
Abstract
Hippocampal CA1 neurons exposed to a nonlethal period (2 min) of ischemia, acquired tolerance to a subsequent lethal 5-min period of ischemia, which usually causes delayed-type neuronal death. Intracelluar Ca2+ movements before and after the 5 min of forebrain ischemia were evaluated in gerbil hippocampal CA1 pyramidal neurons, had acquired tolerance in comparison with nonischemia-tolerant CA1 neurons. Evaluation was performed by observing the ultrastructural intracellular Ca2+ distribution and the Ca2+ adenosine triphosphatase (Ca2+-ATPase) activity using electron microscopic cytochemistry. In comparison with nonischemia-tolerant CA1 neurons, mitochondria of ischemia-tolerant CA1 neurons sequestered more Ca2+ from the cytosomal fraction 15 min after the 5-min period of ischemia, and Ca2+ deposits in these mitochondria were rapidly decreased. Plasma membrane Ca2+-ATPase activities were already significantly elevated before the 5 min of ischemia, and remained at a higher level subsequently compared to nonischemia-tolerant CA1 neurons. Changes in the mitochondrial Ca2+ distribution and Ca2+-ATPase activities in ischemia-tolerant CA1 neurons after the 5-min period of ischemia showed a strong resemblance to those in CA3 neurons, which originally possess resistance to such periods of ischemia. These findings suggest that enhanced or maintained activities of mitochondrial Ca2+ sequestration and plasma membrane Ca2+-ATPase reduced Ca2+ toxicity following 5-min ischemia in terms of time, resulting in escape from delayed neuronal death.
Overloading of intracellular Ca2+ brought about by excessive extracellular release of exitotoxic amino acids during ischemia plays a pivotal role in the pathogenesis of delayed neuronal death of the gerbil hippocampal CA1 neurons following 5-min of transient forebrain ischemia (Benveniste et al., 1984; Simon et al., 1984; Kudo et al., 1986; Choi DW, 1987). It was reported recently that hippocampal CA1 neurons subjected to a nonlethal 2-min period of transient ischemia survived subsequent exposure to a normally lethal 5-min ischemic insult (Kitagawa et al., 1990; Kirino et al., 1991; Furuta et al., 1993). However, pretreatment by nonlethal ischemic loading did not alter the amount of released extracellular glutamate or glutamate receptor binding potential during or after subsequent potentially lethal ischemic insult (Nakata et al., 1992; Kato et al., 1992). These results suggested that the intracellular accumulation of Ca2+ in ischemia-tolerant hippocampal CA1 neurons must be similar to that in the originally vulnerable CA1 neurons after the 5-min ischemic insult. We proposed that the intracellular Ca2+ movement may differ between nonischemia-tolerant and ischemia-tolerant CA1 neurons after forebrain ischemia in terms of the defense mechanism against Ca2+ toxicity. In the present study, we evaluated intracellular Ca2+ movement by observing the ultrastructural distribution of Ca2+, especially the amounts of Ca2+ deposited in the mitochondria, which sequester Ca2+ in some cases under conditions of high intracellular Ca2+ concentration, and the activities of plasma membrane Ca2+ ATPase, which exports intracellular Ca2+ to the extracellular space, before and after 5 min of transient forebrain ischemia in CA1 and CA3 neurons of gerbils that had and had not been subjected to two 2-min periods of ischemia, using electron microscopic cytochemical techniques.
MATERIALS AND METHODS
A total of 94 adult male gerbils (Meriones unguilantus), weighing 65–80 g each, were kept at a constant temperature (24 ± 2°C) in an air-conditioned room under a 12-h light/dark cycle, for at least 14 days prior to the study. Animals were divided into ischemia-tolerant and nonischemia-tolerant groups. Gerbils in the former group were loaded with two 2-min periods of forebrain ischemia separated by a 24-h interval. Animals were then exposed to another 5-min ischemic insult after 48 h of recirculation, according to the method of Kitagawa et al. (1990). Gerbils in the nonischemia-tolerant group were loaded with only one 5-min forebrain ischemic period 48 h after two sham operations performed at an interval of 24 h.
Ischemic operations
To achieve transient ischemia, animals were lightly anesthetized with 1% halothane in a mixture of 70% nitrous oxide and 30% oxygen (vol/vol). Bilateral common carotid arteries were exposed briefly and occluded with miniature aneurysmal clips, and the halothane discontinued. Occlusion and reperfusion of carotid arteries were verified by direct visual inspection under an operating microscope, and skin incisions were sutured. Rectal temperature, monitored with a digital thermometer inserted 7 cm from the anus, was maintained at 36.5–37.5°C throughout this procedure using a heat lamp. Temperature was monitored following 30 min of recirculation.
Histological assessment by light microscopy
Gerbils that survived for 7 days after 5-min occlusion of the carotid arteries (n = 18) were reanesthetized and killed by decapitation. Brains were removed promptly, fixed in 10% formaldehyde, and embedded in paraffin. Then, 5 μm-thick cross sections of specimens containing the bilateral dorsal hippocampi, situated 1.4–1.6 mm caudal to the bregma, were stained with cresyl violet. Surviving pyramidal neurons per 1 mm length of medial CA1 and CA3 regions were counted under a light microscope at 400× magnification.
Intracellular calcium histochemistry
Five gerbils from each group at each time period—before and at 15, 60, 180, and 360 min after 5 min of ischemia—were reanesthetized and perfused through the left ventricle of the heart with a mixture of 2.5% glutaraldehyde, 2% paraformaldehyde, and 90 mM potassium oxalate (pH 7.4) at 4°C. The brain was removed, and small blocks, including the hippocampal CA1 or CA3 sector, were immersed in the same fixative. Tissue blocks were rinsed for 15 min in 7.5% sucrose containing 90 mM potassium oxalate, adjusted to pH 7.4 with potassium hydroxide, and postfixed in 0.01 M acetic acid with 1% osmium tetroxide and 2% potassium pyroantimonate, adjusted to pH 7.4 with 0.1 M potassium hydroxide, for 2 h to transform oxalate-bound calcium into an electron-dense insoluble precipitate. After a 15-min rinse in cold distilled water brought to pH 10.0 with potassium hydroxide, specimens were dehydrated through a graded ethanol series and embedded in Quetol 812. CA1 or CA3 pyramidal neurons were identified on semithin sections stained with 1% toluidine blue, and 70 nm-thick ultrathin sections were cut on an ultramicrotome and examined with a H-800 Hitachi transmission electron microscope (Hitachi Company, Tokyo, Japan). The specificity of the cytochemical reaction was tested by treating mounted ultrathin sections with 5 mM ethylene glycol bis (β-aminoethyl ether) N,N′-tetraacetic acid (EGTA) for 1 h at 60°C.
Morphometric assessment of mitochondrial Ca2+
Electron micrographs of the CA1 and CA3 pyramidal neurons on slices treated for Ca2+ histochemistry were taken of 30 random fields at a magnification of 20,000× in the nonischemia-tolerant and the ischemia-tolerant gerbils, and mitochondria in the cytosol were classified in a manner similar to that described by Hossmann et al. (1985): grade 0, no calcium deposits; grade 1, few dust-like calcium deposits; grade 2, several grains of calcium deposit; grade 3, numerous calcium deposits; and grade 4, massive calcium deposits. Mitochondrial calcium uptake index was also calculated by averaging individual grading scores.
Plasma membrane Ca2+-ATPase histochemistry
Histochemical examination for plasma membrane Ca2+-ATPase was performed according to the method of Ando et al. (1981) before (n = 5/group), and 15 (n = 4/group) and 180 min (n = 4/group) after a 5-min period of ischemia in nonischemia-tolerant and ischemia-tolerant gerbils. While still under anesthesia, gerbils were perfused through the left ventricle of the heart with a mixture of 0.25% glutaraldehyde and 2% paraformaldehyde in 0.1 M cacodylate-HCl buffer (pH 7.4) at 4°C. Then, 70 μm-thick CA1- or CA3-containing sections were cut and immersed in the same fixative for 40 min. Sections were then rinsed with 0.1 M cacodylate-HCl buffer (pH 7.4) at 4°C for 24 h and incubated for 60 min at 37°C in a reaction medium containing 3 mM ATP-2Na, 1 mM CaCl2, 4 mM lead citrate, 8% sucrose, and 2.5 mM levamisole (which inhibits intrinsic alkaliphosphatase), in 250 mM glycine-KOH buffer (pH 9.0). Control sections were incubated in reaction medium devoid of Ca2+. After rinsing with 0.1 M cacodylate-HCl buffer (pH 7.4) at 4°C for 10 min, sections were postfixed with 1% osmium tetroxide in the same buffer for 15 min, rinsed with distilled water at 0°C, and dehydrated through a graded ethanol series. Ultrathin sections were cut, and reaction products were observed by transmission electron microscopy.
Morphometric assessment of plasma membrane Ca2+-ATPase activity
Electron micrographs of CA1 and CA3 pyramidal neurons in sections histochemically treated to detect Ca2+-ATPase were taken of 30 random fields at a magnification of 8,000× in nonischemia-tolerant and ischemia-tolerant gerbils. Areas of enzymatic reaction products on and attached to the cytoplasmic membrane were calculated per unit length of the plasma membranes of CA1 or CA3 neurons using an image analyzer (NIH Research Service Branch, Bethesda, MD, U.S.A.).
Statistical analyses
All values are given as mean ± SD in the text and mean ± SEM in figures. Mitochondrial Ca2+ uptake indices were compared by Fisher's and Schéffe's methods following one- or two-way analysis of variance (ANOVA). Plasma membrane Ca2+-ATPase activities were compared by two-way ANOVA.
RESULTS
Histology
There was no significant difference in rectal temperatures between nonischemia-tolerant and ischemia-tolerant groups within 30 min following the 5-min ischemic insult. Numbers of surviving neurons in the sham-operated control group were 224.3 ± 11.1 and 278.2 ± 12.3/mm length of the CA1 and CA3 pyramidal layers, respectively, the same as those in normal gerbils. The number of surviving neurons in CA1 was markedly decreased to 16.0 ± 3.5/mm in the nonischemia-tolerant group. However, numbers of surviving neurons in CA3 in the nonischemia-tolerant group and in CA1 and CA3 in the ischemia-tolerant group were preserved to 272.9 ± 9.2, 219.9 ± 15.6, and 274.3 ± 18.1/mm, respectively. The two 2-min ischemic pretreatments conferred significant tolerance to delayed neuronal death in the CA1 region after 5 min of ischemia. We confirmed that almost all CA1 and CA3 neurons in the ischemia-tolerant group survived for at least 30 days after a 5-min ischemic insult.
Ca2+ histochemistry
There were no marked differences between nonischemia-tolerant and ischemia-tolerant CA1 neurons before the 5-min period of ischemia, when a little Ca2+ deposition was observed only in the mitochondria, Golgi apparatus, and smooth endoplasmic reticulum. After 15 min of recirculation, many dust-like Ca2+ deposits were diffusely distributed throughout the cytosol, and Ca2+ deposits in the mitochondria were moderately increased compared with those of the preischemic state in nonischemia-tolerant CA1 neurons (Fig. 1A). On the other hand, in ischemia-tolerant CA1 neurons, massive or numerous Ca2+ deposits were noted in mitochondria, with sparse deposits in the cytosol, in contrast to nonischemia-tolerant CA1 neurons (Fig. 1B). There were several Ca2+ deposits in the Golgi apparatus and smooth endoplasmic reticulum in neurons of both groups. In nonischemia-tolerant CA1 neurons, Ca2+ deposits were increased in the mitochondria and decreased in the cytosol at 60 min of recirculation; dense Ca2+ deposits were not only still present in the mitochondria but also gradually reincreased in the cytosol and smooth endoplasmic reticulum at 180–360 min of recirculation (Fig. 1C). In ischemia-tolerant CA1 neurons, these deposits were gradually decreased in the mitochondria, with almost no increase in the cytosol or smooth endoplasmic reticulum at 60–180 min after a 5-min ischemic insult (Fig. 1D), and returned to preischemic state levels at 360 min of recirculation, showing a marked difference in Ca2+ distribution between the two groups.
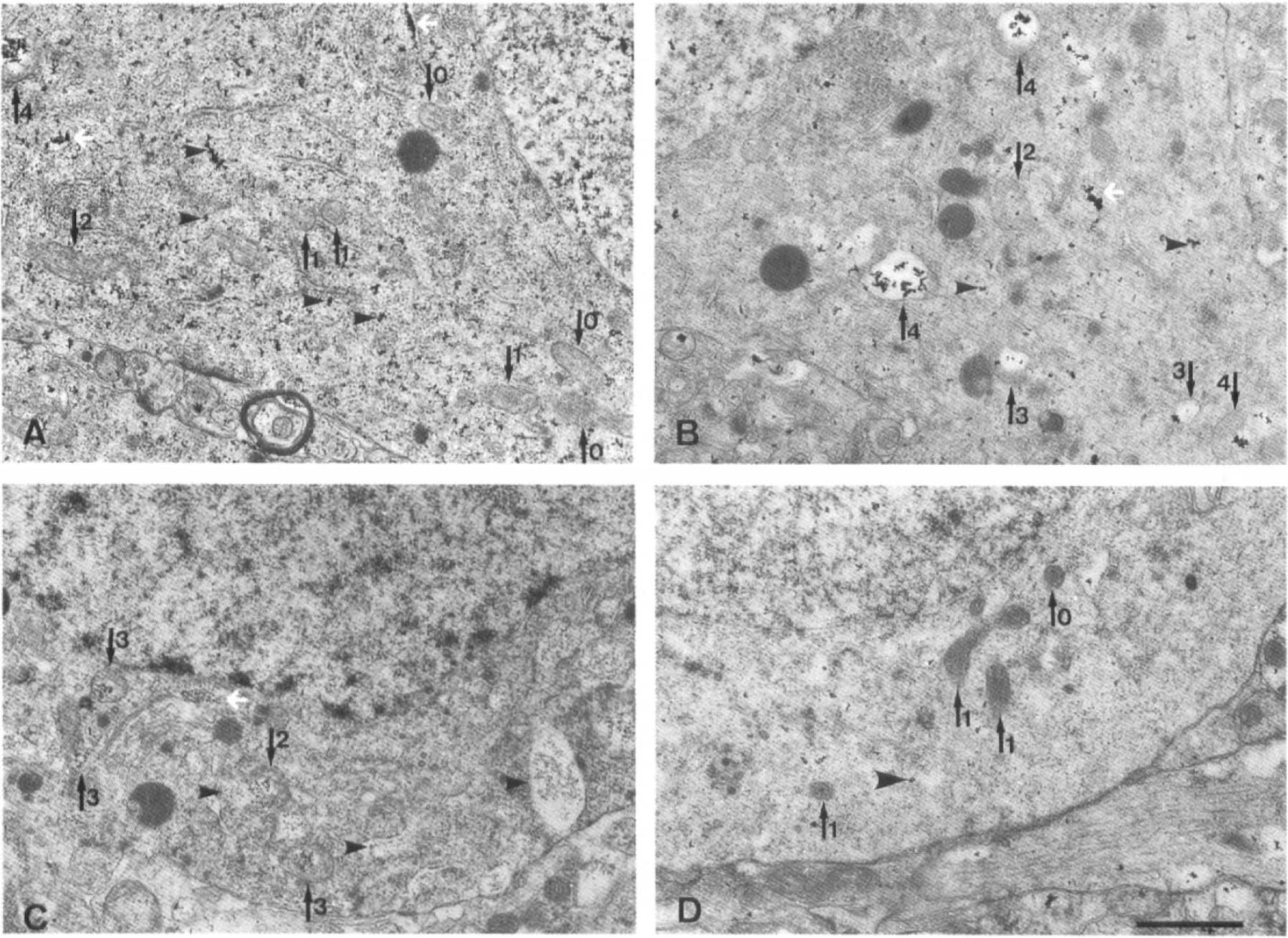
Transmission electron micrographs following Ca2+ histochemical staining by the oxalate-pyroantimonate technique in nonischemia-tolerant and ischemia-tolerant hippocampal CA1 neurons after 15 and 180 min of recirculation. Numbered black arrows on the photographs indicate mitochondria, classified according to the amounts of Ca2+ deposits (described in text), in which the structure, including many Ca2+ dense deposits, was partly destroyed in the process of cytochemical staining. Arrowheads indicate dust-like Ca2+ deposits in the cytosol and white arrows indicate the Ca2+ deposits in the smooth endoplasmic reticulum. At 15 min of recirculation after the 5-min period of ischemia, there were many dust-like Ca2+ deposits (arrowhead) scattered diffusely throughout the cytosol of nonischemia-tolerant CA1 neurons
Electron-dense stained oxalate-pyroantimonate deposits disappeared completely following pretreatment with 5 mM of the calcium-chelating agent, EGTA.
Mitochondrial Ca2+ uptake index
The time course of changes in the mitochondrial Ca2+ uptake index before and after the 5-min period of ischemia are shown in Fig. 2. There were no differences in the Ca2+ uptake index before 5 min of ischemia among nonischemia-tolerant and ischemia-tolerant CA1 or CA3 neurons. After recirculation for 15 min, although the Ca2+ uptake index was markedly increased in all neurons, that of nonischemia-tolerant CA1 neurons was lower than in other neurons (significant difference by Fisher's method—p < 0.05—but not by Schéffe's method). At 360 min after a 5-min period of ischemia, ischemia-tolerant neurons showed significantly lower Ca2+ uptake index than did nonischemia-tolerant CA1 neurons, which had not been preexposed. There were significant differences between nonischemia-tolerant CA1 and other neurons at 60–360 min after a 5-min ischemic insult, as determined by both Fisher's and Schéffe's methods.
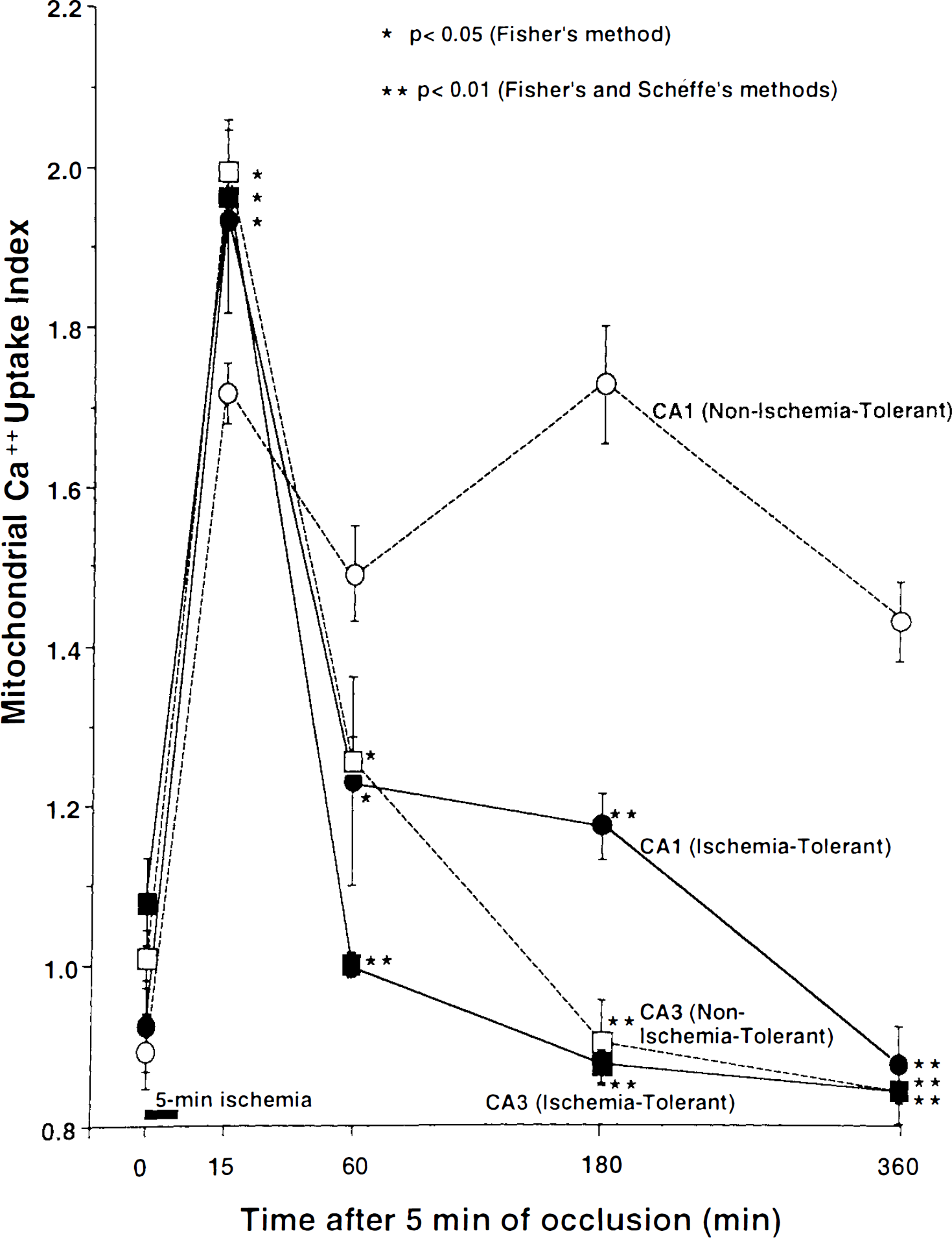
Changes in mitochondrial Ca2+ uptake index after the 5-min period of ischemia in nonischemia-tolerant and ischemia-tolerant CA1 neurons, and originally-resistant CA3 neurons in nonischemia-tolerant and ischemia-tolerant gerbils. Values represent means ± SEM. There were significant differences in the Ca2+ uptake indices at 15 min after the 5-min ischemic insult between nonischemia-tolerant CA1 and other neurons, by Fisher's comparison. Note that the Ca2+ uptake index of ischemia-tolerant CA1 and CA3 neurons in both groups returned to preischemic values 180–360 min following the 5 min of ischemia, although the Ca2+ uptake index of nonischemia-tolerant CA1 neurons remained at a higher level after 60–360 min of recirculation; there were significant differences between nonischemia-tolerant CA1 and other neurons, by Fisher's and Schéffe's comparisons.
Ca2+-ATPase histochemistry
The electron-dense reaction products of Ca2+-ATPase were observed predominantly along the external surface of plasma membranes of pyramidal neurons at all experimental time points for both groups (Fig. 3A,B). Intracellular organelles did not show any specific black deposits, and no reaction products were observed in control sections incubated in a Ca2+-free medium. Smaller positive foci were distributed sparsely along the surface of the plasma membrane before the 5-min period of ischemia in nonischemia-tolerant CA1 neurons (Fig. 3A). On the other hand, greater amounts of reaction products were observed successively in ischemia-tolerant CA1 neurons (Fig. 3B). There were no marked changes in the morphological appearance of reaction products after, compared with before, the 5-min ischemic insult in any neurons examined.
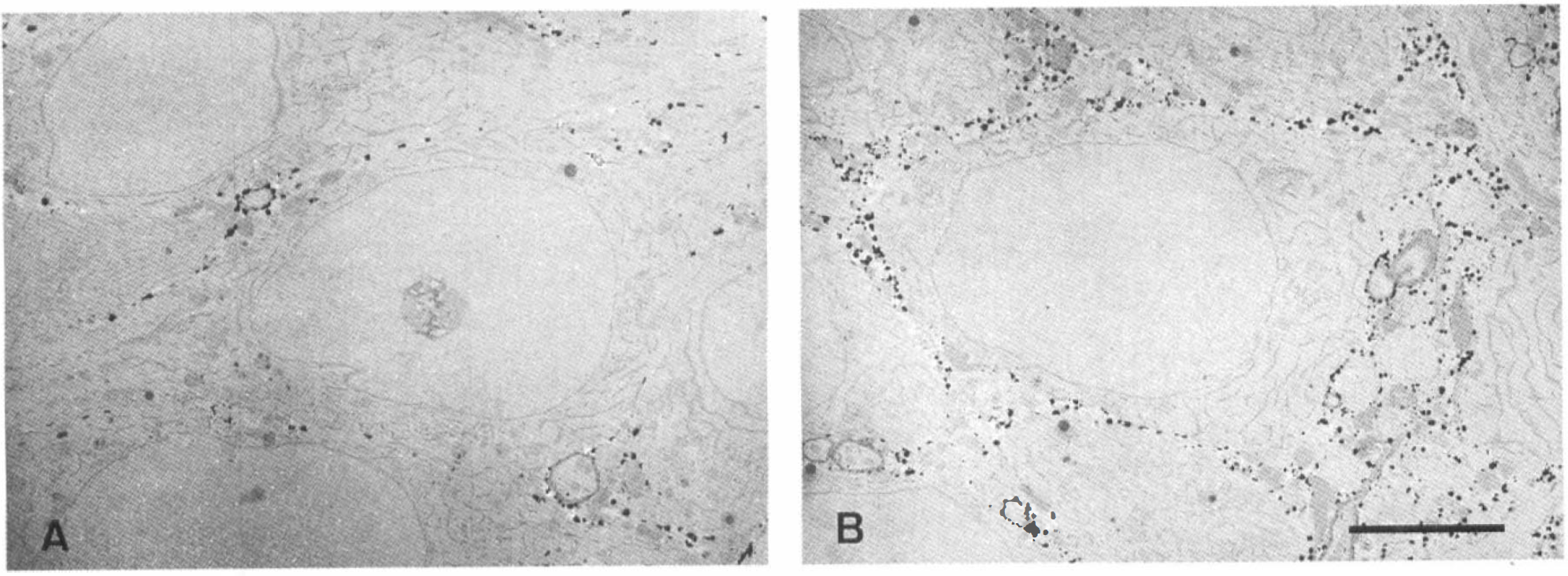
Transmission electron micrographs of Ca2+-ATPase histochemical staining in nonischemia-tolerant
Semiquantitative analysis of plasma membrane Ca2+-ATPase activity
Time courses of changes in Ca2+-ATPase activities, determined semiquantitatively, before and after the 5 min of ischemia are shown in Fig. 4. While the area of reaction products per unit length of plasma membrane before ischemic treatment was 23.98 ± 5.92 nm2/nm in nonischemia-tolerant CA1 neurons, the value was significantly higher, 46.03 ± 9.91 nm2/nm, in ischemia-tolerant CA1 neurons, similar to the values of 44.87 ± 8.39 and 38.68 ± 6.53 nm2/nm of CA3 neurons in nonischemia-tolerant and ischemia-tolerant gerbils. Ca2+-ATPase activities showed little alteration after the 5-min ischemic insult, and those in ischemia-tolerant CA1 and CA3 neurons in both groups remained at significantly higher levels compared with those in nonischemia-tolerant CA1 neurons throughout the duration of the experiment (p < 0.01).
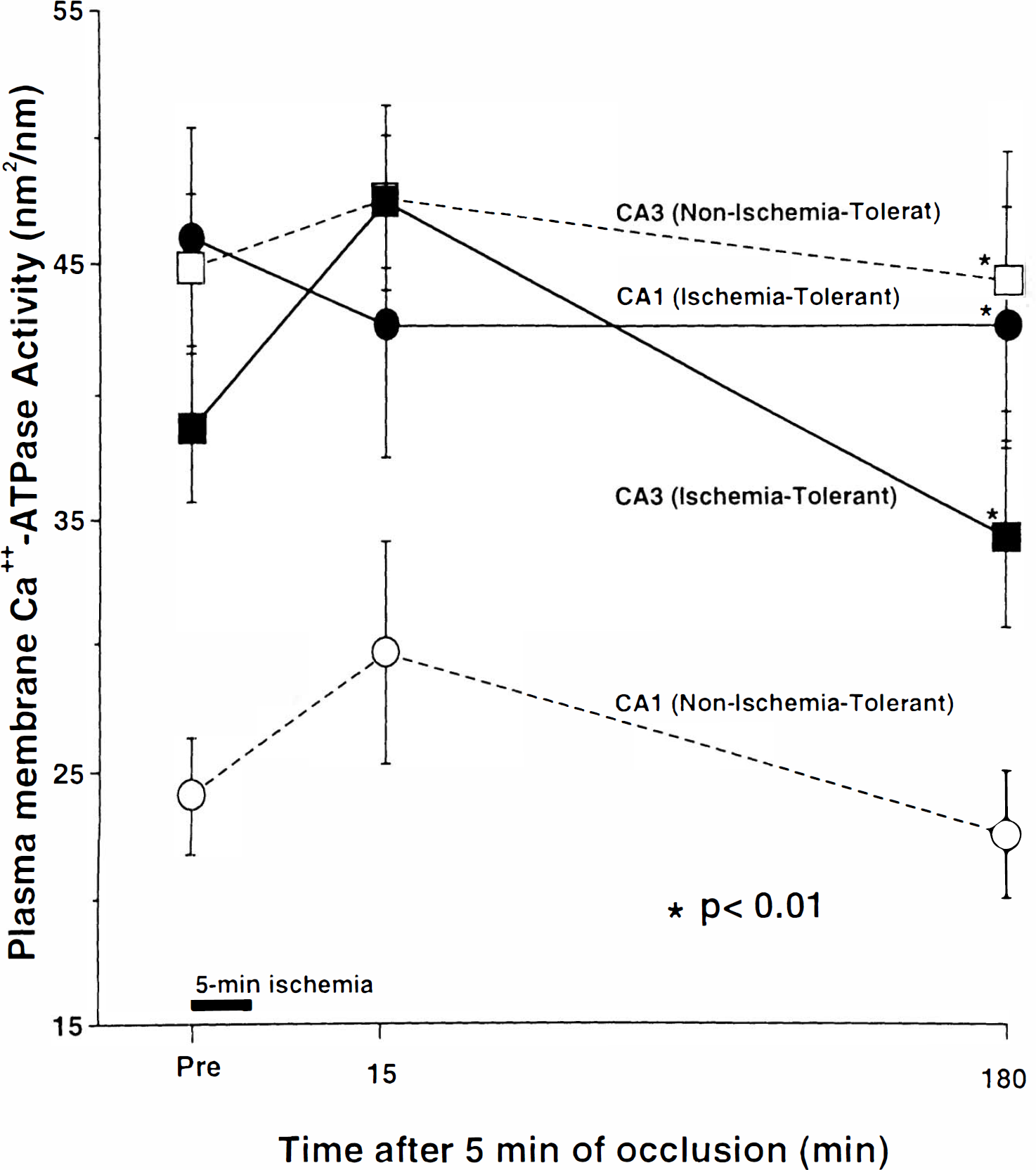
Changes in plasma membrane Ca2+-ATPase activities measured semiquantitatively before and after the 5-min period of ischemia in nonischemia-tolerant and ischemia-tolerant CA1 neurons, and originally-resistant CA3 neurons of both groups. Values represent mean ± SEM. Note that plasma membrane Ca2+-ATPase activities in ischemia-tolerant CA1 neurons and CA3 neurons from both groups were significantly higher than those in nonischemia-tolerant CA1 neurons before the 5-min period of ischemia, and Ca2+-ATPase activities showed little alteration after the 5-min ischemic insult in any neurons. There were significant differences at each time point before and after the 5-min period of ischemia between nonischemia-tolerant CA1 and other neurons.
DISCUSSION
Some organelles, such as the Golgi apparatus, endoplasmic reticulum, and mitochondria, function as part of a defense system against Ca2+ toxicity in cases of excessive accumulation of intracellular Ca2+. When the concentration of intracellular Ca2+ increases to μM proportions in response to physiological stimuli, the endoplasmic reticulum mainly takes up Ca2+ to be buffered (Calafoli, 1987; Blaustein, 1988). However, once it increases to the lethal level >μxM concentrations induced by pathological conditions, such as ischemic insult, the mitochondria actively sequester Ca2+ from the cytosol and store it as calcium phosphate compounds (Siesjo, 1992; Abe, 1992). Plasma membrane Ca2+-ATPase and Na+/Ca2+ antiporter export intracellular Ca2+ to the extracellular space. However, the Na+/Ca2+ antiporter, driven by the Na+ gradient, occasionally imports in the opposite direction when either intracellular Na+ is raised or extracellular Na+ is lowered. Plasma membrane Ca2+-ATPase can steadily export intracellular Ca2+ even with relatively lower transport capacity during prolonged membrane hyperpolarization and under conditions of elevated intracellular Na+ concentration, both states of which are usually observed under ischemic conditions (Abe, 1992; Furukawa et al., 1988). Therefore, Ca2+-ATPase is considered to be more important as a Ca2+ export system when there is long-term and excessive intracellular Ca2+ loading after ischemia.
At present, there are some methods available to measure intracellular Ca2+ content; these include fluorescent calcium indicator dyes (Grynkiewicz et al., 1985), calcium electrodes (Oehme et al., 1976), and oxalate-pyroantimonate histochemistry (Hossmann et al., 1985; Dux et al., 1987; Kohno et al., 1991). Among these methods, the latter is superior for examination of intracellular Ca2+ distribution and is especially useful, as it allows identification of Ca2+ in individual organelles at the electron microscopic level (although it is possible that the Ca2+ deposits visualized by this method could represent not only free Ca2+ but also that already bound to intracellular Ca2+-sequestering substances) (Borgers et al., 1977). Ando's method for Ca2+-ATPase histochemistry (Ando et al., 1981) also allows localization of the enzyme at the electron microscopic level, and the validity of the semiquantitative assessment of plasma membrane Ca2+-ATPase activity used here was verified in our previous study by comparison to a biochemical assay (Wang et al., 1994).
By 15 min of recirculation after 5 min of ischemia, there were many more Ca2+ deposits in mitochondria and correspondingly less cytosolic Ca2+ deposition in ischemia-tolerant CA1 neurons compared to the originally vulnerable nonischemia-tolerant CA1 neurons, in which many Ca2+ deposits remained diffusely scattered throughout the cytosol where they could disturb cellular biochemical integrity. It was suggested that mitochondria rapidly sequester a great deal of Ca2+ from the cytosol after excessive intracellular Ca2+ influx; the effect of this is that of reducing Ca2+ toxicity in terms of time in ischemia-tolerant neurons, similar to what occurs in originally resistant CA3 neurons. Furthermore, in ischemia-tolerant CA1 neurons, mitochondrial distribution (classified according to the amount of Ca2+ deposition) returned to the preischemic value as early as did those in the CA3 neurons, while both the increase and the decrease in Ca2+ deposits in the mitochondria were further delayed in nonischemia-tolerant CA1 neurons.
Numbers of animals in each group for each time

n, number of animals.
Preischemia
Ca2+ sequestered by the mitochondria is eventually released into the cytosol and exported from intracellular to extracellular compartments by plasma membrane Ca2+-ATPase or Na+/Ca2+ antiporter (Lehninger, 1970). It was revealed, in the present study, that plasma membrane Ca2+-ATPase activity was already significantly elevated prior to the 5-min period of ischemia by pretreatment with two 2-min ischemic periods, and its elevated activity was maintained after the 5-min period of ischemia in ischemia-tolerant CA1 neurons. Although we did not study activity of the Na+/Ca2+ antiporter on the plasma membrane, it was suggested that ischemia-tolerant CA1 neurons had the capacity to more efficiently export a larger amount of Ca2+, and could sweep away not only the Ca2+ flood into the cytosol from the extracellular compartment accompanying the ischemic insult but also the Ca2+ subsequently released from the mitochondria, predominantly or partly, through enhanced plasma membrane Ca2+-ATPase activity, compared with nonischemia-tolerant CA1 neurons. Less efficient export of Ca2+ in nonischemia-tolerant CA1 neurons was also supported by observation of a subsequent reelevation of Ca2+ in the cytosol and smooth endoplasmic reticulum at 180–360 min following reinstatement of circulation. Interestingly, plasma membrane Ca2+-ATPase activity of CA3 neurons was, initially, as high as that of ischemia-tolerant CA1 neurons, and this may have been related to the previous finding that the dominant mRNA species (among those encoding the four Ca2+-ATPase isoforms) are naturally different between CA1 and CA3 neurons (Stahl et al., 1992).
There was a possibility that moribund neurons, such as nonischemia-tolerant CA1 neurons, could not accumulate as much Ca2+ in the mitochondria as nonmoribund neurons and might lose plasma membrane Ca2+-ATPase activity (because of compromised Ca2+/Na+ or Ca2+/H+ exchange) across mitochondrial membranes and plasma membrane after 5 min of ischemia. However, we believe that our results indicated that ischemia-tolerant CA1 neurons preloaded with two 2-min periods of sublethal ischemia showed boosted potential of mitochondrial Ca2+ sequestration and plasma membrane Ca2+-ATPase activity, since later increases in grades 3 and 4 mitochondria were observed in nonischemia-tolerant CA1 neurons, and the plasma membrane Ca2+-ATPase activity in ischemia-tolerant CA1 neurons was already elevated before the 5-min ischemic insult. This enhanced buffering system for Ca2+ toxicity may induce tolerance against subsequent potentially lethal 5-min ischemic insult, protecting neurons from cell death. The mechanism of activation or maintenance of this Ca2+ buffering system should be investigated further. Such studies may contribute to the prevention or treatment of delayed neuronal death following ischemia insult.
Footnotes
Acknowledgment:
We thank Mr. Hamai of the Laboratory Animal Center and Mr. Kameda of the Central Research Laboratory, Ehime University School of Medicine, for their technical assistance. This study was carried out in compliance with the Guidelines for Animal Experimentation, Ehime University School of Medicine.