
Abstract
Excessive levels of extracellular glutamate in the nervous system are excitotoxic and lead to neuronal death. Glutamate transport, mainly by glutamate transporter GLT1/EAAT2, is the only mechanism for maintaining extracellular glutamate concentrations below excitotoxic levels in the central nervous system. We recently showed that neuroprotection after experimental ischemic preconditioning (IPC) involves, at least partly, the upregulation of the GLT1/EAAT2 glutamate transporter in astrocytes, but the mechanisms were unknown. Thus, we decided to explore whether activation of the nuclear receptor peroxisome proliferator-activated receptor (PPAR)γ, known for its antidiabetic and antiinflammatory properties, is involved in glutamate transport. First, we found that the PPARγ antagonist T0070907 inhibits both IPC-induced tolerance and reduction of glutamate release after lethal oxygen-glucose deprivation (OGD) (70.1% ± 3.4% versus 97.7% ± 5.2% of OGD-induced lactate dehydrogenase (LDH) release and 61.8% ± 5.9% versus 85.9% ± 7.9% of OGD-induced glutamate release in IPC and IPC + T0070907 1 μmol/L, respectively, n = 6 to 12, P < 0.05), as well as IPC-induced astrocytic GLT-1 overexpression. IPC also caused an increase in nuclear PPARγ transcriptional activity in neurons and astrocytes (122.1% ± 8.1% and 158.6% ± 22.6% of control PPARγ transcriptional activity, n = 6, P < 0.05). Second, the PPARγ agonist rosiglitazone increased both GLT-1/EAAT2 mRNA and protein expression and [3H]glutamate uptake, and reduced OGD-induced cell death and glutamate release (76.3% ± 7.9% and 65.5% ± 15.1% of OGD-induced LDH and glutamate release in rosiglitazone 1 μmol/l, respectively, n = 6 to 12, P < 0.05). Finally, we have identified six putative PPAR response elements (PPREs) in the GLT1/EAAT2 promoter and, consistently, rosiglitazone increased fourfold GLT1/EAAT2 promoter activity. All these data show that the GLT1/EAAT2 glutamate transporter is a target gene of PPARγ leading to neuroprotection by increasing glutamate uptake.
Introduction
Excessive extracellular levels of the excitatory neurotransmitter glutamate are toxic and lead to neuronal death (Rothman and Olney, 1986; Choi and Rothman, 1990). Glutamate uptake by specific transporters is the primary and only mechanism for maintaining extracellular glutamate concentrations below excitotoxic levels in the central nervous system (CNS) (Takahashi et al, 1997; Seal and Amara, 1999; Gegelashvili et al, 2001; Danbolt, 2001; Maragakis and Rothstein, 2001). Among these transporters, GLT-1, also known as EAAT2 (Pines et al, 1992; Shadshidharan et al, 1994), plays a major role being responsible for up to 90% of all glutamate transport in adult tissue (reviewed in Robinson, 1999) and, therefore, its modulation with pharmacologic strategies could have important therapeutic applications. However, in our knowledge, there are no currently approved pharmacologic treatments aimed to upregulate GLT-1/EAAT2 transporter and, experimentally, only a few drugs (estrogen, β-lactam antibiotics, and glucocorticoids) are known to increase GLT-1/EAAT2 expression (Pawlak et al, 2005; Rothstein et al, 2005; Zschocke et al, 2005).
The mechanisms involved in ischemic preconditioning (IPC)-induced cytoprotection in several experimental models of both CNS and heart pathologies have been the center of attention for many scientists in the search for new therapeutic targets (see reversus by Barone et al, 1998; Kirino, 2002; Dirnagl et al, 2003). We have recently shown that IPC causes upregulation of two glutamate transporters, GLT-1/EAAT2 and EAAC1/EAAT3, and a subsequent decrease in ischemia-induced glutamate release (Romera et al, 2004). Our data also showed that EAAT3 is upregulated by the TACE/TNF-α pathway. However, the mechanisms for upregulation of GLT-1/EAAT2 remained unknown.
Recently, cytoprotective, antiinflammatory actions of the peroxisome proliferator-activated receptor (PPAR)γ type of nuclear receptor have been described in different experimental models of stroke (Pereira et al, 2005, 2006; Shimazu et al, 2005; Sundarajan et al, 2005; Zhao et al, 2005a), intracerebral hemorrhage (Zhao et al, 2005b), Alzheimer's disease (Combs et al, 2002), multiple sclerosis (Feinstein et al, 2002), neuroinflammation (Luna-Medina et al, 2005), or stress (García-Bueno et al, 2005). Moreover, a study carried out in humans showed that increased plasma levels of the endogenous PPARγ agonist 15-deoxy-delta-12,14-prostaglandin J2 (15dPGJ2) are associated with better outcome in acute atherothrombotic ischemic stroke (Blanco et al, 2005). Because this prostaglandin (Forman et al, 1995; Kliewer et al, 1995) as well as other PPARγ ligands are endogenously produced, PPARγ activation may occur endogenously. We have therefore studied whether PPARγ activation is involved in IPC, and its possible involvement in the upregulation of glutamate transporters, by using rat cortical cultures exposed to sublethal oxygen-glucose deprivation (OGD) as a model of in vitro IPC.
Materials and methods
Rat Cortical Neuronal-Astrocytic Cocultures
All experimental protocols adhered to the guidelines of the Institutional Review Board for Animal Care of the Universidad Complutense (following DC86/609/EU and 2003/65/CE). Primary pure neuronal cultures were performed as described (Romera et al, 2004) by removing brains from fetal Wistar rats at embryonic day (E) 16. At in vitro days 8 to 10, cultures consisted of 94% ± 6% neurons, as determined by flow cytometry (Hurtado et al, 2002).
Primary astrocyte cultures were prepared from the neonatal (P0) Wistar rat cortex, as described previously (Romera et al, 2004). On day 10 in culture, astrocytes were collected by trypsinization, resuspended, and plated at a density of 1.4 × 105 cells per cm2 in culture plate inserts (1 μm membrane pore size, 4.2 cm2 of effective membrane growth area; Falcon; Becton Dickinson Labware, Franklin Lakes, NJ, USA) in a total volume of 1.5 mL of fresh astrocytic culture medium. Two days after astrocyte plating, coinciding with pure neuronal culture day 7, neurons plated in six-well companion plates (Falcon, Becton Dickinson Labware) had their medium replaced, then astrocyte-containing inserts were transferred to these wells, and 1.5 mL of normal medium was added. Cocultures were transferred to the normoxic incubator, and exposure to the different treatments (IPC or drugs) was performed at least 48 h later.
Exposure of Rat Cortical Cultures to Lethal Oxygen-Glucose Deprivation or Ischemic Preconditioning
Lethal OGD was performed as described (De Cristóbal et al, 2002; Hurtado et al, 2002; Romera et al, 2004).
For IPC, cortical cultures were deprived of oxygen-glucose for 20 mins as described (Romera et al, 2004), an insult that did not induce neuronal death as measured by LDH release, in agreement with previous results (Grabb and Choi, 1999). After that time, OGD solution was replaced with reperfusion medium, and cultures were returned to the normoxic incubator. Simulated reperfusion was performed, as indicated above, for 24 h either in absence or presence of the specific PPARγ antagonist T0070907 (2-chloro-5-nitro-N-4-pyridinyl-benzamide), (0.1 to 1 μmol/L, Tularik; Lee et al, 2002). T0070907 is a potent and selective PPARγ antagonist with an apparent IC50 of 1 nmol/L for the binding inhibition of rosiglitazone. This compound covalently modifies PPARγ on cysteine 313 in helix 3 of human PPARγ2. T0070907 is able to block PPARγ function in both cell-based reporter gene and adipocyte differentiation assays (Lee et al, 2002).
In another set of experiments, cultures were exposed to the PPARγ agonist rosiglitazone (0.03 to 1 μmol/L) for 24 h and cells were then exposed to OGD or collected for different determinations. Rosiglitazone is a potent and selective PPARγ ligand belonging to the thiazolidinedione (TZD) family, that binds to the PPARγ ligand-binding domain with a Kd of 43 nmol/L. This compound activates luciferase-based expression constructs PPARγl and PPARγ2 with EC50 values of approximately 30 and 100 nmol/L, respectively (Lehmann et al, 1995).
A similar protocol was used to assay the effect of the PPARγ agonists cyclopentenone prostaglandin 15dPGJ2 (1 μmol/L) or the non-TZD L-796,449 (3-chloro-4-(3-(3-phenyl-7-propylbenzofuran-6-yloxy) propylthio) phenyl acetic acid; 1 μmol/L) (Castrillo et al, 2001; Straus and Glass, 2001; Pereira et al, 2005, 2006).
In an additional set of experiments, specific phosphorothioate antisense or random oligodeoxynucleotides (ODNs) to GLT-1/EAAT2 glutamate transporter were added to astrocytic cultures either in the presence of rosiglitazone or after exposure to IPC, 24 h before exposure to lethal OGD, and LDH release was measured 24 h after OGD. The sequences for the ODNs used were as follows: antisense GLT-1, 5′-ATATTGTTGGCACCCTCGGTTGAT-3′ and random GLT-1, 5′-AATTGTGTTAGCCCCCTCTGTT GA-3′ (Bristol and Rothstein, 1998). Oligodeoxynucleotides were added at a final concentration of 5 μmol/L in the presence of Lipofectamine 2000 (Invitrogen SA, Barcelona, Spain) following the manufacturer's instructions. As another tool to study GLT1/EAAT2 activity, the effect of the GLT-1/EAAT2 transporter inhibitor dihydrokainate (DHK; 100 μmol/L; Arriza et al, 1994; Tanaka, 1994) on LDH release was measured 24 h after OGD in cells exposed to IPC or rosiglitazone.
Assessment of Cell Viability
As a marker of necrotic tissue damage, LDH activity released from damaged cells was determined. Culture medium and lysate from 0.1% Triton X-100 in phosphate-buffered saline were collected 24 h after the OGD period. Lactate dehydrogenase activity was measured spectrophotometrically at 340 nm following the oxidation of nicotinamide adenine dinucleotide (reduced form) (decrease in absorbance) in the presence of pyruvate (Koh and Choi, 1987) using a Tecan Sunrise microplate reader (Tecan, Salzburg, Austria). Lactate dehydrogenase was measured in at least six different wells per experiment, and the experiment was repeated no less than six times. Lactate dehydrogenase release is expressed as the percentage of total cell LDH and is plotted as the percentage of LDH release induced by OGD. Basal LDH release was 6% ± 1% (n = 12).
High-Pressure Liquid Chromatography Determination of Glutamate Concentration
Samples of incubation solution were collected at the end of the OGD period, a time at which LDH efflux was not significantly different from control values, thus allowing us to study glutamate release caused by OGD and excluding the efflux of this excitatory amino acid attributable to damaged membranes. Analysis of glutamate in each sample was performed by RP-high-pressure liquid chromatography with fluorimetric detection (Binary LC Pump 250 and Fluorescence Detector LC 240; Perkin Elmer, Madrid, Spain) after precolumn derivatization with the o-phtalaldialdehyde procedure (Lindroth and Mopper, 1979) as described (Romera et al, 2004). Glutamate release is expressed as the percentage of net release induced by OGD. Basal release was below the detection limit of our assay (0.17 nmol/106 cells).
Western Blot Analysis of GLT1/EAAT2 Glutamate Transporter
Homogenate containing 10 μg of protein was loaded, and the proteins were size-separated in 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (90 mA). The proteins were blotted onto a polyvinylidine diflouride membrane (Millipore, Madrid, Spain) and incubated with the following specific monoclonal primary antibody against GLT-1/EAAT2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA; 1:1,000) glutamate transporter. Proteins recognized by the antibody were revealed by ECL-kit following the manufacturer's instructions (Amersham Pharmacia Biotech, Piscataway, NJ, USA).
Reverse Transcription-Polymerase Chain Reaction Assay of GLT1/EAAT2 mRNA
Total RNA was extracted from 107 astrocytes per group using Trizol solution (Gibco BRL, NY, USA) in accordance with the manufacturer's instructions and the excess of salt was eliminated by precipitation using sodium acetate and ethanol. RNA samples were resuspended in diethyl pyrocarbonate-treated water and its concentration was determined spectrophotometrically. The RNA purity was confirmed by the relative absorbance at 260 versus 280 nm and RNA samples were stored at −80°C.
For cDNA synthesis, RNA (1 μg) was reverse-transcribed using oligo dT sequence as primer and 50 U of M-MLU and AMV reverse transcriptases (Promega, Madison, WI, USA) in accordance with the manufacturer's instructions. Specific primer sequences (5′–3′ ATTGGTGCAGGCCAG TATTC, left, and AATCGCCCACTACATTGAC, right) against GLT-1 genes were designed from rat sequences available in Genbank using Primer Express Software (PE Applied Biosystems, Foster City, CA, USA). PCR amplification of the reverse-transcribed RNA was carried out with 1 unit of DNA polymerase in a 50 μL reaction mixture containing 200 μmol/L of each dNTPs, 0.3 μmol/L of specific primers, and 1 × reaction buffer using a DNA Thermal Cycler. Cycling conditions were 3 mins at 94°C, 20 secs at 94°C, 20 secs at 52°C, 30 secs at 72°C, 33 cycles, and a final elongation for 5 mins at 72°C. Amplified DNA fragments were analyzed by electrophoresis in a 1.5% agarose gel. Quantification of the bands was performed by laser densitometry after normalization with β-actin.
The absence of genomic contamination in the RNA samples and of DNA contamination in RNA mimics was confirmed with reverse transcriptase-negative controls for each experiment.
[3H]Glutamate Uptake
[3H]Glutamate uptake was determined as described (Romera et al, 2004).
Preparation of Nuclear Extracts
Cells were collected 24 h after IPC in phosphate-buffered saline (106 cells/mL). The homogenate was centrifuged at 4°C, 12,000g. Supernatant was discarded and nuclear extracts were prepared as described (Cárdenas et al, 2000).
Peroxisome Proliferator-Activated Receptor γ Transcriptional Activity Assay
Peroxisome proliferator-activated receptor γ transcription factor activity was assayed on nuclear proteins by using enzyme-linked immunosorbent assay-based PPARγ Transcription Factor Assay Kit to detect and qualify specific transcription PPARγ factor activation (Active Motif Trans-AM, Rinxensart, Belgium). This kit consists of a 96-well plate to which oligonucleotide containing the PPRE has been inmmobilized. Peroxisome proliferator-activated receptors contained in nuclear extracts bind specifically to this oligonuleotide and are detected through use of an antibody directed against PPARγ. Addition of a secondary antibody conjugated to horseradish peroxidase provides sensitive colorimetric readout that is easily quantified by spectrophotometry. Measurement was performed according to the manufacturer's instructions. This assay is specific for PPARγ activation, and it does not crossreact with PPARα or PPARβ.
Middle Cerebral Artery Occlusion and Infarct Size Determination
Adult male Fischer rats weighing approximately 250 g were used. Rats were housed individually under standard conditions of temperature and humidity and a 12-h light/dark cycle (lights on at 08:00) with free access to food and water. All experimental protocols adhered to the guidelines of the Animal Welfare Committee of the Universidad Complutense (following DC 86/609/EU and 2003/65/CE).
Animals were anaesthetized with 1.5% halothane in a mixture of 70% nitrogen/30% oxygen, and the body temperature was maintained at physiologic levels using a heating pad throughout the surgery procedure and during postsurgery recovery. Permanent focal cerebral ischemia was induced by occlusion of the ipsilateral middle cerebral artery as described (Pereira et al, 2006). Rats in which the middle cerebral artery was exposed but not occluded served as sham-operated controls (SHAM). After surgery, subjects were returned to their cages and allowed free access to water and food.
Several groups were used for determinations of infarct size and protein levels of GLT-1/EAAT2: middle cerebral artery occlusion (MCAO) 10 mins before an intraperitoneal (i.p.) injection of dimethylsulfoxide (MCAO; 10% in saline; n = 6) and MCAO 10 mins before an i.p. injection of rosiglitazone 1 mg/kg (RSG; n = 6). An additional group consisted of SHAM-operated animals 10 mins before an i.p. injection of saline (control; n = 6). Injection volume was ≤ 400 μl/250 g body weight. Animals were sacrificed 1 to 2 days after surgery for sample collection.
Infarct outcome was assessed 2 days after the occlusion as described (Pereira et al, 2006).
Bioinformatic Analysis of GLT-1/EAAT2 Promoter
A bioinformatic approach was performed by using PROMO (Messeguer et al, 2002; Farré et al, 2003; http://alggen.lsi.upc.es/), a virtual laboratory for the identification of putative transcription factor binding sites in DNA sequences from a species or groups of species of interest. Transcription factor binding site defined in the TRANS-FAC database (version 8.3; database on eukaryotic transcription factors, their genomic binding sites and DNA-binding profiles; www.gene-regulation.com/pub/databases.html#transfac) are used to construct specific binding sites weight matrices for transcription factor binding site prediction. PPAR response element was recognized as a motif composed of a direct repeat (DR) spaced by one nucleotide (DR1) with a consensus sequence of RGGTGA-A-AGGTCA (Nakshatri and Bhat-Nakshatri, 1998; Tachibana et al, 2005).
Construction of the Plasmid pEAAT2PrLUC
Construction of a plasmid containing a promoter fragment known to keep EAAT2 promoter activity (Su et al, 2003) was performed as described (Mallolas et al, 2006). Briefly, a 773 bp fragment including −742/+ 31 of full-length EAAT2 promoter was amplified by PCR introducing MluI and BglII restriction sites (Fw-MluI: 5′-CTAGACGCGTTT CAGCCGCTCGACTC-3′ and Rv-Bg/II: 5′-ATGAAGATCT CTTGGCGGGGAGCTC-3′). After enzyme digestion and fragment purification, the −742/+ 31 fragment from EAAT2 promoter was fused upstream of the firefly luciferase reporter in pGL3-basic luciferase reporter vector.
Transient Transfection and Luciferase Reporter Gene Assay
Reporter assays were performed on EAAT2-Promoter construction (pEAAT2PrLUC) using the Dual-Luciferase reporter assay system (Promega), with which pRL-thymidine kinase (TK) plasmid (Promega), containing the Renilla luciferase gene under control of the TK promoter, was cotransfected as an internal control. Transient transfection of astrocytes was performed as described (Arima et al, 2003, Mallolas et al, 2006) using Lipofectamine 2000 (Invitrogen SA, Barcelona, Spain). The activities of control Renilla luciferase and firefly luciferase were measured in triplicate.
Chemicals and Statistical Analyses
T0070907 was a kind gift of Tularik Inc. (San Francisco, CA, USA) and rosiglitazone was provided by Cayman Europe (Tallinn, Estonia). Other chemicals were obtained from Sigma-Aldrich Quimica SA, Madrid, Spain or as indicated in the text. Results are expressed as mean ± s.e.m. of the indicated number of experiments; statistical analysis involved one-way analysis of variance (or the Kruskal–Wallis test when the data were not normally distributed) followed by individual comparisons of means (Student–Newman–Keuls, or Dunn's method when the data were not normally distributed). P < 0.05 was considered statistically significant.
Results
Effect of the Peroxisome Proliferator-Activated Receptor γ Antagonist T0070907 on Ischemic Preconditioning-Induced Neuroprotection and Decrease of Glutamate Release After Oxygen-Glucose Deprivation in Neuronal-Astrocytic Cocultures
As previously shown (Romera et al, 2004), OGD caused cell death by an increase in LDH efflux to the incubation medium measured 24 h after the insult (n = 12, Figure 1A), which was decreased by prior exposure to sublethal OGD (IPC) (Figure 1A). Ischemic preconditioning did not alter cell viability by itself (7.5% ± 0.9% versus 6.1% ± 0.6% basal LDH release in IPC and control, respectively; n = 12, P > 0.05). OGD also caused the release of the excitatory neurotransmitter glutamate from rat cortical cultures (1.49 ± 0.19 nmol/106 cells, n = 12) when compared with the control group (below detection limit, see Materials and methods; Figure 1B), an effect that was decreased after exposure to IPC (Figure 1B).
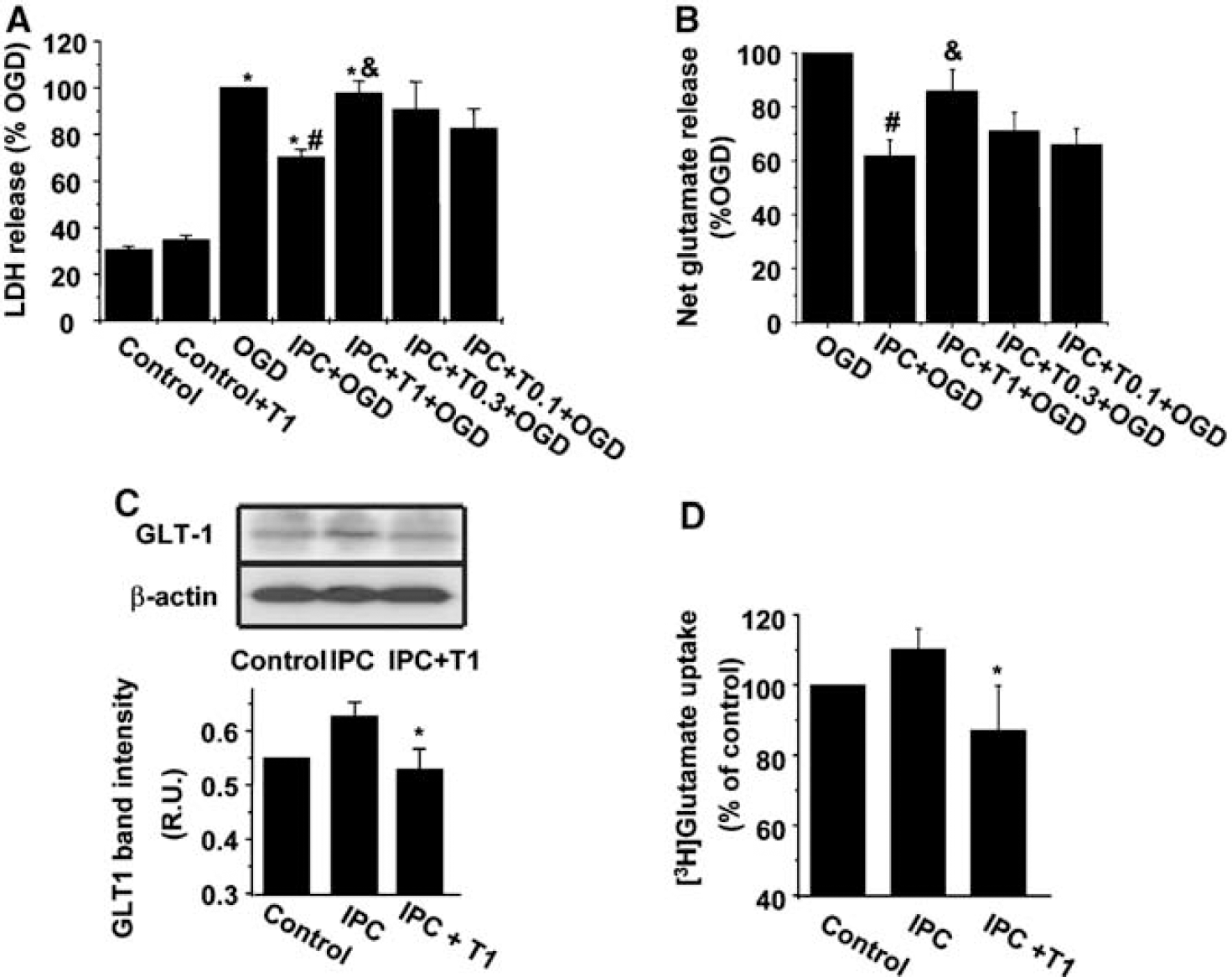
Effect of the PPARγ antagonist T0070907 on the consequences of IPC in rat cortical neuronal-astrocytes co-cultures. (
The specific PPARγ antagonist T0070907 (0.1 to 1 μmol/L) concentration-dependently inhibited the protective effect caused by IPC (Figure 1A). T0070907 (1 μmol/L) alone did not affect cell viability in control cultures (n = 6, P > 0.05, Figure 1A) or after OGD (98% ± 5% OGD-induced LDH release, n = 6, P > 0.05). In addition, T0070907 (0.1 to 1 μmol/L) reversed the inhibitory effect of IPC on OGD-induced glutamate release (Figure 1B). Incubation with T0070907 1 μmol/L alone did not affect OGD-induced glutamate release (1.53 ± 0.21 nmol/106 cells, n = 12, P > 0.05).
Effect of the Peroxisome Proliferator-Activated Receptor γ Antagonist T0070907 on Ischemic Preconditioning-Induced GLT-1/EAAT2 Transporter Upregulation and [3H] Glutamate Uptake in Rat Astrocytes from Cocultures
In agreement with our previous results (Romera et al, 2004), exposure to IPC caused an upregulation of GLT-1 transporter in rat astrocytes, as shown by Western blotting (Figure 1C). Treatment with T0070907 (1 μmol/L) prevented the effect of IPC on GLT-1 expression (Figure 1C).
Incubation with the PPARγ antagonist T0070907 (1 μmol/L) produced a decrease in [3H]glutamate uptake when compared with IPC-exposed cultures (Figure 1D; n = 6, P < 0.05).
Effect of Ischemic Preconditioning on Peroxisome Proliferator-Activated Receptor γ Transcriptional Activity
To check the involvement of PPARγ activation in IPC, we next quantified PPARγ activation by the ability of nuclear proteins to bind a specific oligonucleotide containing the PPRE consensus sequence using an enzyme-linked immunoassay-based PPARγ transcription factor assay kit. Indeed, IPC induced an increase in PPARγ activation mainly in astrocytes (n = 6, P < 0.05) but also in neurons (n = 6, P < 0.05) when measured 24 h after the treatment (Figure 2).
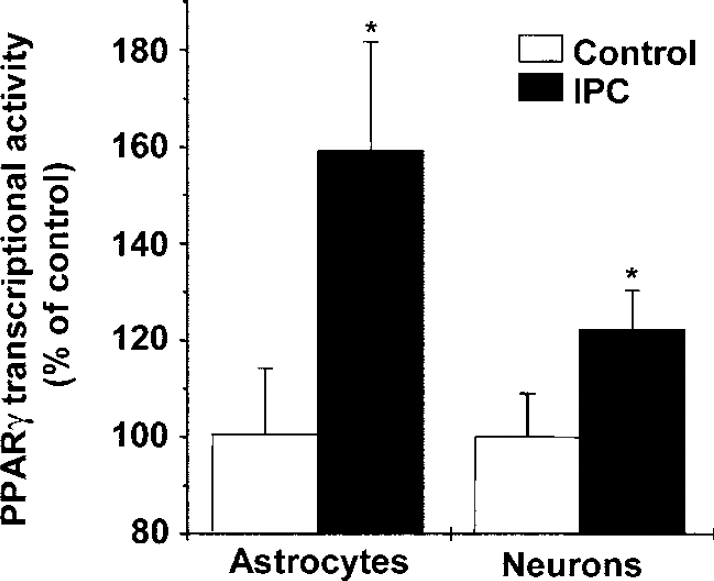
Effect of IPC on PPARγ transcriptional activity in astrocytes and neurons from cocultures. The values represent means ± s.e.m. relative to control, untreated cells, n = 6, *P < 0.05 versus control.
Effect of the Peroxisome Proliferator-Activated Receptor γ Agonist Rosiglitazone on GLT1/EAAT2 Transporter Expression and [3H] Glutamate Uptake in Rat Astrocytic Cultures: Effect of Other Peroxisome Proliferator-Activated Receptor γ Agonists in Astrocytic GLT1/EAAT2 Expression
Because the previous data strongly suggest the implication of PPARγ in IPC-induced GLT-1 upregulation, we then studied the direct effect of PPARγ agonists. The incubation of rat-cultured astrocytes with the PPARγ agonist rosiglitazone (0.1 to 1 μmol/L) caused a concentration-dependent increase in GLT-1 protein expression and [3H]glutamate uptake in rat astrocytes (Figures 3A and 3B). Consistently, rosiglitazone (1 μmol/L) also increased GLT1 mRNA expression (Figure 3C).
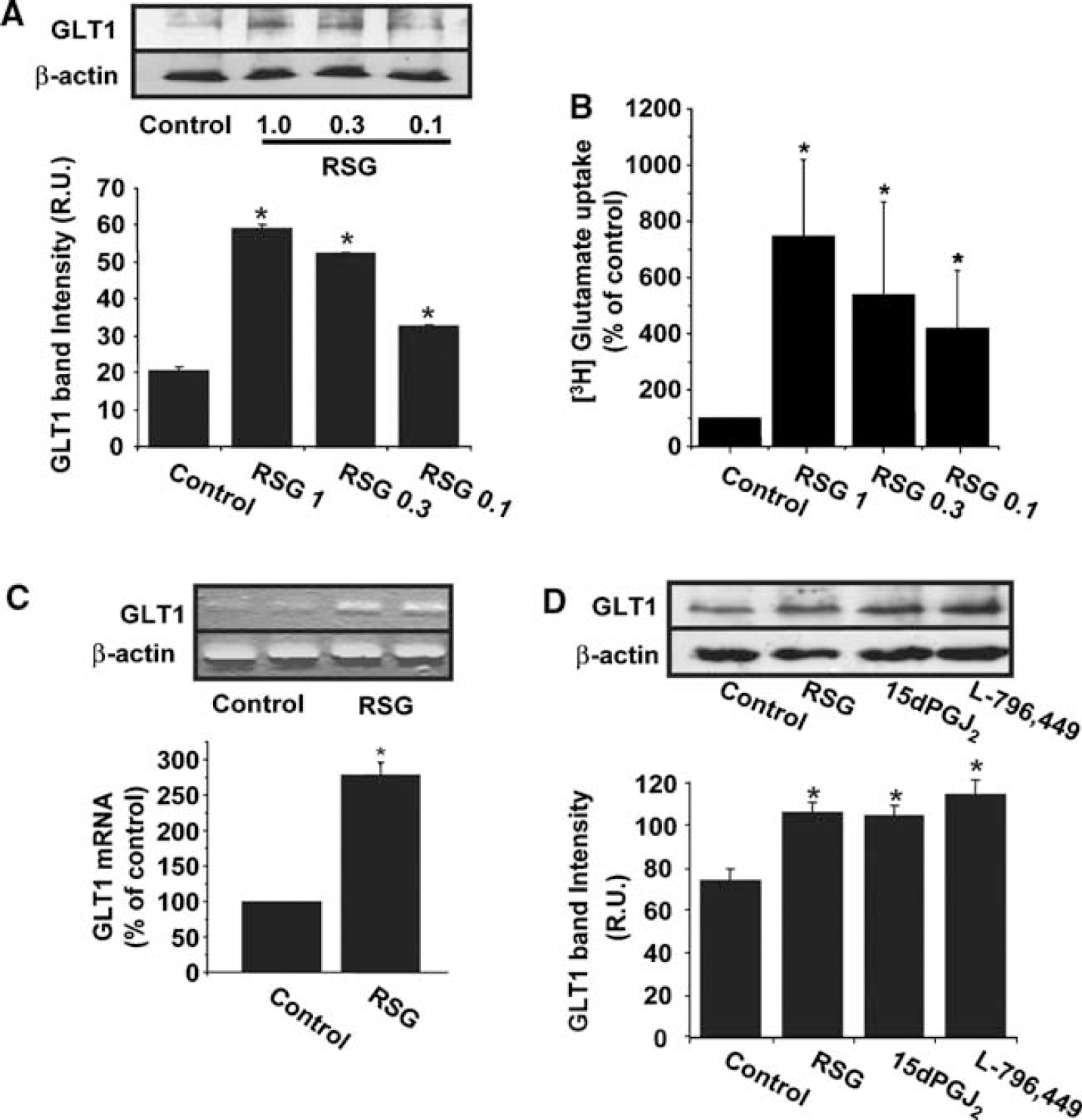
Effect of the PPARγ agonist rosiglitazone (RSG; 0.1 to 1 μmol/L) on GLT-1 protein expression levels (
Incubation with the thiazolidinedione-unrelated PPARγ agonists 15dPGJ2 (1 μmol/L) or L-796 449 (1 μmol/L) for 24 h also increased astrocytic GLT-1 protein expression (Figure 3D).
Effect of the Peroxisome Proliferator-Activated Receptor γ Agonist Rosiglitazone on Oxygen-Glucose Deprivation -Induced Cell Death and Glutamate Release in Neuronal-Astrocytic Cocultures
In agreement with the effect on glutamate transport, OGD-induced cell death was decreased in cells that had been pretreated with the PPARγ agonist rosiglitazone (0.03 to 1 μmol/L) 24 h before the ischemic insult (Figure 4A, n = 6, P < 0.05). In addition, preincubation with the PPARγ agonist rosiglitazone (0.1 to 1 μmol/L) caused an inhibition of OGD-induced glutamate release (Figure 4B).
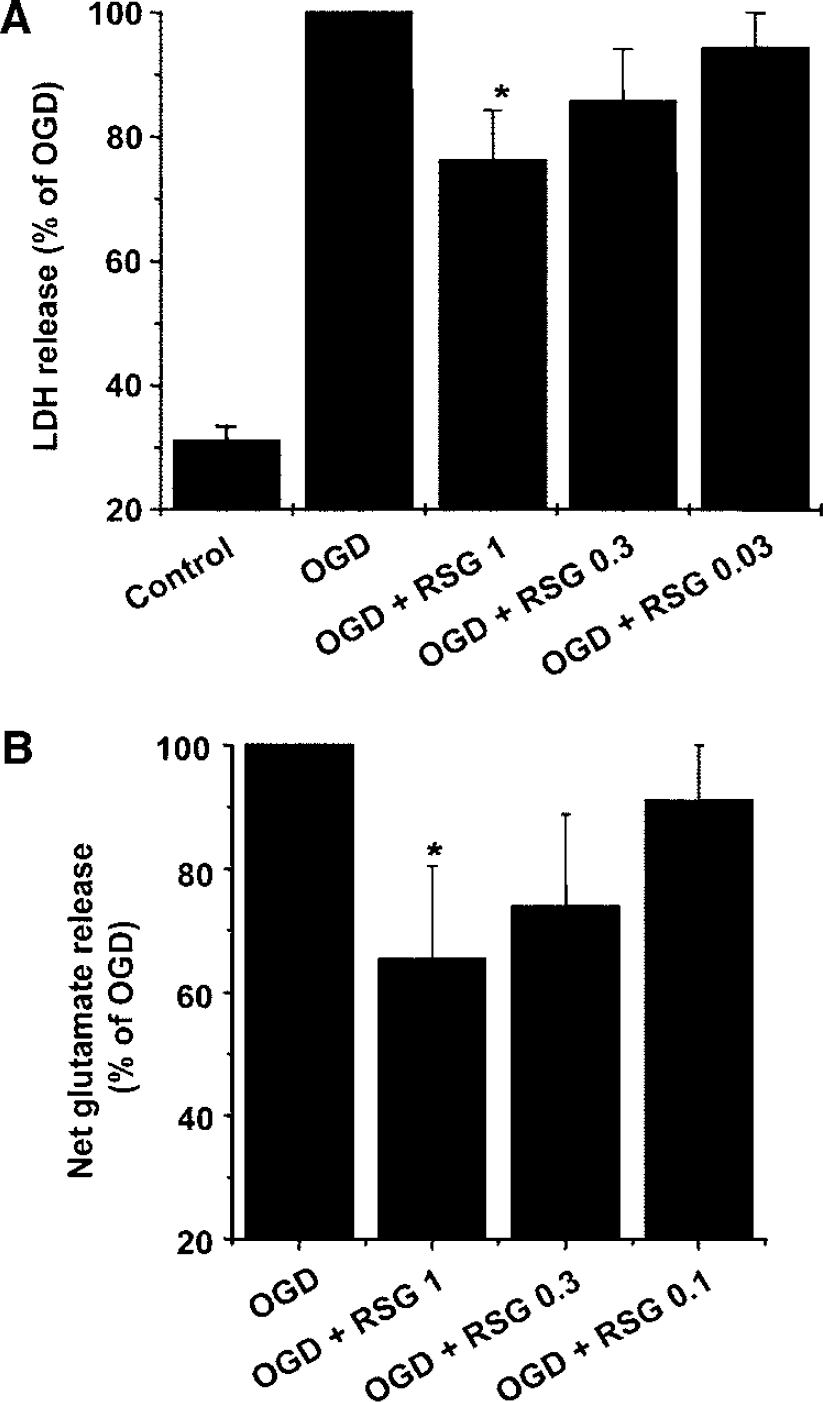
Effect of rosiglitazone (RSG; 0.03 to 1 μmol/L) on cell death determined as LDH efflux (
Effect of GLT1/EAAT2 Inhibition on the Neuroprotective Effect of Ischemic Preconditioning or Rosiglitazone on Neuronal-Astrocytic Cocultures
To confirm that GLT1 upregulation is involved in neuroprotection after IPC or treatment with rosiglitazone, the effect of the glutamate transporter inhibitor DHK or an antisense ODN to GLT1 was tested. Dihydrokainate (100 μmol/L) inhibited the protective effect caused by IPC (70% ± 3% versus 96% ± 2% OGD-induced LDH release in IPC and IPC + DHK, respectively, n = 5-6, P < 0.05) and by rosiglitazone (76% ± 8% versus 96% ± 2% OGD-induced LDH release in RSG and RSG ± DHK, respectively, n = 5 to 6, P < 0.05). In addition, antisense ODN to GLT1 also inhibited neuroprotection caused by IPC (80% ± 1% versus 92% ± 2% OGD-induced LDH release in IPC + random-ODN and IPC + antisense-ODN, respectively, n = 4 to 6, P < 0.05) or by rosiglitazone (79% ± 3% versus 95% ± 1% OGD-induced LDH release in RSG + random-ODN and RSG + antisense-ODN, respectively, n = 5 to 6, P < 0.05).
Effect of the Peroxisome Proliferator-Activated Receptor γ Agonist Rosiglitazone in Infarct Size and GLT1/EAAT2 Brain Expression in Brain After Middle Cerebral Artery Occlusion in Rats
In agreement with our previous observations (Pereira et al, 2006), the administration of rosiglitazone (1 mg/kg) decreased infarct volume when measured 48 h after the occlusion (164.5 ± 13.8 versus 101.1 ± 9.8 mm3 in MCAO and MCAO + rosiglitazone, respectively, n = 6 to 8, P < 0.05). In addition, this treatment also increased GLT1/EAAT2 expression in the brain of MCAO-exposed rats when determined 24 h after the occlusion (Figure 5).
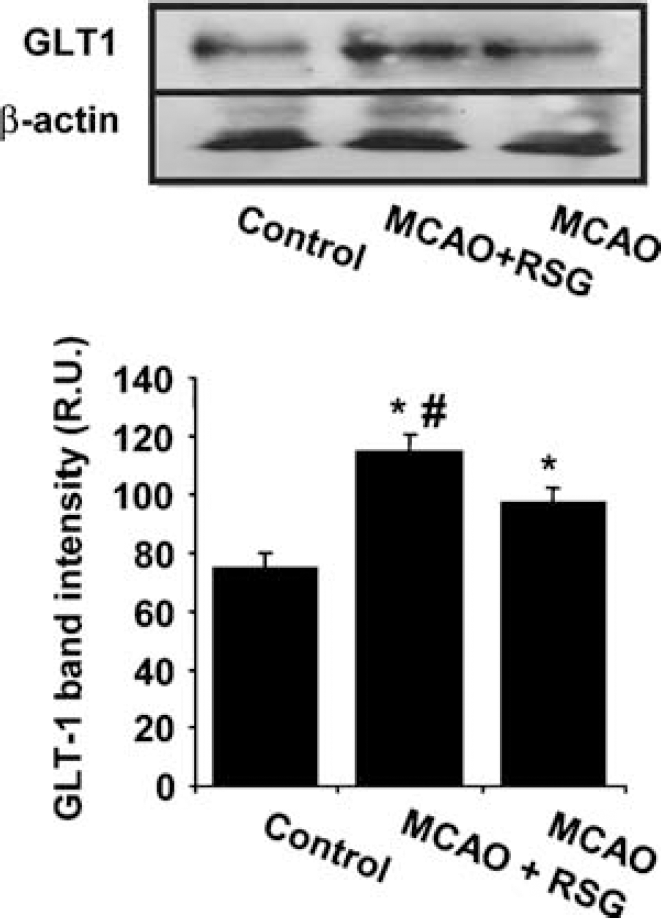
Effect of rosiglitazone (1 mg/kg) on GLT1/EAAT2 expression after MCAO in rat brain. GLT1/EAAT2 was determined by Western blot analysis in brain homogenates. Lower panel shows the densitometric analysis of bands. Data are mean ± s.e.m., n = 6, *P < 0.05 versus control, #P < 0.05 versus MCAO.
Bioinformatic Analysis of the EAAT2 Promoter
A bioinformatic analysis identified six putative PPRE in the EAAT2 promoter region, located as shown in Figure 6 (upper panel). These putative PPREs have a DR1 sequence similar to the consensus PPRE for known PPAR target genes (Nakshatri and Bhat-Nakshatri, 1998; Tachibana et al, 2005). The 773 bp cloned fragment including −742/ + 31 of full-length EAAT2 promoter selected for transient transfection contains two of the six consensus PPRE (see Figure 6, upper panel).
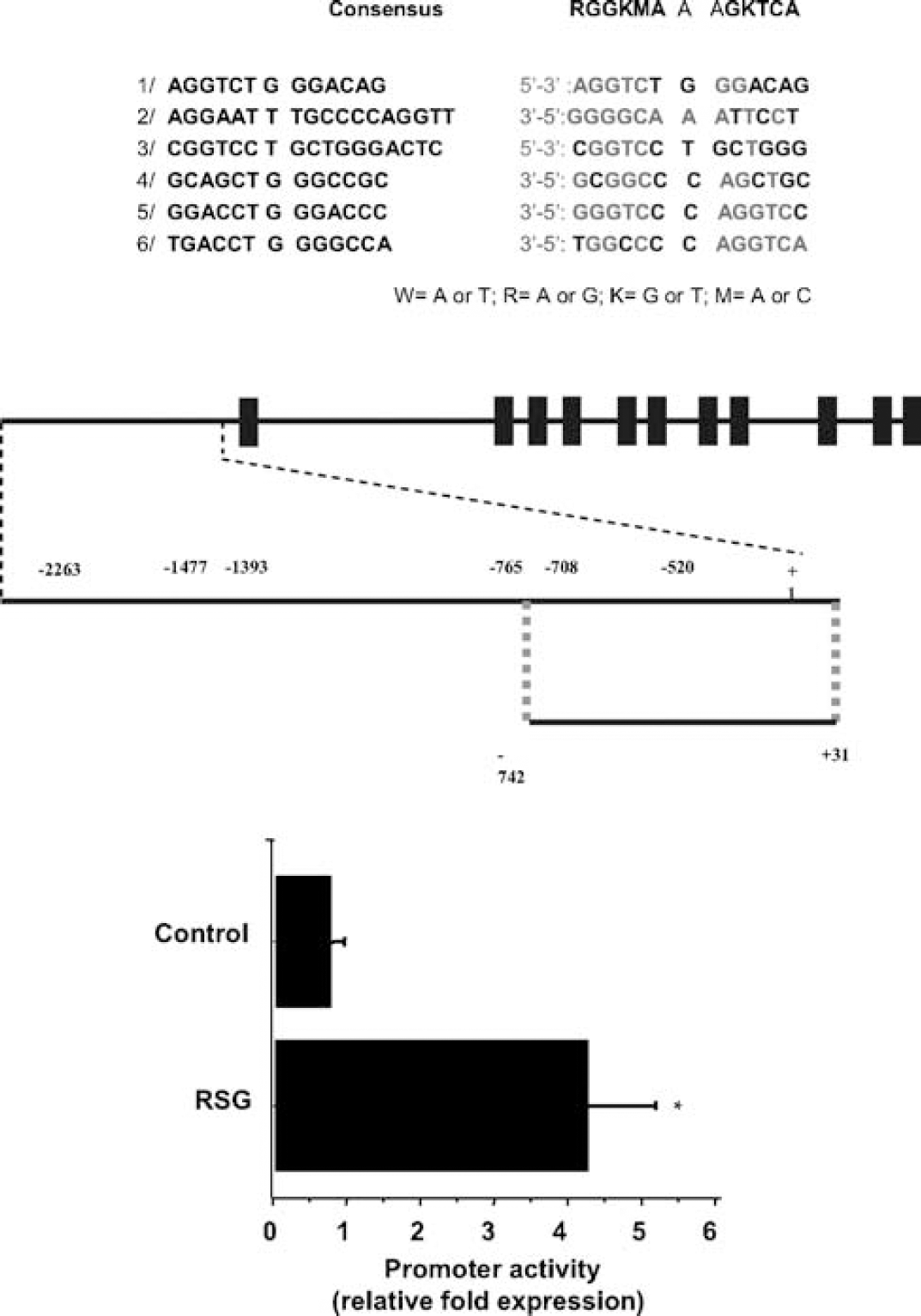
EAAT2 gene promoter. Upper panel: schematic representation of six putative PPRE sites in EAAT2 gene. Location of the binding sites is indicated by gray boxes above the approximate location in the 2.5 kb EAAT2 promoter and in the 773 bp fragment used in our analysis (p–742/ + 31, MluI/Bg/II fragment; cloned into the pGL3-basic vector). Exons are indicated by bold boxes, as described by Su et al (2003). Lower panel: effect of rosiglitazone (RSG; 1 μmol/L) on GLT-1/EAAT2 promoter construction transfected into cultured rat astrocytes. Data are mean ± s.e.m., n = 6, P < 0.05 versus control.
Effect of Rosiglitazone on EAAT2-Promoter Activity Transfected into Rat Astrocytes
Rat astrocytes were transfected with pEAAT2PrLUC (p-742/ + 31, MluI/BglII fragment; cloned into the pGL3-basic vector). Treatment with rosiglitazone (1 μmol/L) for 24 h increased fourfold EAAT2 promoter activity (Figure 6, lower panel; P < 0.001).
Discussion
We hereby report that in vitro IPC in rat cortical neuronal-astrocytic cocultures has allowed us to identify astrocytic GLT1/EAAT2 glutamate transporter as a novel PPARγ target gene.
From very early evidences (Janoff, 1964), it is known that a mild ischemic event can protect the brain against a more severe subsequent ischemia (reviewed in Barone et al, 1998; Kirino, 2002; Dirnagl et al, 2003). In this context, using an in vitro model of IPC by exposing rat cortical cultures to brief periods of OGD, we recently showed that ischemic tolerance is mediated by the upregulation of excitatory amino-acid transporters EAAT2 (GLT-1) and EAAT3 (EAAC1). In the case of EAAT3, we showed that this upregulation was mediated by a TACE/TNF-α/TNFR1 pathway (Romera et al, 2004; Pradillo et al, 2005). However, the mechanisms involved in GLT-1/EAAT2 upregulation in IPC were unknown. Interestingly, recent evidences have shown that agonists of the PPARγ receptor, a ligand-activated transcription factor belonging to the superfamily of nuclear receptors, are neuroprotective in experimental stroke models (Pereira et al, 2005, 2006; Shimazu et al, 2005; Sundarajan et al, 2005; Zhao et al, 2005a). Moreover, a study carried out in humans showed that increased plasma levels of the endogenous PPARγ agonist 15dPGJ2 are associated with better outcome in acute atherothrombotic ischemic stroke (Blanco et al, 2005). We therefore explored whether endogenous activation of the PPARγ receptor is involved in ischemic tolerance.
As previously shown by several groups including ours (Bruer et al, 1997; Grabb and Choi 1999; Liu et al, 2005; Romera et al, 2004), experimental IPC caused ischemic tolerance to subsequent lethal OGD exposures, an effect that we showed to be due, at least partly, to an overexpression of the astrocytic GLT1/EAAT2 glutamate transporter and a subsequent decrease in glutamate release after OGD (Romera et al, 2004). More interestingly, our new data show that the selective PPARγ antagonist T0070907 (Lee et al, 2002) (1) inhibits IPC-induced neuroprotection, (2) blocks IPC-induced reduction in glutamate release, (3) inhibits IPC-induced astrocytic EAAT2/GLT1 increased expression, and (4) decreases astrocytic [3H] glutamate uptake. This drug did not affect by itself cellular viability. Taken together, these results strongly support the involvement of PPARγ in the tolerance afforded by IPC.
To confirm the involvement of PPARγ activation on IPC-induced effects on glutamate transport, we studied the transcriptional activity of PPARγ in this setting by measuring the ability of nuclear proteins to bind a specific oligonucleotide containing the consensus PPRE sequence with a commercial kit. Indeed, our data show a marked increase in PPARγ transcriptional activity in astrocytes and also in neurons, 24 h after IPC, thus indicating a ligand-induced activation of this receptor. To our knowledge, this is the first report on the involvement of PPARγ in ischemic tolerance. Several endogenous molecules may account for this effect, such as the cyclopentenone prostaglandin 15dPGJ2 (Forman et al, 1995; Kliewer et al, 1995), fatty acids including oleic, linoleic, linolenic, and arachidonic acids (Kliewer et al, 1997), as well as nitrolinoleic acid (Schopfer et al, 2005) and epoxyeicosatrienoic acids (Liu et al, 2005), oxidized alkyl phospholipids (Davies et al, 2001), lysophosphatidic acid (McIntyre et al, 2003), and cyclopentenone isoprostanes (Musiek et al, 2005), among others. Further studies are needed to clarify the molecules involved in this setting.
The most important consequence of our results is the possibility to obtain a simple pharmacologic strategy to upregulate GLT-1 glutamate transporter. Therefore, we studied whether PPARγ agonists could have this effect. To this aim, we first used the PPARγ agonist rosiglitazone, a thiazolidinedione with a high selectivity for PPARγ, with a Kd of 40 nmol/L (Lehmann et al, 1995). Indeed, our data show for the first time that the PPARγ agonist rosiglitazone increases astrocytic GLT-1/EAAT2 expression both at protein and mRNA levels; this drug also augments [3H]glutamate uptake in astrocytes, thus showing that this enhanced expression is functional and may exert neuroprotective effects by preventing the extracellular glutamate levels from rising to neurotoxic values. This was further confirmed by our results showing that rosiglitazone decreases cell death after exposure to lethal OGD, in parallel to a reduction in OGD-induced glutamate release. Several groups including ours have shown neuroprotective actions of PPARγ agonists in different paradigms of CNS pathologies ranging from cerebral ischemia, Alzheimer's disease, or multiple sclerosis, among others (Combs et al, 2000; Feinstein et al, 2002; Pereira et al, 2005, 2006; Shimazu et al, 2005; Sundarajan et al, 2005; Zhao et al, 2005a, b). Apart from the previously described antiinflammatory actions of these agents, antiexcitotoxic actions via GLT-1/EAAT2 overexpression are likely to participate in the neuroprotection reported. This is further confirmed by the data showing that either inhibition of GLT-1/EAAT2 transporter expression using antisense ODN or inhibition of the transporter activity using the nontransported glutamate transporter analogue DHK (Arriza et al, 1994; Tanaka, 1994) significantly decrease IPC- and rosiglitazone-induced neuroprotection.
These data strongly suggest that the GLT1/EAAT2 glutamate transporter is a PPARγ target gene. To explore this hypothesis further, we have tested the effect of other chemically unrelated PPARγ agonists, the cyclopentenone prostaglandin 15dPGJ2 (reviewed in Straus and Glass, 2001) and the non-TZD L-796,449 (Castrillo et al, 2001). Similarly to rosiglitazone, both compounds increased the astrocytic expression of GLT1/EAAT2. Moreover, we tested the effect of rosiglitazone in a different scenario, an in vivo model of stroke by middle cerebral artery occlusion in rats. Apart from confirming the neuroprotective effect of this molecule as shown by a decrease in infarct volume, our results show that the treatment with this PPARγ agonist increased the expression of GLT1/EAAT2 in brain homogenates after the ischemic injury. These data suggest that an increased uptake of cerebral extracellular glutamate levels after the occlusion may represent an additional mechanism for the neuroprotection exerted by PPARγ activation in this model, a hypothesis that deserves further studies.
To understand better the mechanism of action of rosiglitazone on GLT-1/EAAT2 expression, we searched for the presence of PPREs in the promoter region of the EAAT2 gene. Bioinformatic analysis of this promoter showed the presence of at least six putative consensus PPRE sites that may be responsible for the effect observed, supporting the hypothesis of a direct effect of PPARγ on GLT-1/EAAT2 expression. To explore whether these sites are functional, we studied the activity of a EAAT2 promoter construct consisting of a fragment linked to the firefly luciferase, which has been shown to keep the whole promoter activity (Su et al, 2003), and that contains two of the putative PPREs. In these experiments, rosiglitazone increased fourfold the EAAT2 promoter activity, thus confirming that GLT-1/EAAT2 is a novel PPARγ target gene.
Apart from their cytoprotective, antiinflammatory effects of these compounds previously described in stroke models, our findings show novel, additional neuroprotective actions of PPARγ agonists and open new possibilities in the treatment and/or prevention of cerebral ischemia and other neuropathologies. Indeed, impaired glutamate transport is known to induce neurotoxicity associated with numerous neurologic processes. In the case of GLT-1/EAAT2, reductions in its protein expression have been described to take place in ischemia (Torp et al, 1995), epilepsy (Rothstein et al, 1996; Tanaka et al, 1997), Alzheimer's disease (Li et al, 1997; Dabir et al, 2006), Huntington's disease (Lipton and Rosenberg, 1994), and amyotrophic lateral sclerosis (Rothstein et al, 1995; Lin et al, 1998), among others. In addition, we have recently identified a novel and highly prevalent polymorphism in the EAAT2 promoter, which is associated with increased plasma glutamate concentrations and with a higher frequency of early neurologic worsening in human stroke (Mallolas et al, 2006) and which could be involved in CNS disorders associated to excitotoxicity. All these evidences highlight the importance of the findings described in this paper, not only on stroke outcome, but also in other neuropathologies. The involvement of other effects of rosiglitazone in neuroprotection, such as an increased mitochondrial biogenesis as recently described in other systems (Wilson-Fritch et al, 2004), remains to be investigated.
Taken together, these data show that GLT-1/EAAT2 is a PPARγ target gene, with important potential neuroprotective applications in a vast range of CNS pathologies. Considering that several PPARγ agonists are already commercially available, our findings may have immediate therapeutic repercussions. In addition, given the role of the pathway described in endogenous neuroprotection, plasma levels of endogenous PPARγ agonists may serve as powerful prognostic markers in several CNS pathologies apart from stroke.
Footnotes
Acknowledgements
We are grateful to Tularik Inc. for kindly providing T0070907.