 ng/d of EGF (C) during 7 days after 3-minutes of forebrain ischemia. (
TUNEL staining revealed that many TUNEL-positive neurons were present in the hippocampal CA1 field of 3-minute ischemic gerbils with vehicle infusion at the seventh day after ischemic insult, suggesting that irreversible neuronal degeneration was in progress at this period (Fig. 6A). The 7-day infusion of EGF in a dose of 120 ng/d but not 24 ng/d starting either before or just after 3-minute ischemia not only prevented delayed neuronal death in the hippocampal CA1 field at the seventh day after ischemic insult but also reduced the number of TUNEL-positive neurons that were in the course of a more delayed degeneration (Fig. 6B, C). The count of TUNEL-positive cells in the vehicle-infused ischemic gerbils indicated that without EGF treatment nearly half of the CA1 neurons surviving at the seventh day after 3-minute ischemia would undergo a further degeneration within a few days (Fig. 7). EGF treatment precluded the late onset of ischemia-induced neuronal death reminiscent of apoptosis (120 ng/d EGF versus vehicle infusion starting before ischemia: U = 0, P < .01; 120 ng/d EGF versus vehicle infusion starting after ischemia: U = 0, P < .01).
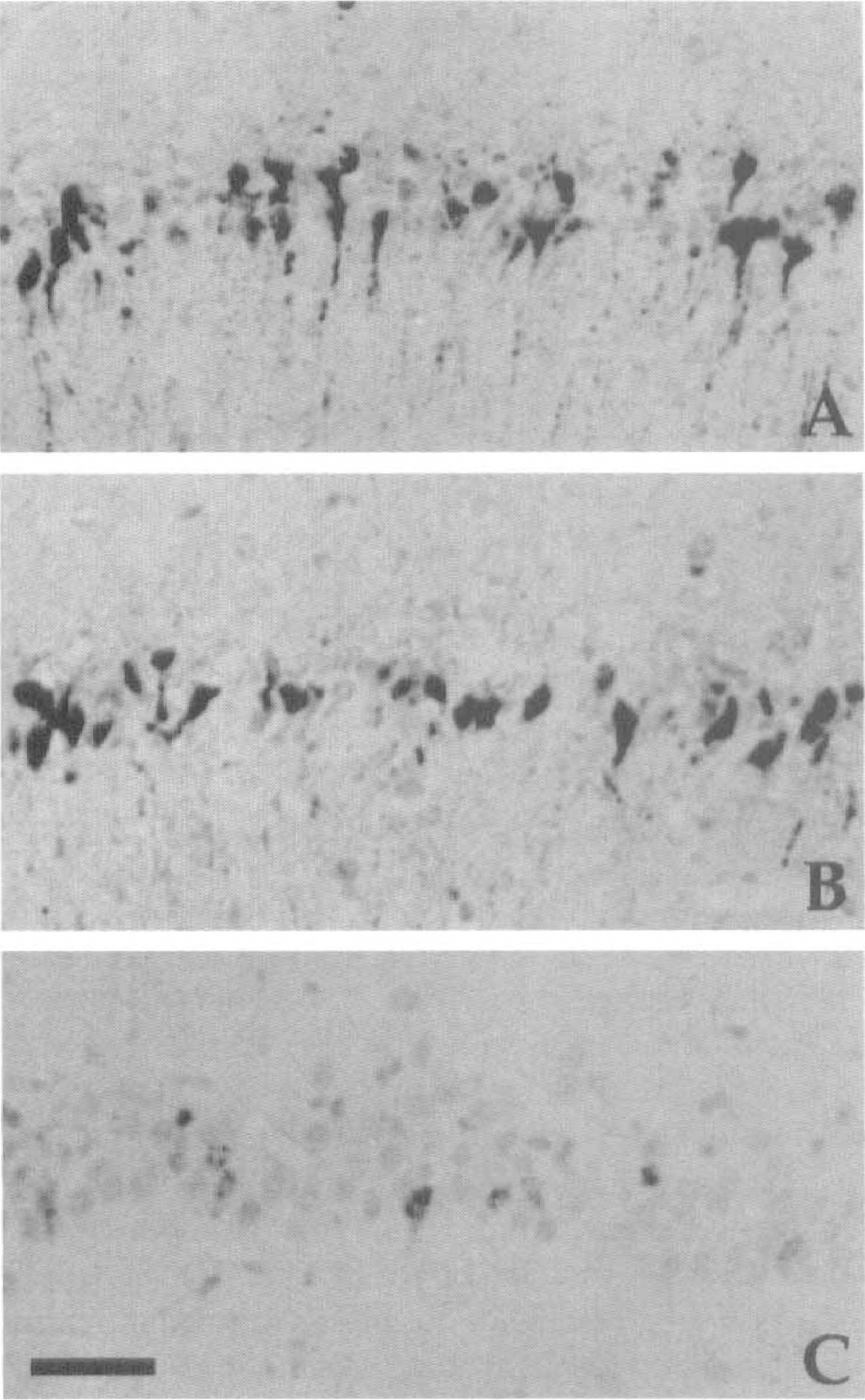
Photomicrographs of the TUNEL-positive neurons in the hippocampal CA1 field of 3-minute ischemic gerbils after 7-day infusion of vehicle or EGF. (
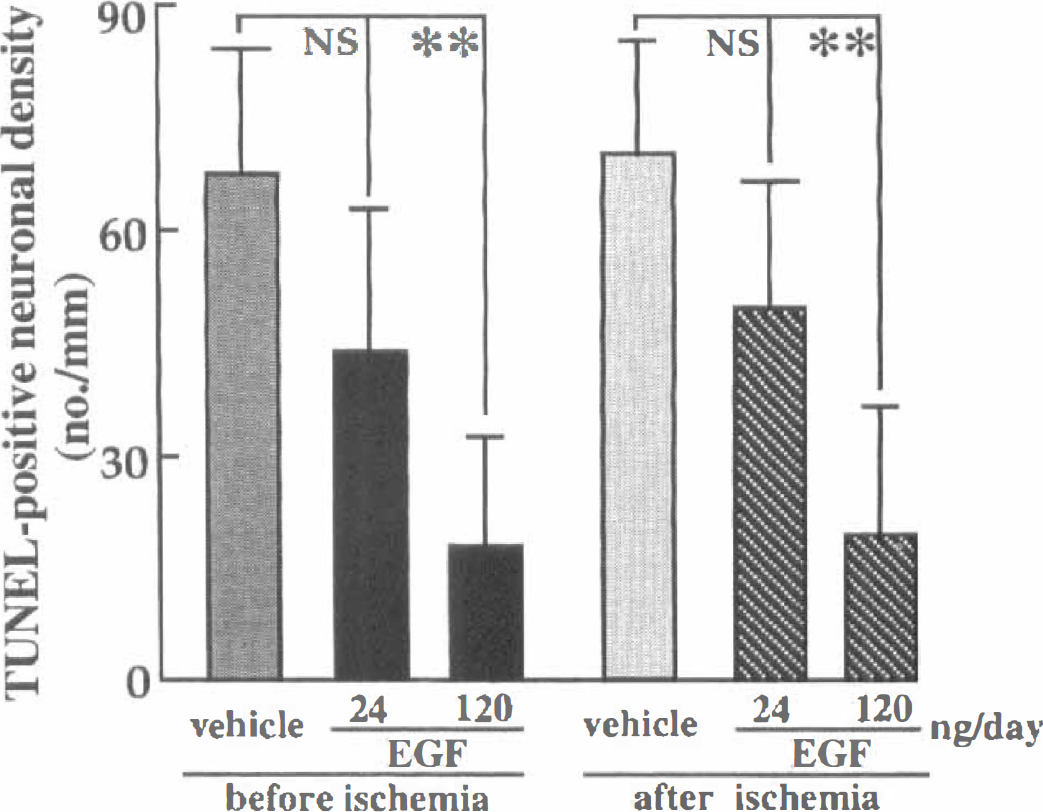
Number of TUNEL-positive neurons in the hippocampal CA1 field of ischemic gerbils. The infusion of EGF (120 ng/day) into the cerebral ventricles of ischemic gerbils, starting either before or after 3-minutes of forebrain ischemia, caused a significant reduction in the number of TUNEL-positive neurons in comparison with those in the corresponding vehicle-treated ischemic gerbils. Each value represents mean ± SD (n = 8). **P < 0.01, significantly different from the corresponding vehicle-treated ischemic groups (statistical significance tested by the two-tailed Mann-Whitney U-test).
In vitro culture experiments
When EGF at concentrations of 0.048 to 6.0 ng/mL (0.008 to 1 nmol/L) was added to the culture medium on the second day of culture, hippocampal neurons became more numerous than those without EGF treatment on the fifth day of culture (Fig. 8A, B). Moreover the neurons with EGF treatment gave rise to many processes (Fig. 8B). As the hydroxyl radical-producing agent FeSO4 was added to the culture medium, the processes of hippocampal neurons were fragmented within 16 hours (Fig. 8C) and they were no longer visible within 24 hours. However, when the cultures were pretreated with EGF, the damage to hippocampal neurons as caused by FeSO4 was apparently attenuated (Fig. 8D).
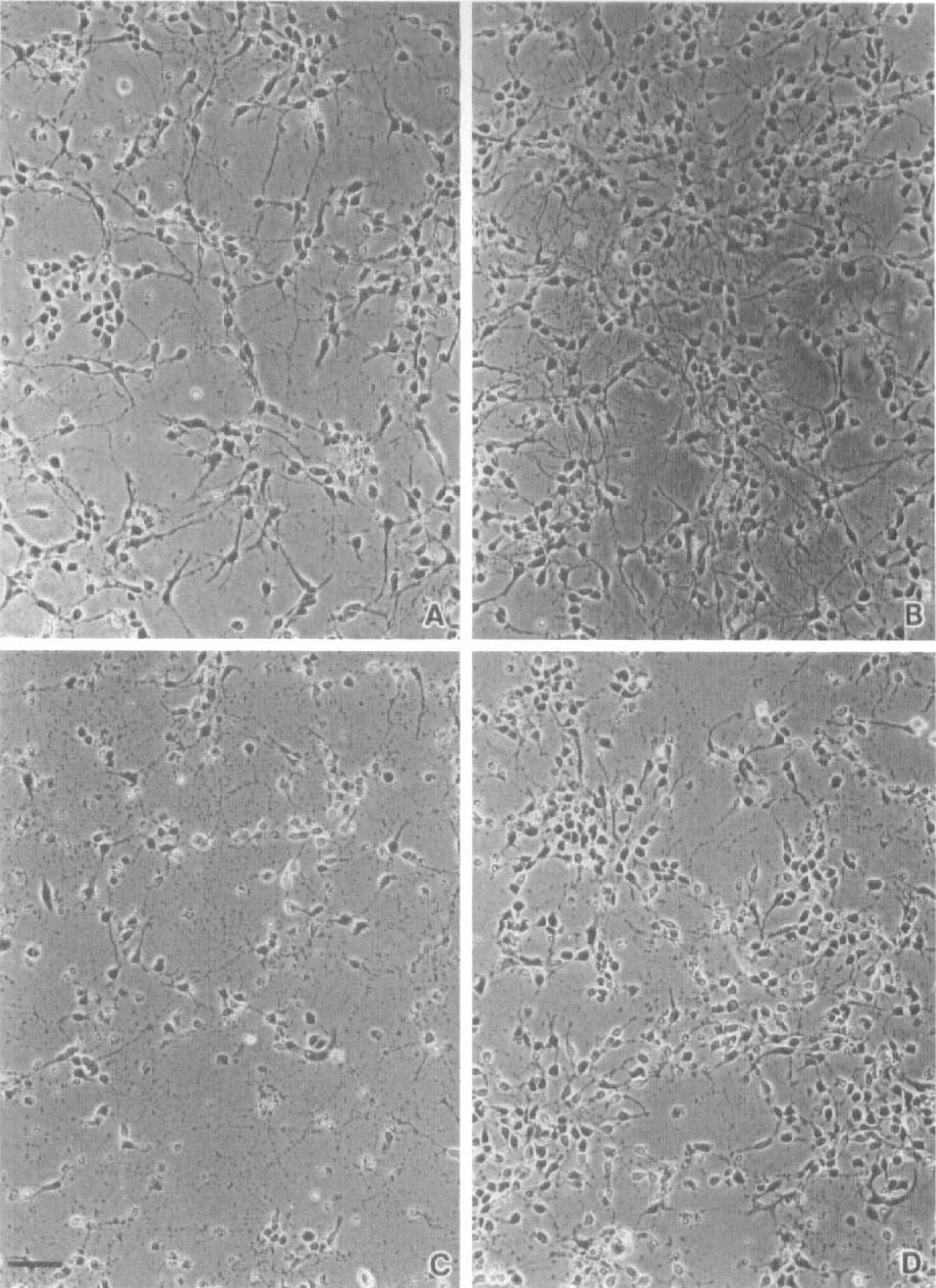
Photomicrographs of cultured hippocampal neurons. (
To quantify the trophic action of EGF on cultured hippocampal neurons, the neurons were solubilized, electrophoresed, and immunoblotted with a monoclonal antibody against MAP2. The control hippocampal neurons not exposed to FeSO4 exhibited a weak MAP2 band, and the MAP2 band increased in intensity in response to EGF treatment (Fig. 9A). The most effective concentration of EGF was 1.2 ng/mL (P < .01)(Fig. 9A). In FeSO4-exposed neurons, the MAP2 band was almost invisible without EGF treatment (Fig. 9B). When EGF at concentrations of 0.048 to 6.0 ng/mL was added to the culture medium beforehand, the MAP2 band significantly increased in intensity (Fig. 9B), suggesting that FeSO4-induced neuron death was prevented by EGF. Pretreatment with EGF at concentrations of 0.048 to 6.0 ng/mL also protected hippocampal neurons against SIN-1-induced damage in a dose-dependent manner (Fig. 10). Similar results were obtained when cortical neurons were used instead of hippocampal neurons in the above culture studies, except that the concentration of FeSO4 inducing oxidative injury to cortical neurons was 10 μmol/L (data not shown).
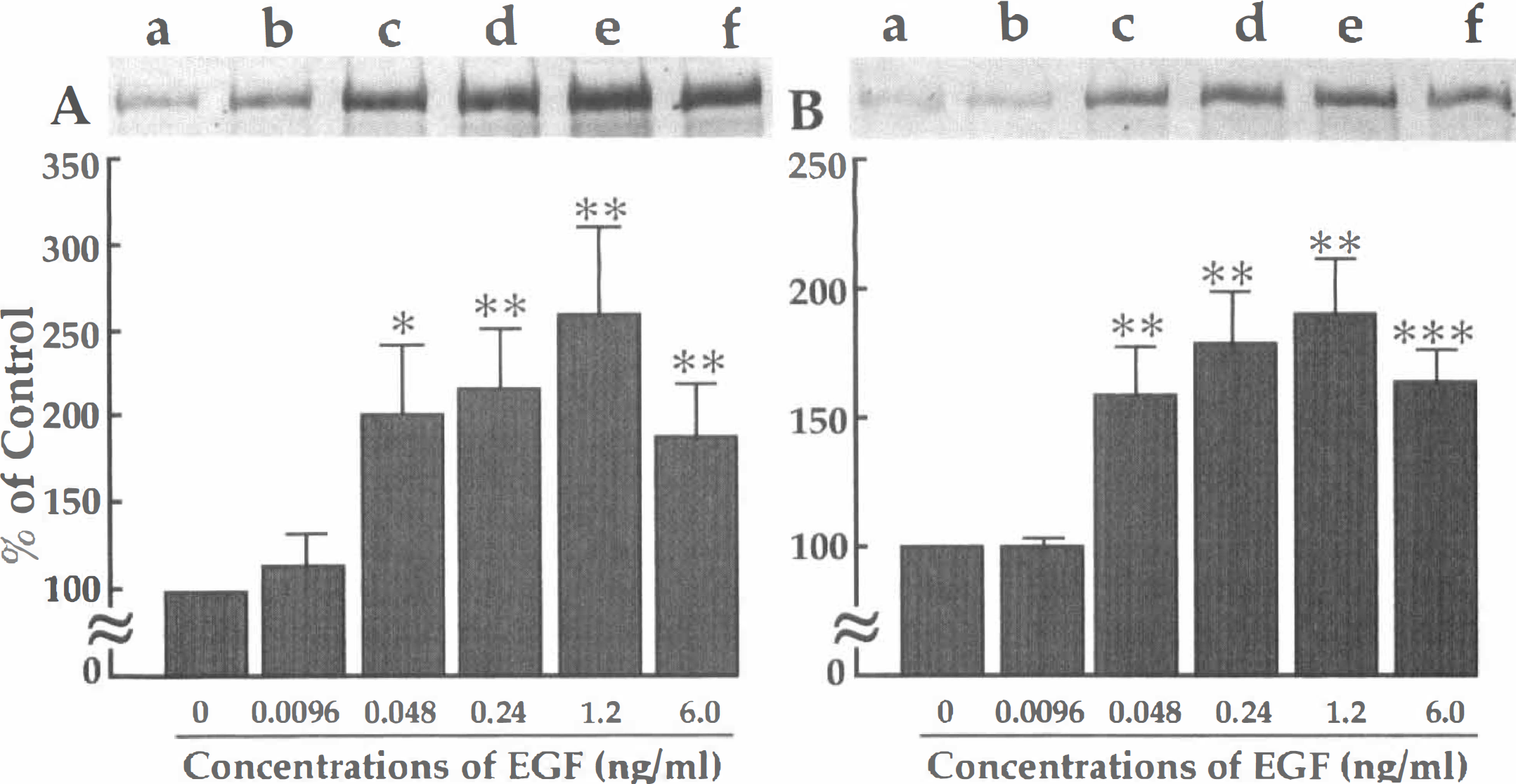
MAP2-immunoblot analysis of EGF-treated hippocampal neurons not exposed to (
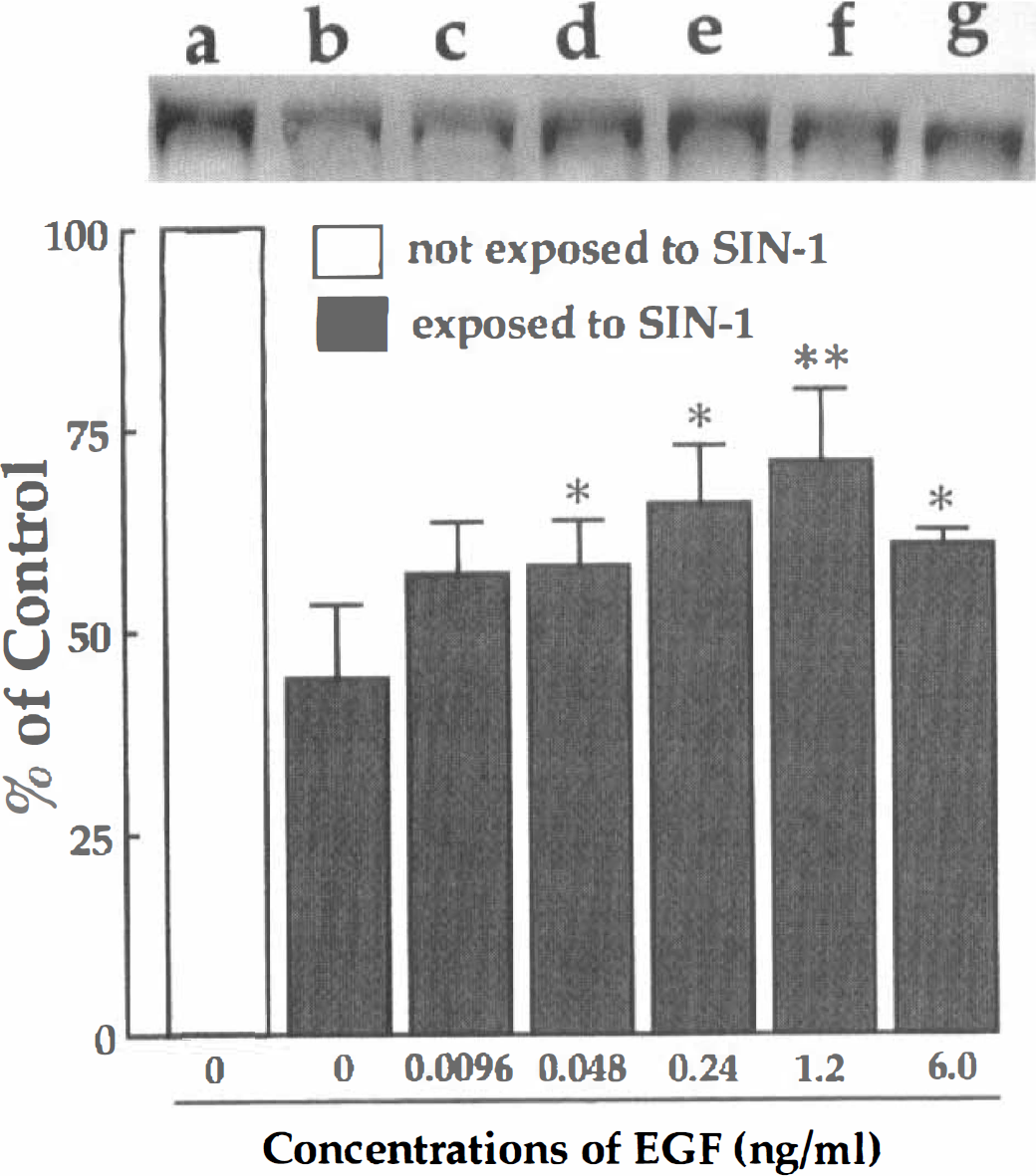
MAP2-immunoblot analysis of EGF-treated hippocampal neurons exposed to SIN-1. Samples cultured with 0 to 6.0 ng/mL of EGF were immunoblotted with a monoclonal antibody against MAP2. In control samples, an intense MAP2-positive band was observed (a). The cultured sample with SIN-1 treatment but without EGF pretreatment showed a weak immunoreactive band (b). In samples treated with 0.048–6.0 ng/mL EGF and then with SIN-1, the intensity of MAP immunoreactive bands increased (d-g). The densitometric analysis of MAP2-immunoreactive bands showed that EGF protected the neurons against SIN-1-induced oxidative injuries in a dose-dependent manner. The data were obtained from four separate cultures and expressed as percentage of the control culture which was not exposed to SIN-1. Each column indicates mean ± SD. *P < 0.05, **P < 0.01, significantly different from the SIN-1-treated control value (tested with one-way analysis of variance followed by Fisher's PLSD).
For the assays of lipid peroxidation, we used cortical neurons instead of hippocampal neurons because of the need of more neurons. The exposure of cortical neurons to 10 μmol/L FeSO4 for 2 hours resulted in a significant increase in thiobarbituric acid-reactive substances (Fig. 11) due to the induction of lipid peroxidation. Preincubation with EGF significantly attenuated the FeSO4-induced lipid peroxidation in a dose-dependent manner (Fig. 11).
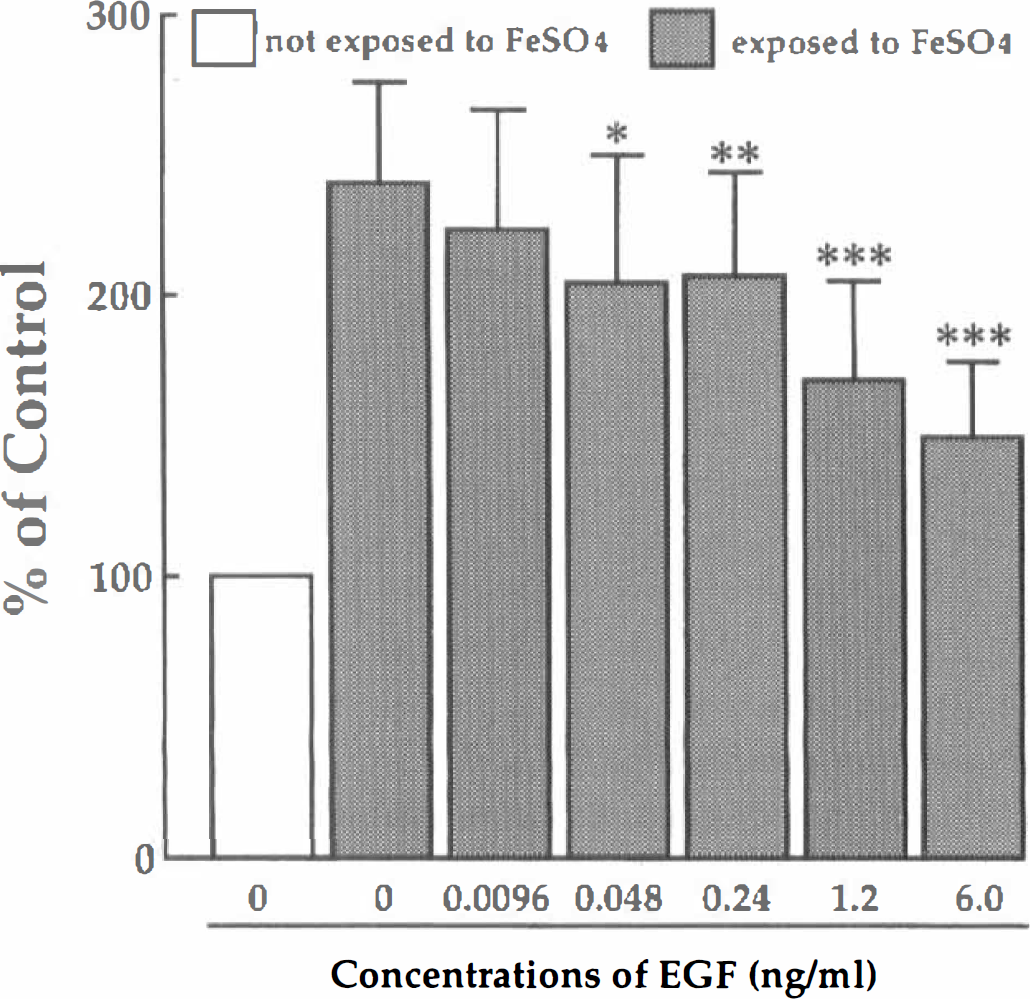
The effect of EGF on lipid peroxidation induced by FeSO4. Cultures were pretreated with 0 to 6.0 ng/mL of EGF and then exposed to 10 μM FeSO4 for 2 hours. Neurons were then fixed with TBA and light absorbance at 535 nm was measured. The data were obtained from 5 separate cultures and expressed as percentage of the control culture which was not exposed to FeSO4. *P < 0.05, **P < 0.01, ***P < 0.001, significantly different from the FeSO4-treated control value (tested with one-way analysis of variance followed by Fisher's PLSD).
DISCUSSION
The present in vivo study showed that the continuous infusion of EGF into the lateral ventricle starting either before or just after forebrain ischemia prolonged response latency time in the passive avoidance task. The improvement in behavioral score correlated well with the results of subsequent histologic examinations showing the suppression by EGF of delayed neuronal death in the hippocampal CA1 field. These results suggest that EGF can act as a neuroprotective agent in brain ischemia. Although EGF has been known as a neuroprotective agent in vitro, this is the first report showing that EGF can protect hippocampal neurons against ischemia-induced damage in vivo. In vitro experimental results, however, do not always reflect in vivo events in the ischemic brain. In fact, all cytokines or growth factors with neurotrophic actions in vitro do not necessarily exhibit neuroprotective effects on ischemic brain in vivo. For example, granulocyte/macrophage colony-stimulating factor has been reported to have a neuroprotective activity in vitro (Kamegai et al. 1990), whereas in the ischemic gerbil model we did not obtain results in support of the in vitro experiments (Peng et al. unpublished observations, August, 1996). Perhaps, the amount of growth factors or cytokines that reach the hippocampus after their infusion into the cerebral ventricles varies among growth factors/cytokines, and endogenous growth factors/cytokines as well as their receptors may respond differentially to ischemic insult in the brain. Accordingly, the in vivo action mechanisms of exogenous growth factors/cytokines are rather difficult to determine at this moment.
EGF is produced in neuronal cells including hippocampal neurons (Schaudies, et al. 1989; Birecree, et al. 1991). EGF receptor is also expressed in neurons within a variety of brain regions including the hippocampus (Werner et al. 1988; Birecree et al. 1991). It is not clear whether these observations imply that EGF acts on neuronal cells in an autocrine fashion. Production of EGF itself appears to be rather poor in amount within the hippocampal formation, but EGF receptor is highly expressed in the hippocampus (Birecree et al. 1991). Further, EGF is present in the CSF (Hirata et al. 1982). Therefore, it is plausible that EGF is produced by certain populations of neurons and plays a neuroprotective role in the ischemic brain in a paracrine manner. We speculate that under normal conditions the activities of the hippocampal CA1 pyramidal neurons are maintained, at least in part, by EGF derived from other neurons, but in the ischemic brain the supply of EGF becomes insufficient. If this is true, the infusion of EGF may be experimentally a favorable medication to attenuate the ischemia-induced neuronal damage. Indeed, as shown in the present study, exogenous EGF infusion into the cerebral ventricles of ischemic gerbils significantly rescued CA1 neurons from delayed neuronal death.
In the conventional 5-minute forebrain ischemia technique for which brain temperature is not maintained at 37.0 ± 0.2°C during ischemia, TUNEL-positive CA1 neurons have been shown to peak at the fourth day after ischemic insult, and only few ones are visible at the seventh day after ischemia possibly because of the so-called delayed death of almost all CA1 neurons (Nitatori et al., 1995). In contrast, the normothermic gerbils with 3-minute forebrain ischemia exhibited many TUNEL-positive neurons in the hippocampal CA1 field even at the seventh day after ischemia. This finding suggests that the hippocampal CA1 neurons after 3-minute ischemia undergoes a progressive degeneration at a slower rate than those after 5-minute ischemia. Thus, the present animal model with an episode of 3-minute ischemia has a wider therapeutic window than the conventional 5-minute ischemia model and may be useful for the screening of neuroprotective agents. Indeed, EGF treatment, starting either before or after 3-minute ischemia, reduced dramatically the number of TUNEL-positive neurons in comparison with those of 3-minute ischemic gerbils with vehicle infusion. This indicates that the slowly progressive degeneration as observed in the vehicle-treated gerbils at the seventh day after 3-minute ischemia is markedly inhibited by EGF even after the termination of its infusion. In situ detection of DNA fragmentation (TUNEL-staining) appears to be a reliable tool to assess the effects of neuroprotective agents on the late degeneration of hippocampal CA1 neurons in 3-minute ischemic gerbils. We speculate that a brief ischemic insult similar to 3-minute ischemia in gerbils is occasionally loaded to the human brain leading to sustained damage of the ischemic focus. If this is the case, continuous treatment with peptide growth factors (Sano et al., 1994; Wen et al., 1995a,b; Kotani et al., 1996; Matsuda et al., 1996) or nonpeptide neuroprotective agents (Wen et al., 1996; Lim et al., 1997; Matsuda et al., 1997) may be needed to lessen slowly progressive ischemic neuronal damage in patients with mild or brief brain ischemia.
The mechanism(s) by which EGF prevents delayed neuronal death in vivo is not clear at present, but the ability of EGF to protect oxidative injuries and to suppress accumulation of lipid peroxides may account for the action mechanism of EGF in part, as suggested by the present in vitro study. Free radicals and lipid peroxides overproduced after brain ischemia and reperfusion are responsible for brain edema and neuronal damage (Flamm et al., 1978; Siesjö, 1981; Chan et al., 1984; Kogure et al., 1985; Bolanos et al., 1997; Iadecola et al., 1997; Samdani et al., 1997). Lipid peroxides in the hippocampal CA1 region are increased after cerebral ischemia in the rat (Arai et al., 1987; Bromont et al., 1989). Hara et al. (1990) have reported that post-ischemic administration of α-tocopherol, a free radical scavenger, prevents delayed neuronal death in the ischemic hippocampal CA1 region. Thus, inhibition of free radical generation and/or lipid peroxidation is likely to prevent the occurrence of brain edema and neuronal damage. In the present study, pretreatment of neurons in cultures with EGF successfully prevented neuronal death and lipid peroxidation induced by FeSO4 which has been used to generate hydroxyl radicals in culture (Zhang et al. 1993; Cheng et al. 1995). Because the simultaneous addition of EGF and FeSO4 into the culture medium did not attenuate FeSO4 neurotoxicity (data not shown), EGF itself dose not appear to act as a radical scavenger. In addition, pretreatment with EGF significantly protected hippocampal neurons against oxidative damage caused by SIN-1 which is known to generate superoxide radicals and nitric oxide causing to the formation of peroxynitrite (Groot et al., 1993). Furthermore, the present in vitro results on the dose-dependency of EGF showed that EGF displayed its neurotrophic and neuroprotective effects on hippocampal neurons at concentrations of 0.008 to 1 nmol/L. This effective concentration of EGF is in the range of the Kd value (0.02 to 0.05 nmol/L) of EGF receptor. Therefore, it is likely that EGF acts through binding to its receptor on neuronal cell surface.
Basic fibroblast growth factor, nerve growth factor, and brain-derived neurotrophic factor, which are all well-known neurotrophic factors, reduce lipid peroxidation induced by glutamate exposure (Mattson et al., 1995). It has also been shown that the three neurotropic factors enhance more or less the activities of antioxidant enzymes such as superoxide dismutase, glutathione peroxidase, and catalase (Mattson et al., 1995). Because receptors for EGF, basic fibroblast growth factor, brain-derived neurotrophic factor, and nerve growth factor use similar transduction pathways via tyrosine phosphorylation (Schlesinger and Ulrich, 1992), EGF may rescue ischemic neurons by enhancing the activities of the antioxidant enzymes. On the other hand, Kinoshita et al. (1990) reported that EGF rescued neurons from anoxic injury in vitro. Under the anoxic environment, the enhancement of antioxidant enzymes by EGF may not be responsible for the neuroprotective effect. The antianoxic action of EGF may play a significant role in rescuing ischemic neurons together with the anti-oxidant activity.
Footnotes
Abbreviations used
Acknowledgments
The authors thank Dr. K. Ikoma for his encouragement throughout this work and Miss M. Fujimoto for her secretarial assistance.