
Abstract
We tested the hypothesis that quinolinic acid, a tryptophan-derived N-methyl-
Traumatic brain injury (TBI) initiates a cascade of responses that may produce secondary damage to the brain and contribute to the ultimate degree of neurologic impairment. One proposed mechanism of this secondary injury is excitotoxicity via the N-methyl-
Quinolinic acid is a metabolite of tryptophan metabolism produced by infiltrating macrophages and activated microglia as part of the inflammatory response in the CNS (Blight et al., 1995; Heyes et al., 1992, 1996). Outside of the CNS, quinolinic acid has no known role in mediating inflammation; however, within the CNS, quinolinic acid binds to the NMDA receptor and is neurotoxic (Misztal et al., 1996a; Portera-Cailliau et al., 1997; Susel et al., 1991; Yamada et al., 1990). The normal level of quinolinic acid in human CSF is less than 50 nmol/L (Heyes et al., 1995). Quinolinic acid concentrations as low as 100 nmol/L can be neurotoxic (Giulian et al., 1993). Portera-Cailliau et al. (1997) reported that intrastriatal injection of 60 nmol of quinolinic acid in rats produced neuronal necrosis that was inhibited by treatment with the NMDA antagonist MK-801. Heyes (1993) reported increases in concentrations of quinolinic acid in CSF of patients with selected CNS inflammatory disorders, such as acquired immunodeficiency syndrome dementia, meningitis (Heyes et al., 1995), and polio (Heyes et al., 1993). In these studies, some patients had CSF quinolinic acid concentrations greater than 10,000 nmol/L.
Blight et al. (1995, 1997), using a model of spinal cord injury in guinea pigs, showed that macrophage-derived quinolinic acid contributes to secondary damage. High levels of quinolinic acid were detected at and around the injury site where there was significant macrophage accumulation. In these studies, functional recovery was improved in guinea pigs treated with either a selective inhibitor of quinolinic acid synthesis (4-chloro-3-hydroxyanthranilate) or depletion of circulating macrophages.
Reports from our laboratory (Bell et al., 1997; Clark et al., 1996) and studies by Giulian et al. (1989), Holmin et al. (1997), Kossmann et al. (1996), McClain et al. (1987), and Balasingam and Yong (1996), among others, have demonstrated the occurrence of a local inflammatory response to TBI in both laboratory models and in humans. Neutrophil accumulation peaks at 24 hours (Clark et al., 1996) and is followed by macrophage infiltration detected between 2 and 7 days (Balasingam and Yong, 1996; Holmin et al., 1997). Inflammatory markers have been associated with severity of injury and outcome (Bell et al., 1997; McClain et al., 1987). A possible role for the macrophage-derived inflammatory metabolite quinolinic acid in the development of delayed excitotoxicity after TBI, however, has not been investigated in either animal models or in clinical studies.
Finally, we reported that therapeutic hypothermia (24 hours at 32°C versus normothermia) improved outcome and attenuated the increase in the CSF concentration of the proinflammatory cytokine interleukin-1β in the initial 36 hours after severe TBI in humans (Marion et al., 1997). Hypothermia also attenuated leukocyte influx after TBI in rats (Whalen et al., 1997). These studies suggest important attenuation of the inflammatory response by therapeutic hypothermia.
In this study, we tested the hypothesis that quinolinic acid concentration would be increased in ventricular CSF after severe TBI in humans and that this increase would be associated with mortality. We also tested the hypothesis that therapeutic hypothermia would reduce CSF quinolinic acid concentration after severe TBI.
METHODS
Study population
This study included 39 patients admitted to the University of Pittsburgh Medical Center from October 1991 through April 1995 with severe closed head injury. Patients were included in this study if they were between 16 and 75 years of age with an initial Glasgow Coma Scale (GCS) score of 3 to 7, and if they were admitted within 6 hours of injury. Exclusion criteria included clinical brain death (GCS score of 3 with absent brain stem reflexes), prolonged hypoxia or hypotension (oxygen saturation < 94% or systolic blood pressure < 90 mm Hg for 30 minutes or more), gunshot wound, pregnancy, unknown time of injury, normal result on a computed tomographic scan of the head, or family refusal to consent to the study. Patients were prospectively randomized to hypothermia or normothermia study groups as part of an ongoing trial of therapeutic hypothermia (Marion et al., 1997).
Neurointensive care
All patients were intubated and mechanically ventilated. Invasive monitors included intraventricular, jugular venous, and arterial catheters. The management goals for all patients included maintenance of cerebral perfusion pressure > 70 mm Hg by maintaining MABP between 90 and 110 mm Hg, and intracranial pressure < 20 mm Hg. Intracranial hypertension was treated with pharmacologic paralysis and sedation. If unsuccessful, ventricular CSF was drained intermittently and bolus intravenous infusions of mannitol were used, followed by hyperventilation and barbiturates if no mass lesion was found on computed tomography scan.
Study protocol
The patients randomized to the hypothermia group were cooled immediately after enrollment in the study using cooling blankets and nasogastric lavage with iced saline. Once the patients' rectal temperatures reached 33°C, they were maintained between 32° and 33°C for 24 hours, then allowed to gradually increase over the next 12 hours to temperatures of 37° to 38.5°C. The temperatures of patients in the normothermic group were maintained between 37° and 38.5°C during the entire 5-day monitoring period.
Measurement of CSF quinolinic acid
Cerebrospinal fluid samples were taken from the ventricular catheter between 1 and 120 hours after TBI and were stored at −70°C. Quinolinic acid concentration was measured by gas chromatography/mass spectroscopy as previously described (Heyes and Markey, 1988). Aliquots of CSF were mixed with 200 μL of deionized water containing 30 pmol of [13C7]-quinolinic acid as internal standard and freeze-dried overnight. Quinolinic acid and labeled quinolinic acid were derivatized to their dihexafluoroisopropanol esters, washed with 500 μL of water, and extracted into 100 to 3000 μL of heptane. Extracts were injected directly (1 or 2 μL) onto a 1-m deactivated silica precolumn attached to a 15-m DB5 analytical column (J & W Scientific, Folsom, CA, U.S.A.; 110°C isothermal). Quinolinic acid was quantified using a Hewlett-Packard Model 5988 quadruple mass spectrometer operated in the electron capture negative chemical ionization mode with methane as reagent gas. The molecular anions of quinolinic acid (m/z 467) and [13C6]- quinolinic acid (m/z 473) were monitored, and each peak area at the appropriate retention time was quantified.
Statistical analysis
Cerebrospinal fluid samples were prospectively grouped into 12-hour epochs for comparison. If there was more than one sample per epoch, the values were averaged to produce a single value per patient in a given epoch. Because our data consisted of repeated observations on individuals, statistical analysis was performed using a generalized linear model that controls for the correlation between individuals (Rosner, 1984). This model was used to assess the relationship between quinolinic acid concentrations and clinical characteristics including age, gender, initial GCS score, and treatment (hypothermia versus normothermia), as well as mortality. Univariate and multivariate analyses were performed. In addition, a multiple regression model was used to assess the relationship between CSF quinolinic acid and mortality.
RESULTS
Demographics of the patient population are provided in Table 1. From the evaluation at 12 months after trauma, 26 of the 39 patients survived. Two patients were lost to follow-up and were categorized on the basis of their discharge evaluation. Of the 39 patients from whom CSF was available for analysis, 23 had been randomized to the normothermia group and 16 to the hypothermia group. Between 2 and 10 CSF samples were assessed per patient. A total of 230 samples were assessed with approximately 80% obtained during the first 3 days after TBI because CSF became less available at delayed time points owing to patient death or ventricular catheter removal. However, of the 39 patients, 19 had samples taken beyond 3 days after injury. Normal levels of quinolinic acid in CSF have been reported as less than 50 nmol/L (Heyes et al., 1995). Cerebrospinal fluid quinolinic acid concentration was 32 ± 8 nmol/L (mean ± SEM) in the first 12 hours after injury, with only one value greater than 75 nmol/L in our samples for this epoch. Cerebrospinal fluid levels of quinolinic acid increased steadily after 12 hours and were maximal at 72 to 83 hours, 463 ± 128 nmol/L (Fig. 1).
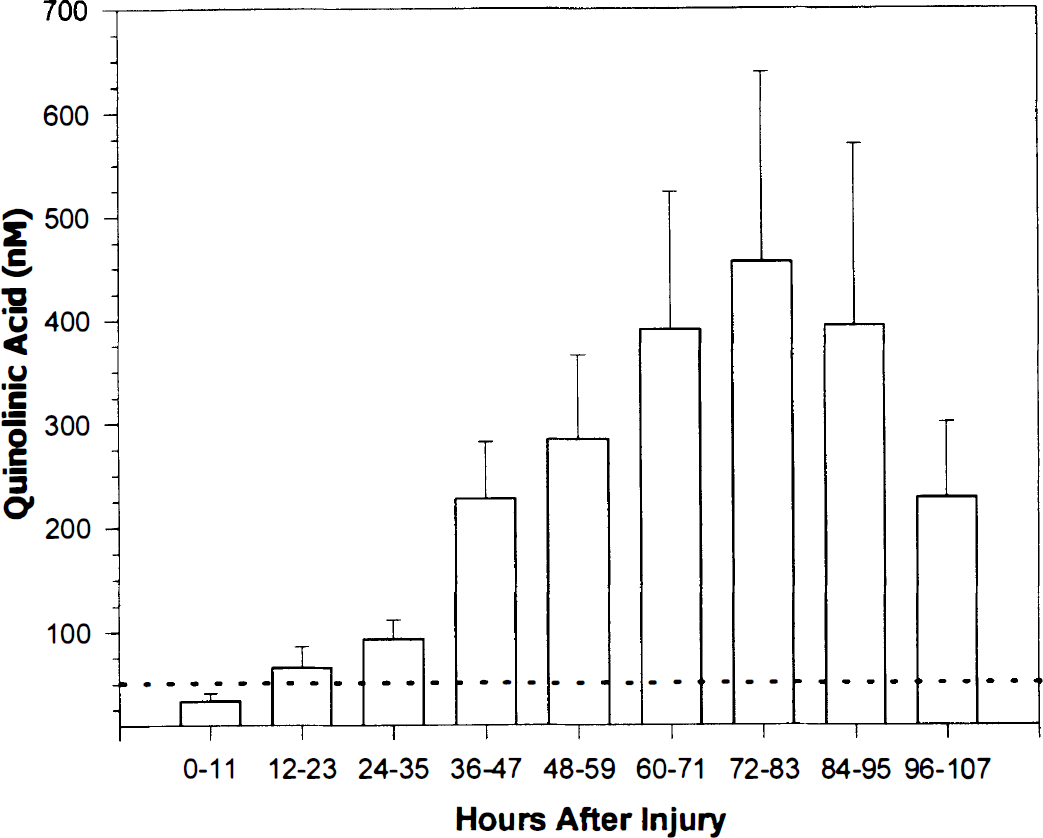
Quinolinic acid concentration (nM) in ventricular CSF versus hours after traumatic brain injury (TBI) for all patients. There was a powerful association between CSF quinolinic acid and time after injury (P < 0.0001). Cerebrospinal fluid quinolinic acid concentration was maximal at 72 to 83 hours. The dotted line represents the upper limit of normal for CSF quinolinic acid (50 nM). Data are mean + SEM.
Clinical and demographic information on the 39 patients in this study
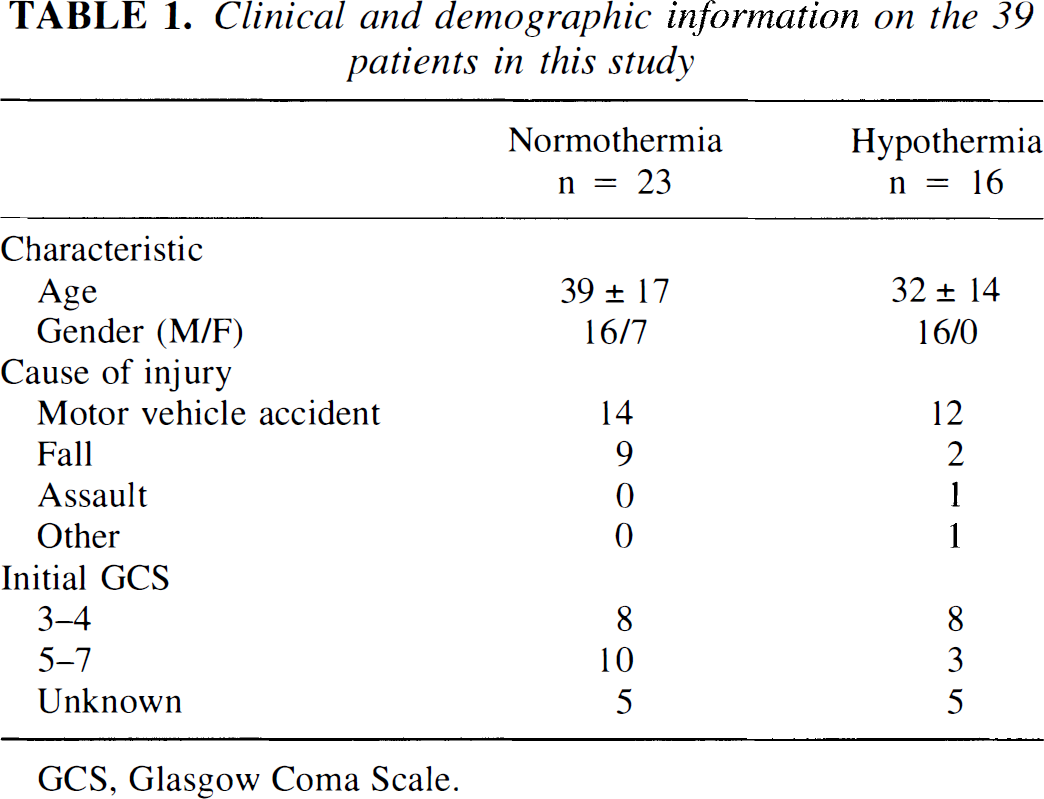
GCS, Glasgow Coma Scale.
The univariate analyses indicated that age, gender, initial GCS score, and treatment (hypothermia versus normothermia), were not associated with quinolinic acid concentration. There was, however, a powerful association between time after TBI and increased CSF quinolinic acid (P < 0.0001) (Table 2). When all variables were entered into a multivariate regression model, again only time was associated with increased CSF quinolinic acid. A multiple regression model was used to assess the difference in quinolinic acid between those who lived and those who died, while controlling for the time that quinolinic acid was measured. This revealed that patients who died had higher levels of quinolinic acid versus survivors (P = 0.003) after controlling for the effect of time (Fig. 2).
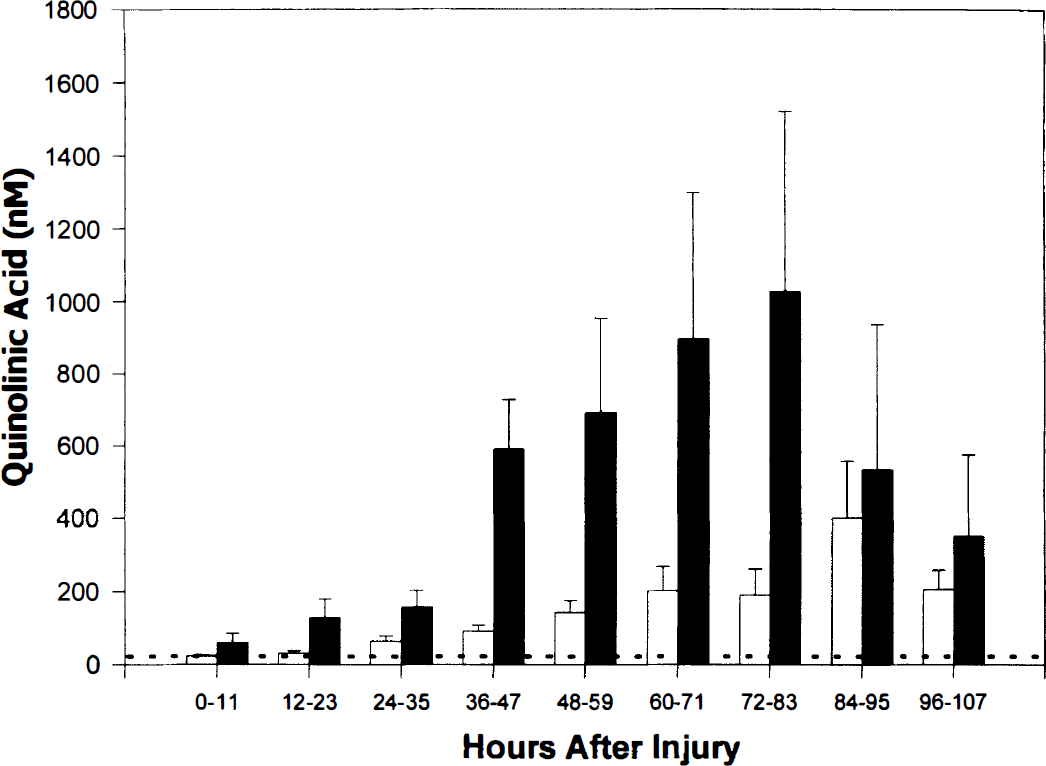
Quinolinic acid concentration (nM) in ventricular CSF versus hours after TBI for survivors (□) and nonsurvivors (■). Non-survivors had higher levels of quinolinic acid versus survivors (P = 0.003). The dotted line represents the upper limit of normal for CSF quinolinic acid (50 nM). Data are mean + SEM.
Univariate and multivariate regression for CSF quinolinic acid and clinical parameters
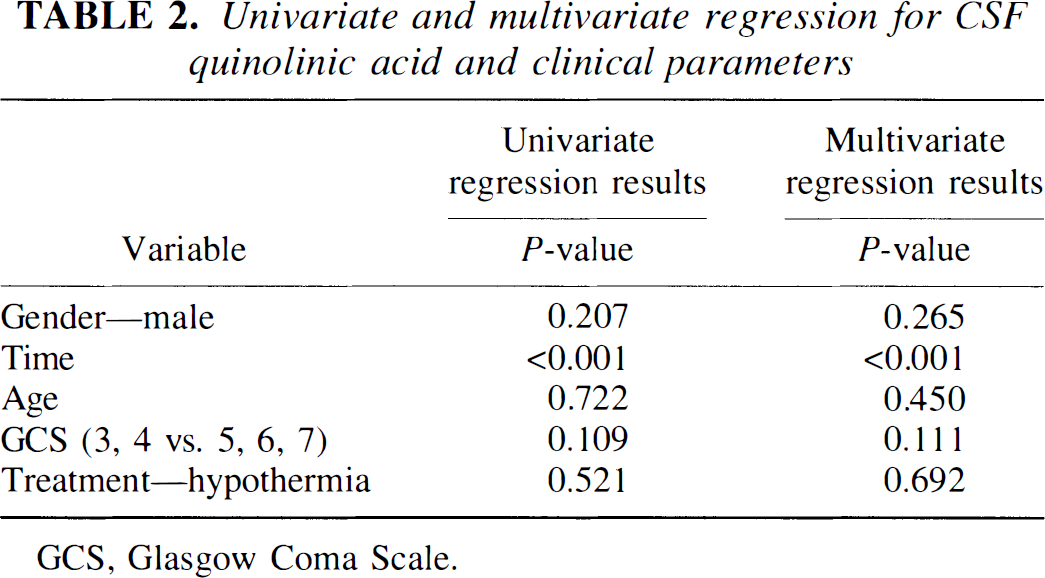
GCS, Glasgow Coma Scale.
Despite the positive effect of hypothermia on outcome previously reported in these patients (Marion et al., 1997), multivariate analysis revealed no difference in quinolinic acid levels between groups. Individual values of CSF quinolinic acid in patients in the normothermia and hypothermia groups at all sampling times after injury are shown in Fig. 3.
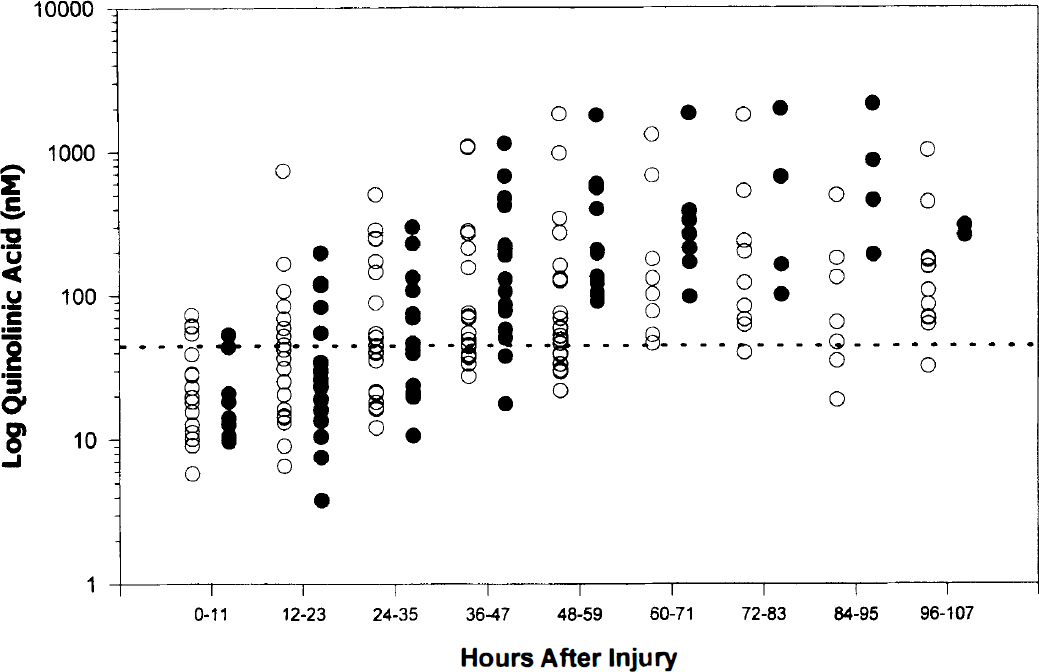
Log plot of quinolinic acid concentration (nM) in ventricular CSF versus hours after TBI for patients in normothermia (37°C, ○) and hypothermia (32°C for 24 hours, •) treatment groups. There was no effect of hypothermia on CSF quinolinic acid concentration. The dotted line represents the upper limit of normal for CSF quinolinic acid (50 nM). Because of the limited sample size at delayed times after injury in the hypothermia group, a log plot of all individual data points is shown.
DISCUSSION
This is the first report of quinolinic acid in CSF after TBI in humans. Immediately after trauma, CSF quinolinic acid levels were typically normal, but then increased by a factor of 5 to 50 during a period of 2 to 4 days. The time course of this increase mirrors that of infiltration of macrophages into the injured brain (Balasingam and Yong, 1996; Holmin et al., 1997). Furthermore, it has been previously shown in vitro that quinolinic acid is produced by macrophages (Heyes et al., 1992) and microglia (Heyes et al., 1996) stimulated by interferon-γ. Central nervous system trauma initiates release of inflammatory stimuli, which may induce indolamine-2,3-dioxygenase (Blight et al., 1993), the rate-limiting enzyme in the kynurenine pathway. This would lead to increased production of quinolinic acid from tryptophan, which occurs in conditions of CNS inflammation (Heyes et al., 1995). Thus, infiltrating macrophages and possibly activated resident microglia are likely sources of quinolinic acid in human TBI (Heyes et al., 1992, 1993).
The neurotoxic effect of quinolinic acid is purported to be caused by its agonist effect at the NMDA receptor (Stone, 1993). Quinolinic acid is a potent agent for initiating seizures. As little as 4 nmol quinolinic acid injected directly into the hippocampus of unanesthetized rats will produce EEG activity consistent with seizures. Models of neurologic degeneration have been developed using low-dose intrastriatal (Bazzett et al., 1994; Susel et al., 1991) or intraventricular (Misztal et al., 1996a; Yamada et al., 1990) infusion of quinolinic acid. These models produce measurable learning deficits. Quinolinic acid is increased after experimental spinal cord injury, peaking in the lesion core at 12 days (Blight et al., 1995). The quinolinic acid synthesis inhibitor, 4-chloro-3-hydroxyanthranilate-3,4-dioxygenase, attenuated quinolinic acid accumulation in spinal cord after injury and reduced the severity of functional deficits, suggesting a substantial contribution of quinolinic acid to the development of secondary damage in CNS trauma (Blight et al., 1995). Saito et al. (1993) also reported accumulation of quinolinic acid in the brains of gerbils after transient ischemia.
Neurotoxicity has been reported at quinolinic acid concentrations as low as 100 nmol/L (Giulian et al., 1993). All but 5 of our 39 patients had at least one CSF sample with a measured quinolinic acid level greater than 100 nmol/L, and several had levels greater than 1,000 nmol/L. In comparison, other human studies have shown the highest CSF quinolinic acid levels in CNS infection, with levels of 500 to 5,000 nmol/L; high levels are also found in patients after intracranial hemorrhage (Heyes et al., 1995; Heyes, 1993). Consistent with a putative neurotoxic role, we found a significant association between mortality and increased CSF quinolinic acid concentrations after TBI.
It may be important that the level of this excitotoxin gradually increased and remained elevated for a prolonged time—more than 5 days in some patients. In fact, the subacute peak of quinolinic acid in CSF 3 days after TBI in these patients may underestimate the true duration of quinolinic acid production. Several severely injured patients died in the initial 3 days, and ventricular catheters were maintained in place more than 5 days after trauma in a limited number of patients. Thus, the duration of elevated levels of quinolinic acid after TBI in humans is not yet known. Peak tissue levels of quinolinic acid occurred at 12 days after spinal cord injury in guinea pigs (Blight et al., 1995). If quinolinic acid plays a role in the development of secondary damage after TBI in humans, this suggests a rationale for trials of therapies directed at excitotoxic mechanisms, even at relatively delayed times after injury. It also suggests the possibility that for interventions such as NMDA antagonists or inhibitors of quinolinic acid synthesis, prolonged treatment may be required. This concept is supported in studies by Dietrich et al., (1995) in a model of transient forebrain ischemia in rats showing improved efficacy using prolonged versus short-term treatment with MK-801. Similarly, Misztal et al. (1996b) showed that administration of NMDA-receptor antagonists prevented the learning deterioration seen with long-term intraventricular infusion of quinolinic acid.
In our study there was no correlation between CSF quinolinic acid concentrations and treatment with 24 hours of moderate hypothermia (32°C), despite the suggested benefit of hypothermia on both CSF interleukin-1β concentration and outcome for these patients (Marion et al., 1997). This lack of effect may represent a failure of the early transient application of hypothermia to influence the delayed nature of quinolinic acid production. The failure of hypothermia to attenuate CSF quinolinic acid levels despite a beneficial effect on outcome could also relate to the fact that quinolinic acid was increased predominantly in patients with mortality from severe injuries, whereas the benefit of hypothermia was largely confined to the subset of patients with GCS between 5 and 7 (Marion et al., 1997). Alternatively, quinolinic acid may not play a major role in the development of secondary damage after TBI in humans.
There are several possible limitations of this study. Because plasma levels of quinolinic acid were not measured, we cannot prove that the increase in CSF quinolinic acid concentration was caused by production in the CNS. We also were unable to measure brain parenchymal levels in these patients, and it is possible that another source of quinolinic acid, such as the meninges, could contribute to the large increases seen in CSF. The issue of local versus systemic production has been studied extensively in animal models of CNS injury, and in these models systemic quinolinic acid production is not responsible for the increase found in CSF (Heyes and Morrison, 1997). Because CSF quinolinic acid is likely to have formed in brain parenchyma, CSF measurements may actually underestimate parenchymal concentrations of quinolinic acid (Heyes et al., 1996; Heyes and Morrison, 1997).
It is not a coincidence that this first report of increased quinolinic acid associated with TBI is from human subjects. This likely results from the fact that the vast majority of animal studies of TBI are performed in rat and mouse models. Rats and mice do not produce an appreciable quantity of quinolinic acid. Macrophages from rats and mice preferentially produce nitric oxide, rather than quinolinic acid, in response to immune stimulants, and nitric oxide is a potent inhibitor of the rate-limiting enzyme in the kynurenine pathway to quinolinic acid synthesis, indolamine-2,3-dioxygenase (Thomas et al., 1994). In contrast, indolamine-2,3-dioxygenase is actually upregulated in guinea pig spinal cord after trauma (Blight et al., 1993). Because quinolinic acid is detected in large quantities in human CSF after TBI, it will be important to study its effects in appropriate animal models in species that produce quinolinic acid (i.e., guinea pigs, gerbils, or primates). An important role has already been reported for quinolinic acid in the pathology of spinal cord injury in guinea pigs (Blight et al., 1995).
In conclusion, the excitatory neurotoxin, quinolinic acid, markedly increases in CSF after severe TBI in humans and is strongly associated with mortality. This unique molecule appears to represent a bridge between inflammatory and excitotoxic mechanisms of damage. Additional studies are needed to address the source of production and the duration of effect, and also to determine the quantitative contribution of quinolinic acid to secondary damage after TBI.
Footnotes
Acknowledgments
The authors thank Linda Amick and Marci Provins for assistance with manuscript preparation, and Lisa Goetz for editorial assistance.