
Abstract
Inflammatory responses are accountable for secondary injury induced by acute ischemic stroke (AIS). Previous studies indicated that O-GlcNAc modification (O-GlcNAcylation) is involved in the pathology of AIS, and increase of O-GlcNAcylation by glucosamine attenuated the brain damage after ischemia/reperfusion. Inhibition of β-N-acetylglucosaminidase (OGA) with thiamet G (TMG) is an alternative option for accumulating O-GlcNAcylated proteins. In this study, we investigate the neuroprotective effect of TMG in a mouse model of experimental stroke. Our results indicate that TMG administration either before or after middle cerebral artery occlusion (MCAO) surgery dramatically reduced infarct volume compared with that in untreated controls. TMG treatment ameliorated the neurological deficits and improved clinical outcomes in neurobehavioral tests by modulating the expression of pro-inflammatory and anti-inflammatory cytokines. Additionally, TMG administration reduced the number of Iba1+ cells in MCAO mice, decreased expression of the M1 markers, and increased expression of the M2 markers in vivo. In vitro, M1 polarization of BV2 cells was inhibited by TMG treatment. Moreover, TMG decreased the expression of iNOS and COX2 mainly by suppressing NF-κB p65 signaling. These results suggest that TMG exerts a neuroprotective effect and could be useful as an anti-inflammatory agent for ischemic stroke therapy.
Introduction
Cerebral ischemic stroke leads to a loss of neuronal function, damage to multiple tissues, and inflammatory responses. 1 However, deterring neuroinflammation during the course of stroke distinctly improves the likelihood of a better outcome.2,3 Therefore, controlling harmful inflammatory responses is a promising therapeutic approach for victims of stroke. Activation of microglia/macrophages plays a crucial part in initiating the immune response. 4 Once activated, microglia undergoes marked morphologic and genetic changes. During that process, microglia/macrophages become polarized to one of two mutually antagonistic subpopulations, either the classical M1 phenotype, which produces pro-inflammatory cytokines that mediate neuronal damage in the ischemic brain, or the alternative M2 phenotype, which secretes anti-inflammatory cytokines that suppress immune responses and contribute to recovery after stroke. However, how these phenotypes shift is a topic trapped in controversy.
O-linked β-N-acetylglucosamine (O-GlcNAc) modification, which is widely expressed on Ser/Thr residues of proteins, is closely linked with protein stability and functions as well in cellular cascades. A unique pair of enzymes that catalyze O-GlcNAc cycling are O-linked N-acetylglucosaminyltransferase (OGT) and the antagonistic glucosidase β-N-acetylglucosaminidase (OGA). 5 Dysregulation of O-GlcNAc modification is a factor in numerous diseases, including neurodegenerative afflictions, diabetic mellitus, cancer, and cardiovascular maladies.6,7 Additionally, an increase of O-GlcNAcylation is known to be tightly linked to the stress response. 8 Accumulated evidence suggests that O-GlcNAc modification is closely associated with vascular ischemic/reperfusion injury. 9 However, other studies showed that an increase of O-GlcNAc levels improved cardiac function and perfusion of critical organ systems and also reduced the amount of inflammatory cytokines in plasma.10–13 Treatment with glucosamine (GlcN) and O-(2-acetamido-2-deoxy-D-glucopyranosylidene)amino N-phenylcarbamate (PUGNAc) during resuscitation dramatically decreased the trauma-hemorrhage-induced increase of TNF-α, IL-6, NF-κB, IκB-α phosphorylation, and NF-κB DNA binding activity in cardiac tissues. In injured arteries, GlcN and PUGNAc also inhibited the infiltration of neutrophils and monocytes and hindered acute inflammatory responses in rat carotid arteries. Possibly, then, augmenting O-GlcNAc modification of proteins in the vasculature represents a novel anti-inflammatory and vasoprotective mechanism. 14 Interestingly, others found that increases of O-GlcNAcylation suppressed TNF-α-induced vascular dysfunction via suppression of iNOS expression. 15 Similarly, Liu et al. documented a correlation between activated O-GlcNAcylation post stroke and functional recovery in mouse brain. 16 Moreover, administration of glucosamine to increase O-GlcNAcylation levels in the brain was reported to suppress inflammatory responses in a rat model of ischemic stroke. 17
In the light of existing evidence, we speculated that ameliorating brain injury and promoting functional repair could be attributed to the elevation of O-GlcNAcylation. Thiamet G (TMG) is a highly selective, blood–brain barrier-permeable inhibitor of OGA. Furthermore, treatment with TMG in cell lines and animals caused a notable accumulation of O-GlcNAcylation on nucleoplasm and cytoplasm proteins.18,19 Subsequently, inhibitors of OGA provided demonstrable cardioprotective functions. 20 However, the effect of OGA inhibition on increasing O-GlcNAc modification has not been explored in a murine model of stroke. Therefore, we investigated the neuroprotective effect and mechanism of TMG by evaluating inflammatory responses and microglia/macrophage polarization in mice with middle cerebral artery occlusion (MCAO).
Materials and methods
MCAO in mice
All the experiments were performed according to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and approved by the Animal Experiments Ethical Committee of Tianjin Medical University General Hospital. The report of animal experiments is in compliance with the ARRIVE guidelines (www.nc3rs.org.uk). The mice were treated during a 12-h light–12-h dark cycle and temperature control with water and food provided ad libitum. These conditions were in effect for one week before the start of experiments.
A total of 216 male C57BL/6 mice (aged 10–12 weeks) were randomly separated into four groups by lottery drawing: sham operation (Sham), preventative treatment (MCAO + TMG-P), therapeutic treatment (MCAO + TMG-T), or placebo administration (MCAO). Sample size was determined by a sample size calculation (α = 0.05, β = 0.2, a 30% decrease of infarct volume [TMG-treated group vs. MCAO group] and a 20% standard deviation of the mean within each group were estimated) and by our own previous experiences, resulting in a sample size of nine mice per group. Focal cerebral ischemia was simulated by occlusion of the left middle cerebral artery (MCAO) based on the method introduced by Longa et al. 21 The stroke surgeon was blind to the group identity of each mouse. Mice were anesthetized with chloral hydrate (30 mg/kg, intraperitoneally), then received a midline neck incision to expose the left external carotid artery and common carotid artery, which were isolated and ligated. Into the internal carotid artery, a monofilament line (Xinong, Beijing, China) was inserted (9–10 mm) through the common carotid artery until resistance was felt, which indicated occlusion of the blood flow from the posterior cerebral artery and the anterior artery. One hour after occlusion, the monofilament line was removed to restore blood flow. The body temperature of these mice was maintained at 37.0 ± 0.5℃ during surgery, and they were kept in a well-ventilated room at 25 ± 3℃ in individual cages, with water provided until they fully regained consciousness. We used Buprenorphine (Sigma, Saint Louis, USA) 0.03 mg/kg intraperitoneally (i.p.), every 12 h for 24 h for postop analgesia. All efforts were made to minimize animal suffering and reduce the number of animals used. The mortality rate of mice subject to MCAO in control group was ∼10% and the TMG groups were ∼7%, in the sham operation group, there was no death.
TMG treatment
TMG (≥98%, purified by HPLC) (Merck Millipore, Darmstadt, Germany) was freshly resuspended in PBS. Each mouse in the placebo administration group (MCAO) received an intraperitoneal injection of PBS; mice in the preventative treatment (MCAO + TMG-P) group were intraperitoneally injected with TMG at a dose of 20 mg/kg per day for three days before the induction of MCAO; mice in therapeutic treatment (MCAO + TMG-T) group were given an intraperitoneal injection of TMG at 0.5 h after MCAO and continuous injections once a day until the third day after surgery. Sham-operated mice (Sham) were also included as controls. Doses and injection time points were optimized based on previous studies and preliminary experiments in our laboratory.22,23
Neurobehavioral testing
All mice received three days of training before neurobehavioral testing. Neurological functions of each mouse were evaluated at 24 h after MCAO. The modified neurological severity score (mNSS), adhesive-removal test, foot-fault test and inclined plane test were used to detect motor coordination, feeling and balance functions, respectively, of the mice.24–27 The person performing these evaluation tests was blinded to the treatment each mouse received.
Infarct volume analysis
Mice from each group were anesthetized with chloral hydrate (30 mg/kg, intraperitoneally), and perfused intracardially with 4℃ PBS for 5 min. The brains of ischemia/reperfusion (I/R) mice were immediately sliced into coronal sections (2 mm thick) from the frontal tips using scalpels. The sections were stained with TTC (Sigma, St. Louis, MO, USA) and immersed in normal saline at 37℃ for 20 min. Brain sections were then fixed in 4% paraformaldehyde at 4℃ overnight before being photographed. Intact mitochondrial function can be stained deep red in viable tissue, whereas infarcts remain unstained. The infarcted regions in each section were evaluated by using Image-Pro® Plus v 4.0 image analysis software (Media Cybernetics, Washington, DC, USA). Statistics for each total infarct volume were calculated by combining the infarct volume of each section. The infarcted area and the percentage of brain swelling for edema correction were determined by subtracting the area of healthy tissue in the ipsilateral hemisphere from the area of the contralateral hemisphere on each section. Infarct volumes were calculated by integrating infarcted areas on each brain slice as quantified with a computer-assisted image analyzer, as previously described. 24
Immunofluorescence and cell counting
Immunofluorescence was performed on 8-µm frozen sections as previous described. 28 Primary antibodies included rabbit anti-Iba1 (Wako, Tokyo, Japan), rat anti-CD16/32 (BD Pharmingen, CA, USA), goat anti-CD206 (Santa Cruz, TX, USA), rabbit anti-iNOS (Cell Signaling Technology, Danvers, MA, USA), rabbit anti-COX2 (Cell Signaling Technology, Danvers, MA, USA), and mouse anti-O-GlcNAc (RL2) (Abcam, Cambridge, UK). DAPI solution (Thermo Fisher, Waltham, MA, USA) was used to stain nuclei. All images were processed with Image J (NIH Image, Bethesda, MD, USA) for counting of recognized cells. The means were calculated from three randomly selected microscopic fields in peri-infarct cortex of each section, respectively, and three consecutive sections were analyzed for each brain (three fields per section × three sections per mouse). Data are expressed as mean numbers of cells per visual field or mean fluorescence intensity (MFI).
Quantitative real-time PCR
Total RNA was extracted from the ischemic hemisphere using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The concentration of RNA was quantified by ultraviolet spectrophotometry at 260/280 nm. cDNA was transcribed using Trans-Script First-Strand cDNA Synthesis SuperMix Kit (Transgen, Beijing, China) in accordance with its explanatory memorandum. PCR was performed on the Opticon 2 Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA) using corresponding primers and SYBR gene PCR Master Mix (Roche, Basel, Switzerland). Samples were run in duplicate and normalized to GAPDH using the 2–ΔΔCt method. Expression levels of the mRNAs were then reported as fold changes vs. control. Primers used in this work were as follows:
GAPDH: (F) TCATTGACCTCAACTACATGGT (R) CTAAGCAGTTGGTGGTGCAG iNOS: (F) ACTTCCGAGTGTGGAACTCG (R) TGGCTACTTCCTCCAGGATG COX-2: (F) GCTGTACAAGCAGTGGCAAA (R) GTCTGGAGTGGGAGGCACT IL-1β: (F) GGAGAAGCTGTGGCAGCTA (R) GCTGATGTACCAGTTGGGGA IL-6: (F) CCGGAGAGGAGACTTCACAG (R) TGGTCTTGGTCCTTAGCCAC TNF-a: (F) GACCCTCACACTCAGATCAT (R) TTGAAGAGAACCTGGGAGTA MCP-1: (F) ACGCTTCTGGGCCTGTTGTT (R) CCTGCTGCTGGTGATTCTCT CD16: (F) TTTGGACACCCAGATGTTTCAG (R) GTCTTCCTTGAGCACCTGGATC CD32: (F) AATCCTGCCGTTCCTACTGATC (R) GTGTCACCGTGTCTTCCTTGAG CD206: (F) CAAGGAAGGTTGGCATTTGT (R) CCTTTCAGTCCTTTGCAAGC Arg-1: (F) TCACCTGAGCTTTGATGTCG (R) CTGAAAGGAGCCCTGTCTTG YM-1: (F) CGAGGTAATGAGTGGGTTGG (R) CACGGCACCTCCTAAATTGT IL-10: (F) AAATAAGAGCAAGGCAGTGG (R) GTCCAGCAGACTCAATACACA TGF-β: (F) TGCGCTTGCAGAGATTAAAA (R) CGTCAAAAGACAGCCACTCA
Western blot analysis
Ischemic brain tissues were pooled and homogenized by sonication in ristocetin-induced platelet aggregation (RIPA) buffer containing protease and phosphatase inhibitors (Complete Protease Inhibitor Cocktail and PhosSTOP Phosphatase Inhibitor Cocktail; both from Roche), and culture cells were also treated with RIPA buffer containing protease and phosphatase inhibitors. For cytoplasmic and nuclear protein extraction, ischemic brain tissue or culture cells were first mixed with Buffer A (KeyGene Biotech, Beijing, China) and resuspended using a shaker. After placement of the cells on ice for 10 min, Buffer B (KeyGene Biotech, Beijing, China) was added and mixed well. Cytoplasmic proteins were obtained by centrifuging with 16,000 r/min at 4℃ for 30 s. Subsequently, we added pre-cold Buffer C (KeyGene Biotech, Beijing, China) to the precipitate and mixed well. After icing for 30 min, nuclear proteins were harvested by centrifuging with 16,000 r/min at 4℃ for 10 min. Protein concentrations were assessed using a Protein Assay Kit (Beyotime, Hangzhou, China), and 10 µg protein was loaded per lane. Proteins were separated on a 10% gel at 80 V and transferred onto a nitrocellulose membrane for 1.5 h at 4℃ with a current of 220 mA. Membranes were blocked for 1 h at room temperature in 5% fat-free milk, incubated with primary antibody overnight at 4℃, washed three times for 20 min each at room temperature, incubated with an HRP-coupled secondary antibody (1:5000; Transgen) for 2 h, and washed three times for 20 min each at room temperature. Finally, the images were captured by densitometry (Bio-Rad, Hercules, CA, USA). All incubations were performed in TBS buffer with 0.1% Tween-20 and 5% fat-free dry milk powder. The following primary antibodies were used: rabbit anti-p65 (1:500; Abcam); and mouse anti-GAPDH (1:1000; Sigma).
Enzyme-linked immunosorbent assay (ELISA)
Cytokines in supernatants from ischemic brain tissues were measured with a quantitative ELISA by using a Multi-Analyte ELISArray Kit (Qiagen, Duesseldorf, Germany) as previously described. 24 The kit detects 12 cytokines simultaneously (interleukin [IL]-1α, IL-1β, IL-2, IL-4, IL-6, IL-10, IL-12, IL-17 A, tumor necrosis factor [TNF]-α, interferon-γ [IFN-γ], granulocyte colony-stimulating factor [G-CSF], and granulocyte-macrophage colony-stimulating factor [GM-CSF]). We used the kit applying an ELISA protocol conducted under uniform conditions according to the manufacturer’s instructions. Assays were performed in duplicate on individual mouse brains, and the results were expressed as mean optical density (OD) values.
Cell culture and treatment
BV2 cells, which are a murine cell line derived from primary microglial cell cultures and immortalized by infection with a v-raf/v-myc oncogene carrying retrovirus (J2), were cultivated in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Carlsbad, CA, USA), supplemented with 10% fetal bovine serum (FBS; Gibco), 100 U/mL penicillin and 100 µg/mL streptomycin (Gibco) at 37℃ in a humidified cell incubator. For lipopolysaccharide (LPS) stimulation, BV-2 cells were incubated in the culture with LPS (100 ng/mL, Sigma) in the presence and absence of TMG (200 ng/mL, Millipore) for 12 h. BV2 cells were then polarized into an M1 or M2 phenotype with LPS (100 ng/mL, Sigma) or IL-4 (20 ng/mL, PeproTech, Rocky Hill, NJ, USA), respectively, by incubation for 12 h with or without TMG.
Evaluation and statistical analysis
GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA, USA) was used. Data are presented as means ± SEM except for neurobehavioral test scores. The Mann–Whitney U test was used for comparison of clinical scores, which is depicted with median (interquartile range). The two-tailed Student’s t test was applied for comparisons between two groups. One-way or two-way ANOVA was applied with Bonferroni post hoc testing for multiple comparisons. P < 0.05 was taken as a significant difference.
Results
TMG protects against brain damage after MCAO
To assess the effect of TMG on the brain injury after focal ischemia in mice, infarct volumes and neurological deficit scores were quantified at several doses and multiple time points after MCAO. Higher doses (10–40 mg/kg) significantly reduced infarct volumes (Figure 1(a)). These decreases of infarct volume reached significance at 72 h after I/R injury, although the decreased mNSS reached significance at 24 h (Supplemental Figure 1). The following experiments were performed at a dose of 20 mg/kg TMG, and infarct volume was evaluated at 72 h after MCAO.
Effect of TMG on behavioral deficits and brain infarct volume in mice with cerebral I/R injury. Multiple tests were performed in mice that underwent 1 h of ischemia followed by 24 h or 72 h of reperfusion. (a) Titering assays were performed to compare changes of infarct volume caused by different doses of TMG. (b) Coronal brain sections were stained with 1.5% TTC. Mice were subjected to 1 h of ischemia followed by 72 h of reperfusion; the infarct area is white; (c) Quantitative analysis of brain infarct volume. (n = 9 per group) (d) mNSS were used to evaluate neuronal deficit after I/R with or without TMG treatment. (n = 9 per group) (e–g) Neurobehavioral tests, including foot-fault testing (e), adhesion-removal test (f), and inclined plane test (g) were applied to examine post-stroke motor coordination. (n = 9 per group) Sham: sham operation; MCAO: MCAO with placebo administration; MCAO + TMG-P: MCAO with TMG administration before surgery; MCAO + TMG-T: MCAO with TMG administration after surgery. Data are expressed as means ± SEM except for neurological testing, which is depicted with median (interquartile range). *P < 0.05, **P < 0.01, vs. the MCAO group.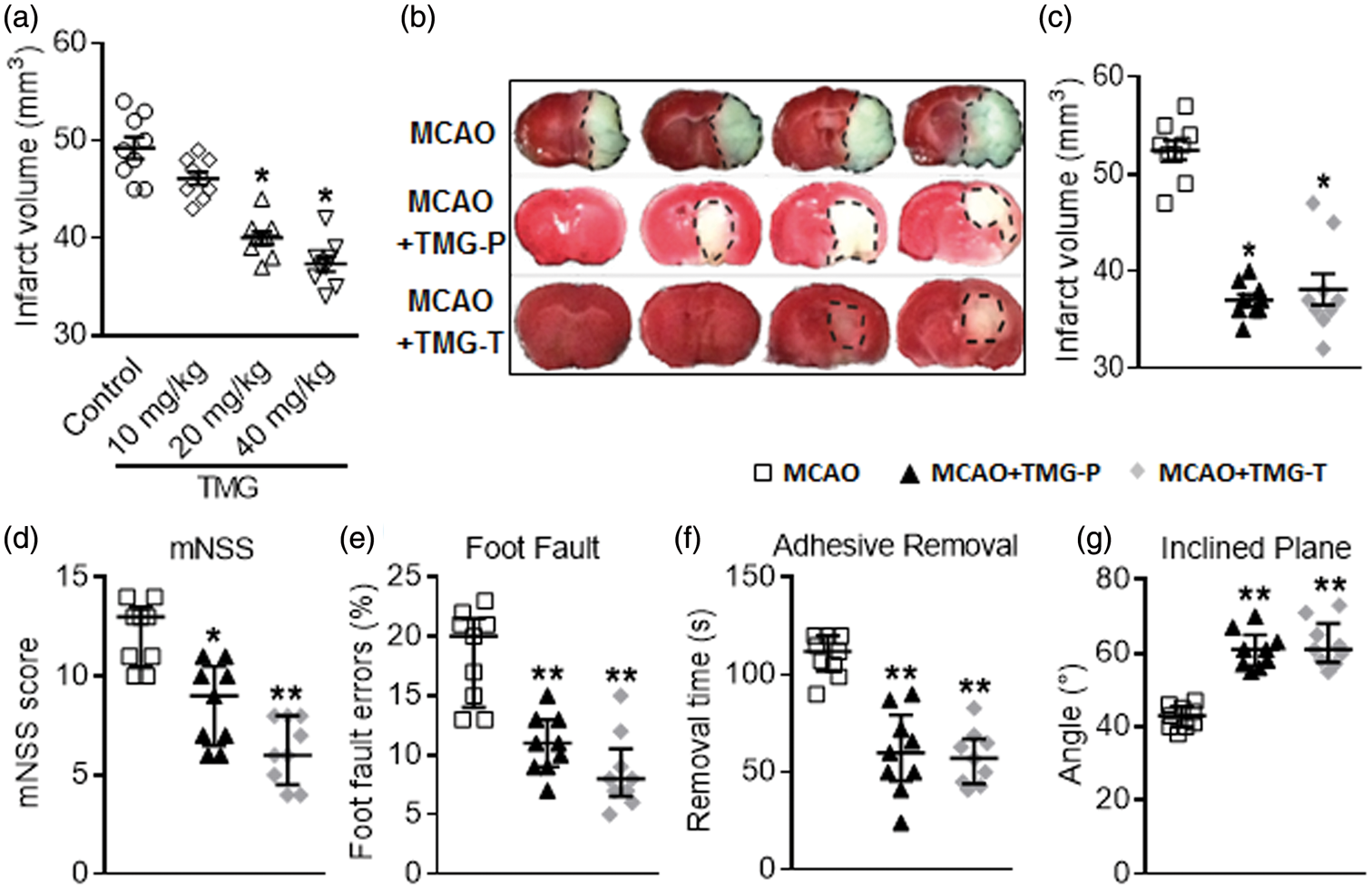
To evaluate the effect of TMG on neuronal function of mice after MCAO, we executed a variety of behavior function tests. All these tests were performed at 24 h after MCAO. Results from mNSS testing exemplified a dramatic change in the MCAO group compared to both TMG-treated groups: 9 (6.5–10.5) in the preventative treatment group vs. 13 (10.5–13.5) in the MCAO group, P < 0.05, n = 9 per group; 6 (4.5–8) in the therapeutic treatment group vs. 13 (10.5–13.5) in MCAOs, P < 0.01, n = 9 per group (Figure 1(d)). Further functional analyses with the foot-fault test, adhesion-removal test, and inclined plane test also indicated markedly improved outcomes after treatment with TMG in MCAO mice (Figure 1(e) to (g)). Together, these results support the presumption that treatment with TMG is neuroprotective in MCAO mice. Results shown in Figure 1(b) and (c) indicate that treatment with TMG before ischemic injury significantly reduced the infarct size (37.00 ± 0.60 mm3 in the preventative treatment group vs. 52.44 ± 1.00 mm3 in the MCAO group, P < 0.05, n = 9 per group). The therapeutic treatment group yielded virtually the same result (38.11 ± 1.60 mm3 vs. 52.44 ± 1.00 mm3 in the MCAO group, P < 0.05, n = 9 per group).
TMG inhibits the inflammatory response in post-ischemic brain
Inflammatory cytokines are a proven predisposing factor for neuronal injury after stroke. In this study, a panel of inflammatory cytokines, including IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-10, IL-12, IL-17A, IFN-γ, TNF-α, G-CSF, and GM-CSF were detected using ELISArray Kits and quantitative real-time PCR (qRT-PCR). As expected, expression of the pro-inflammatory cytokines IL-1α, IL-1β, and IL-6 decreased in the preventative treatment group and the therapeutic treatment group compared with that in the MCAO group. Mixed elevations of GM-CSF expression and downregulation of G-CSF occurred among the TMG-treated groups (Figure 2(a)). qRT-PCR also showed that mRNA expression of TNF-α, IL-6 and IL-1β was reduced by TMG treatment, which actually upregulated IL-10 expression (Figure 2(b) to (e)). Nevertheless, these data suggest that TMG mediated the anti-inflammatory effect in this experimental model of stroke.
Effect of TMG on inflammatory cytokines in ischemic brain. (a) The supernatants of brain tissues from mice that underwent 1 h of ischemia followed by 72 h of reperfusion were measured by ELISA. IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-10, IL-12, IL-17A, IFN-γ, TNF-α, G-CSF, and GM-CSF were included in the panel. (n = 9 per group) (b–e) qRT-PCR were used to detect mRNA expression for TNF-α (b), IL-10 (c), IL-6 (d), and IL-1β (e) in untreated controls and TMG-treated groups. (n = 9 per group) MCAO: MCAO with placebo administration; MCAO + TMG-P: MCAO with TMG administration before surgery; MCAO + TMG-T: MCAO with TMG administration after surgery. Results are presented as mean ± SEM. *P < 0.05, **P < 0.01, vs. MCAO group.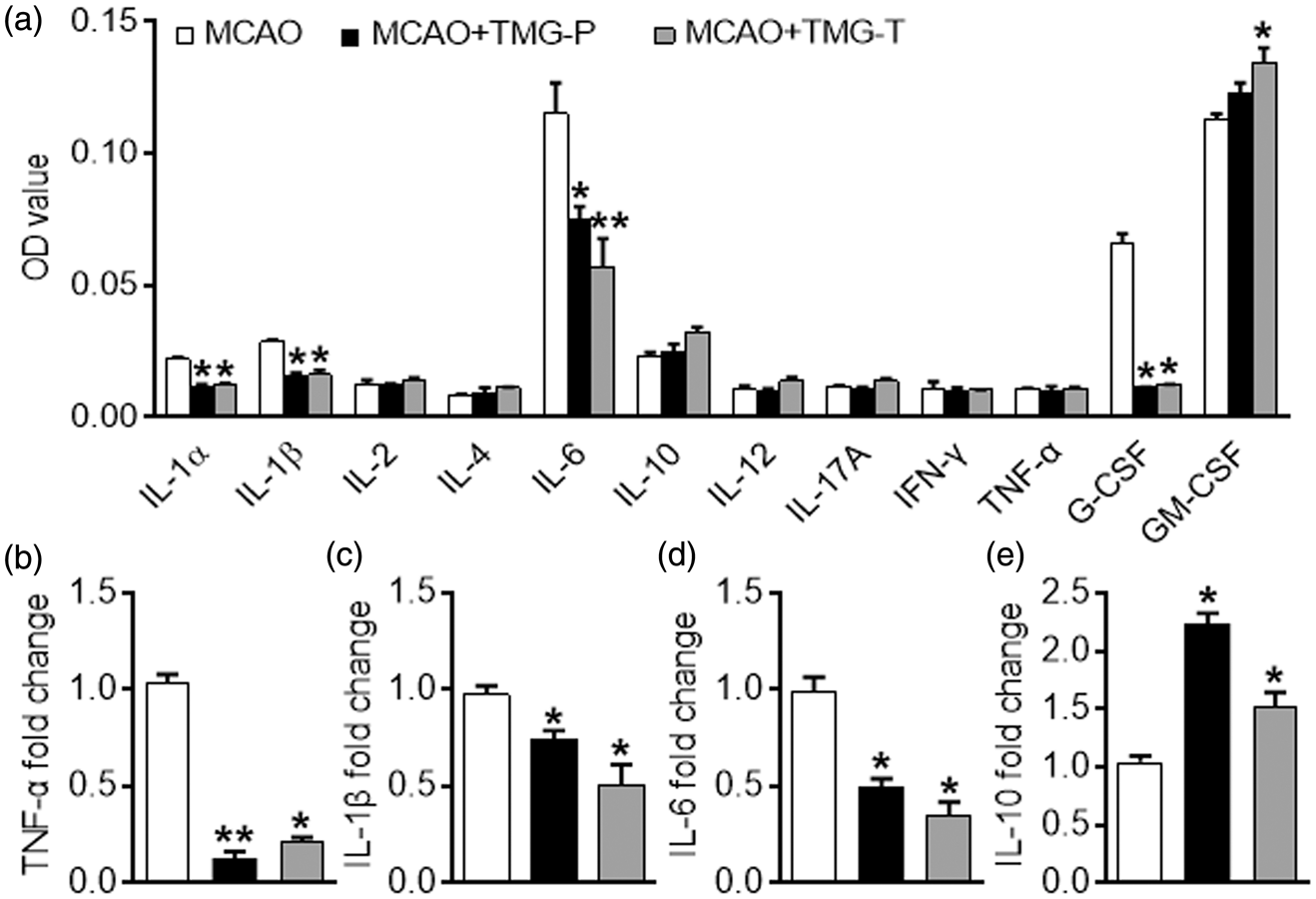
TMG decreases the number of microglia/macrophages in post-ischemic brain and suppresses its LPS-stimulated inflammatory responses
The activation of microglia/macrophage plays an essential role in the ischemic reperfusion injury after stroke. In this study, we found that the number of Iba-1+cells per field in the peri-infarct cortex was significantly reduced in therapeutic treatment and preventative treatment groups compared with that in the MCAO group (Figure 3(a) and (b), Iba-1-positive cells per field: 68.50 ± 2.31 in the preventative treatment group vs. 87.50 ± 2.01 in MCAOs, P < 0.05, n = 9 per group; 58.67 ± 2.45 in the therapeutic treatment group vs. 87.50 ± 2.01 in the MCAO group, P < 0.01, n = 9 per group). We also investigated the expression of pro-inflammatory cytokines in LPS-stimulated microglia in vitro. Notably, the mRNA expression of TNF-α, iNOS, IL-1β, IL-6, and MCP-1 was reduced in both groups to which we added TMG (Figure 3(c). This latter result was in absolute accord with the data produced in vivo. These outcomes indicate that TMG suppresses the activation of microglia in vivo and in vitro.
Effect of TMG on microglia/macrophage inflammatory responses in vivo and in vitro. (a) Representative images from triple independent experiments show the expression of Iba-1 in the peri-infarct area of cortex in all groups (upper panel); morphological characteristics in the MCAO group are amoeboid with small branches (lower panel: partial magnification of upper panel). (b) Quantitative analysis of the number of Iba-1-positive cells per visual field in the peri-infarct cortex of mice from the TMG-injected groups and untreated controls. (n = 9 per group) MCAO: MCAO with placebo administration; MCAO + TMG-P: MCAO with TMG administration before surgery; MCAO + TMG-T: MCAO with TMG administration after surgery. Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01 vs. MCAO group. (c) qRT-PCR for mRNA expression of TNF-α, iNOS, IL-1β, IL-6 and MCP-1 in BV2 cells after stimulation with LPS. (n = 9 per group) Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01 vs. Control group.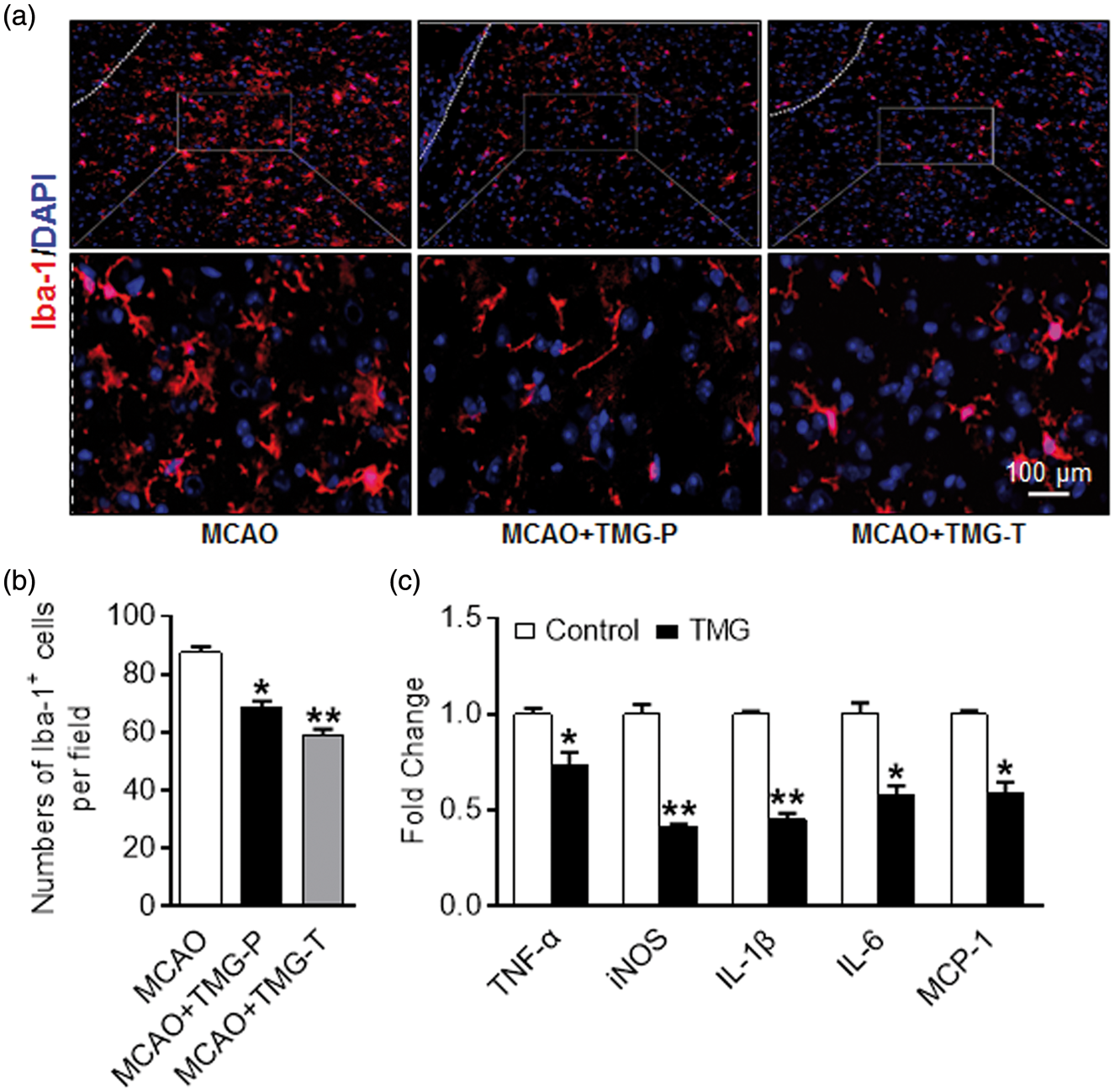
TMG promotes M2 marker expression and inhibits M1 marker expression in the ischemic brain
Activated microglia/macrophages conduct agonistic functions with differing expressions of genetic features for surface markers. As is well known, CD16 and CD32 are cell surface markers for the M1 microglial phenotype, whereas CD206 marks the M2 phenotype. The use of immunofluorescence revealed that CD16/32 co-expressed with Iba-1 in the peri-infarct cortex was reduced by treatment with TMG: 55.50 ± 3.43% in the preventative treatment group vs. 68.44 ± 1.45% in the MCAO group, P < 0.05, n = 9 per group; 52.75 ± 3.28% in the therapeutic treatment group vs. 68.44 ± 1.45% in the MCAO group, P < 0.01, n = 9 per group (Figure 4(a) and (b)). On the contrary, amounts of CD206 co-expressed with Iba-1 increased after TMG treatment: 34.75 ± 3.82% in the preventative treatment group vs. 23.75 ± 2.69% in the MCAO group, P < 0.05, n = 9 per group; 34.25 ± 3.33% in the therapeutic treatment group vs. 23.75 ± 2.69% in the MCAO group, P < 0.05, n = 9 per group (Figure 4(c) and (d)).
Co-expression of Iba-1 and M1/M2 phenotype markers. Brain slices were prepared at 72 h after MCAO. Immunofluorescent images were captured microscopically in the peri-infarct area of cortex. (a) Cortex co-stained for Iba-1 (microglia marker) (green) and CD16/32(M1 marker) (red) in the peri-infarct area. (b) Quantification of CD16/32- and Iba-1-positive cells in each group. (n = 9 per group) (c) Cortex co-stained for Iba-1 (microglia marker) (green) and CD206(M2 marker) (red) in the peri-infarct area. (d) Quantification of CD206- and Iba-1-positive cells in each group. (n = 9 per group) (e) qRT-PCR for mRNA expression of M1 cytokines (TNF-α, IL-1β, MCP-1, CD16, and CD32) on ischemic cortex. (n = 9 per group) (f) qRT-PCR for mRNA expression of M2 cytokines (Arg-1, CD206, TGF-β, IL-10, and YM-1) on the ischemic cortex in treatment groups and MCAO group. (n = 9 per group) MCAO: MCAO with placebo administration; MCAO + TMG-P: MCAO with TMG administration before surgery; MCAO + TMG-T: MCAO with TMG administration after surgery. Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01 vs. MCAO group.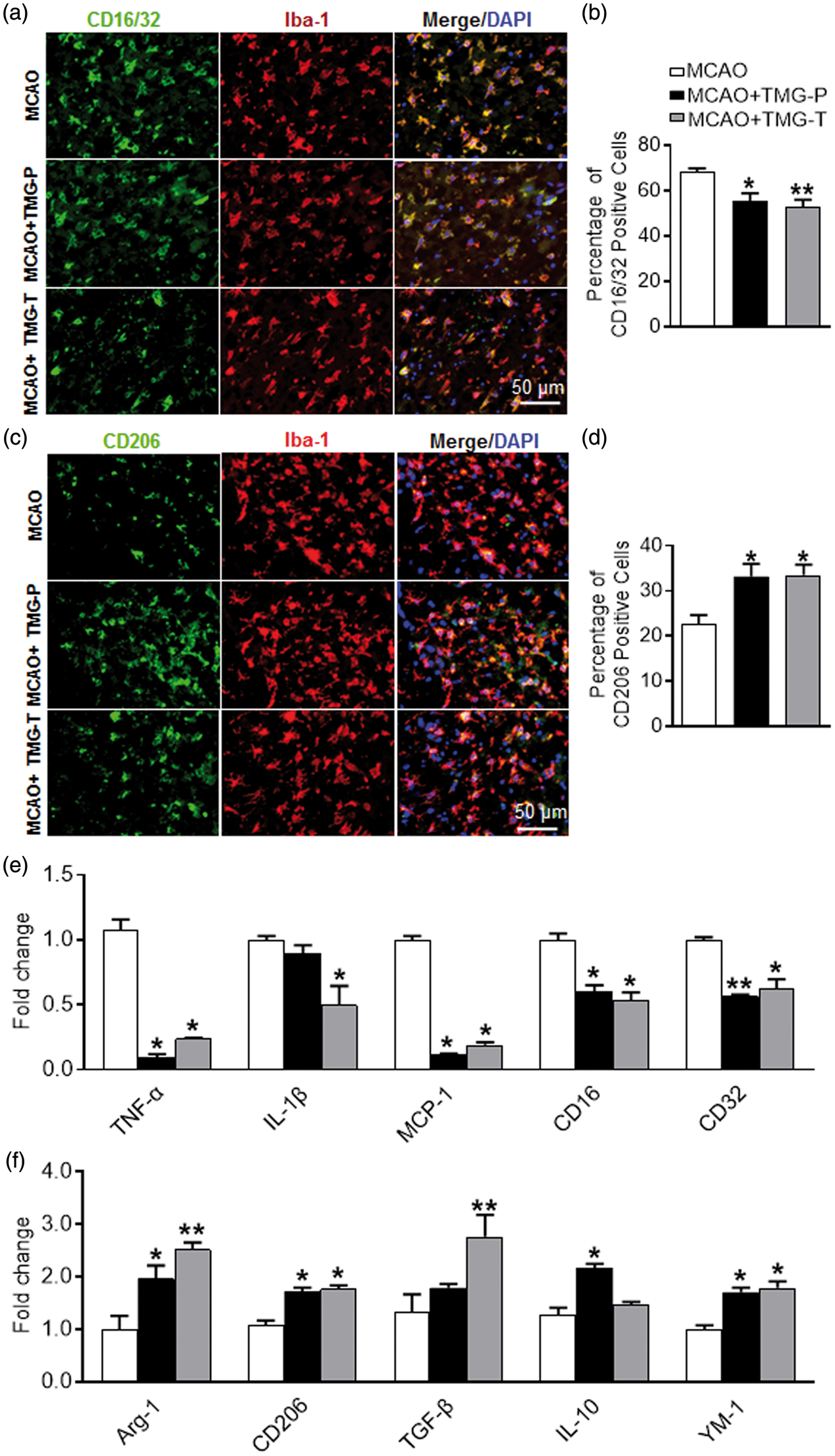
Consistent with the results of immunofluorescent staining, mRNA expression of the M1 genes (TNF-α, IL-1β, MCP-1, CD16, and CD32) was markedly reduced by treatment with TMG in mice, especially in the therapeutic treatment group (Figure 4(e)). However, IL-1β marginally decreased in the group given preventative treatment. Additionally, the expression of M2 genes (Arg-1, CD206, TGF-β, YM-1, and IL-10) increased significantly in TMG-treated groups, compared with the MCAO group (Figure 4(f)). Only TGF-β in the preventative treatment group and IL-10 in the therapeutic treatment group were exceptions. These results suggest that TMG treatment inhibited microglia/macrophage polarization to the M1 phenotype and, instead, promoted a shift to the M2 phenotype.
TMG suppresses the polarization of M1 in vitro
To further confirm the effect of TMG on polarization of microglia, a polarization experiment was carried out in BV2 cells with or without TMG. BV2 cells were cultivated in medium containing M1 or M2 polarization cytokines in the presence of TMG. After 12 h, no significant change of M2 was observed in TMG-treated groups compared with the MCAO group, although M1 polarization was suppressed by TMG (Figure 5(a) to (d)). These results provide further evidence to assert that TMG influences microglial polarization.
Effect of TMG on microglia polarization in vitro. BV2 cells were cultured in growth medium with LPS (100 ng/mL) or IL-4 (20 ng/mL). The phenotype of BV2 cells was examined by the co-expression of M1 marker CD16/32 (green) and M2 marker CD206 (green) with microglia/macrophage marker Iba-1 (red). Representative images of proportions of M1 (a) or M2 (c) phenotype cells in each group. (n = 9 per group). (b) and (d) Statistics for BV2 phenotypes in the presence or absence of TMG. (e–h) Amount of NF-κB p65 in cytoplasmic (e) and nuclear (f) was detected using immunoblotting and quantitated in the bar graph (g–h). (n = 9 per group) Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01 vs. control group.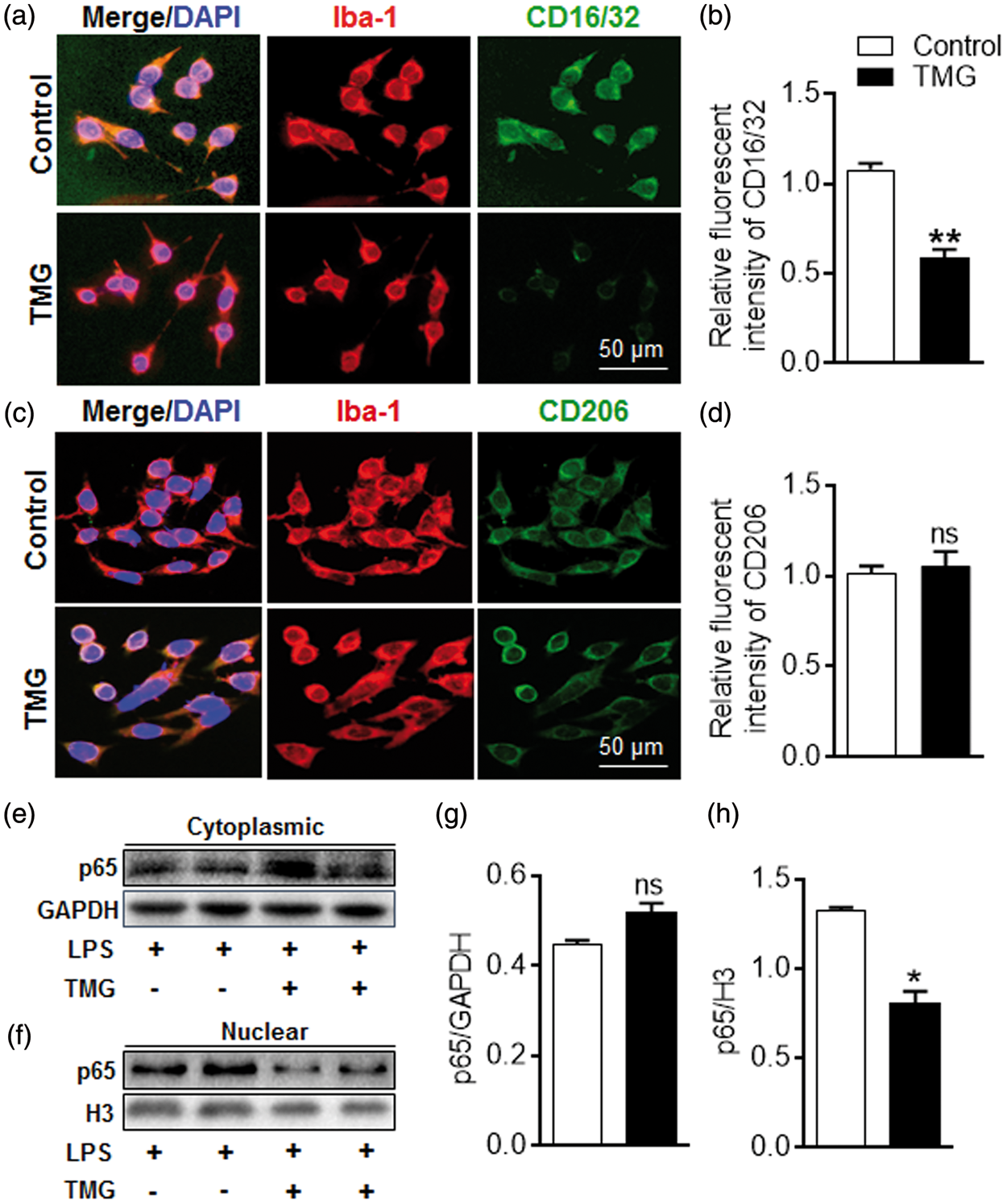
The transcriptional activity of NF-κB was also examined during polarization. Our data showed that NF-κB was suppressed along with the decreased M1 phenotype trigged by TMG treatment (Figure 5(e) to (h)). After intervention with TMG, less p65 translocated into nuclei even when the amount of p65 in the cytoplasm did not change dramatically, which indicated that the transcriptional activity of p65 was suppressed by TMG (Figure 5(e) and (g), cytoplasmic: 0.52 ± 0.02 in the TMG group vs. 0.45 ± 0.01 in the Control group, ns: not significant, n = 9 per group; Figure 5(f) and (h), nuclear: 0.81 ± 0.06 in TMG group vs. 1.33 ± 0.01 in the Control group, P < 0.05, n = 9 per group). The implication was that TMG may shift the polarization of microglia/macrophages by inhibiting NF-κB activation.
TMG inhibits the activation of NF-κB p65 signaling
The NF-κB pathway is activated in microglia during cerebral ischemic stroke and is associated with the inflammatory cytokines and nitric oxide (NO) production. Using RL2 immunofluorescent staining on brain sections of MCAO mice, we found that O-GlcNAcylation level was upregulated by TMG (Supplemental Figure 2). Moreover, the gene expression of COX-2 and iNOS, which are regulated by NF-κB, decreased sharply in TMG-treated mice as well (Figure 6(a) and (c), COX-2-positive cells per field: 26.5 ± 2.03 in the preventative treatment group vs. 36.3 ± 1.43 in the MCAO group, P < 0.05, n = 9 per group; 23.5 ± 1.67 in the therapeutic treatment group vs. 36.3 ± 1.43 in the MCAO group, P < 0.01, n = 9 per group. Figure 6(b) and (d), iNOS-positive cells per field: 24.7 ± 1.56 in the preventative treatment group vs. 33.5 ± 2.17 in the MCAO group, P < 0.01, n = 9 per group; 22.7 ± 1.33 in the therapeutic treatment group vs. 33.5 ± 2.17 positive cells per field in the MCAO group, P < 0.01, n = 9 per group.). Interestingly, injection with TMG after MCAO seemed to be more effective than pretreatment.
Effect of TMG on NF-κB transcriptional activity in MCAO mice. Brain sections were prepared at 72 h after stroke. Immunofluorescent images were captured microscopically. (a) Cortex splices co-stained with COX-2 (green) and DAPI (blue) in the peri-infarct area. (b) Cortex splices co-stained with iNOS (green) and DAPI (blue) in the peri-infarct area. (c) Quantification of COX-2 expression in ischemic brain. (d) Quantification of iNOS expression in ischemic brain. (n = 9 per group) *P < 0.05, **P < 0.01 vs. MCAO group. (e) and (f) mRNA expression of (e) iNOS and (f) COX-2 in mouse brain after MCAO. (n = 9 per group) *P < 0.05, **P < 0.01 vs. MCAO group. (g–j) Western blots show the distribution of p65 in cytoplasm and nuclei after stroke in the absence or presence of TMG. n = 9 per group. MCAO: MCAO with placebo administration; MCAO + TMG-P: MCAO with TMG administration before surgery; MCAO + TMG-T: MCAO with TMG administration after surgery. Data are expressed as mean ± SEM. ns: not significant, *P < 0.05, **P < 0.01 vs. MCAO group.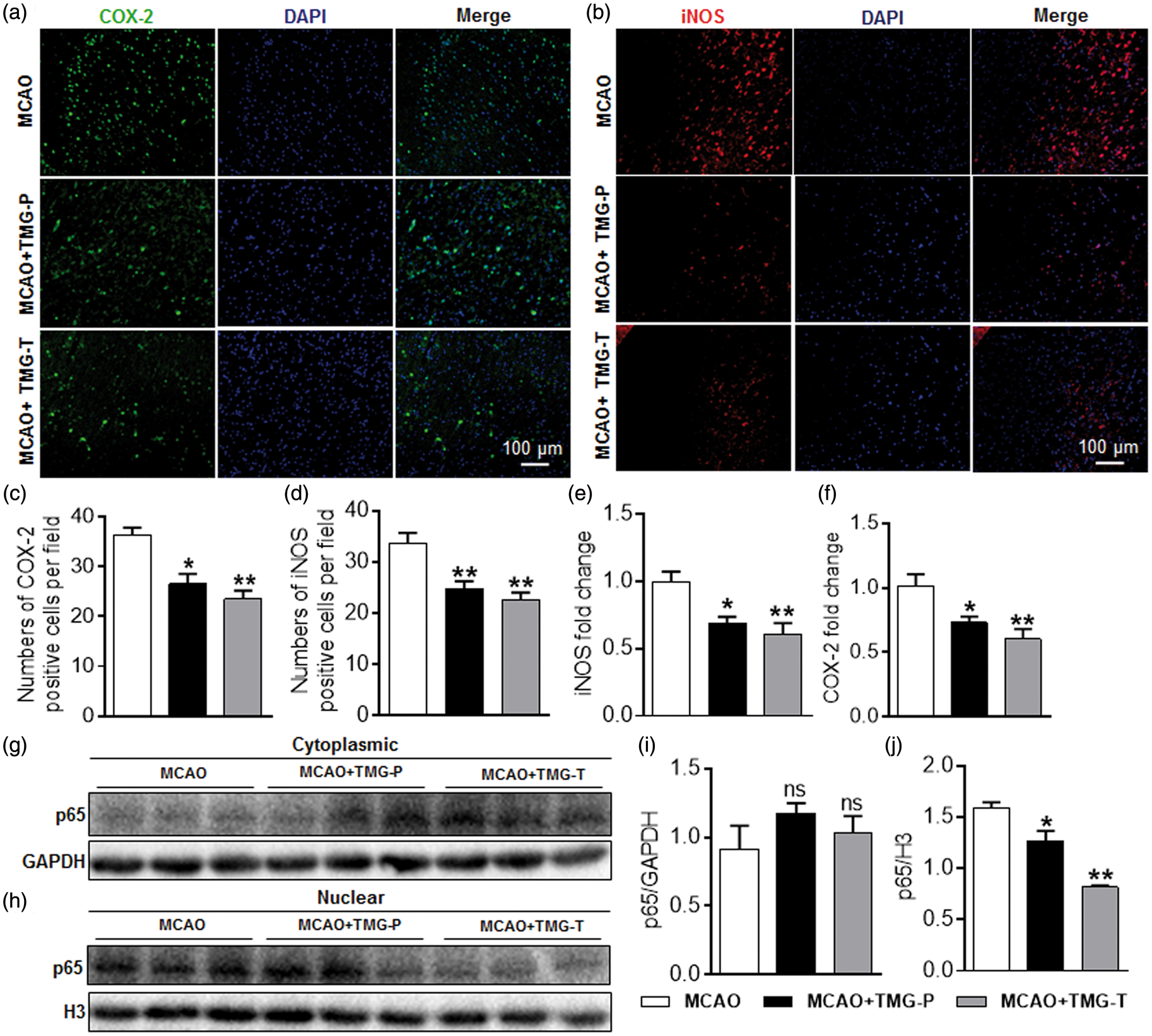
Consistent with immunofluorescent staining, the mRNA expression of COX-2 and iNOS showed similar results to those in previous experiments (Figure 6(e) and (f)). In addition, Western blots also depicted an inhibited translocation of p65, indicating that the transcriptional activity of NF-κB was suppressed by TMG in vivo (Figure 6(g) to (j)). These results further confirm that TMG impeded the NF-κB pathway via suppressing the translocation of p65 into nuclei.
Discussion
In this study, we confirmed the neuroprotective effect of TMG, a novel inflammation antagonist, in mice imbued with transient MCAO. This set of experiments showed that TMG significantly reduced infarct size and diminished the neurological deficits and motor coordination impairment in subjects of experimental stroke. Our results achieved in vivo and confirmed in vitro indicated that the neuroprotective effect of TMG is associated with a shift of microglia/macrophage polarization and inhibition of NF-κB p65 signaling (Figure 7).
Schematic diagram of TMG effect on brain damage post stroke. TMG exerts neuroprotection by phenotypic modulation of the microglia/macrophage shift and suppression of NF-κB p65 signaling. TMG promoted O-GlcNAcylation of p65 for binding to IκB and delayed its translocation into nuclei, which activates downstream gene transcriptions. Thus, expression of pro-inflammatory cytokines was suppressed and the polarization of microglia/macrophages was shifted to the M2 phenotype. I/R: ischemic/reperfusion; OGT: O-linked N-acetylglucosaminyltransferase; OGA: β-N-acetylglucosaminidase; IKK: inhibitor of nuclear factor κB kinase; IκB: inhibitor of nuclear factor κB. “G” indicates the moiety of GlcNAc.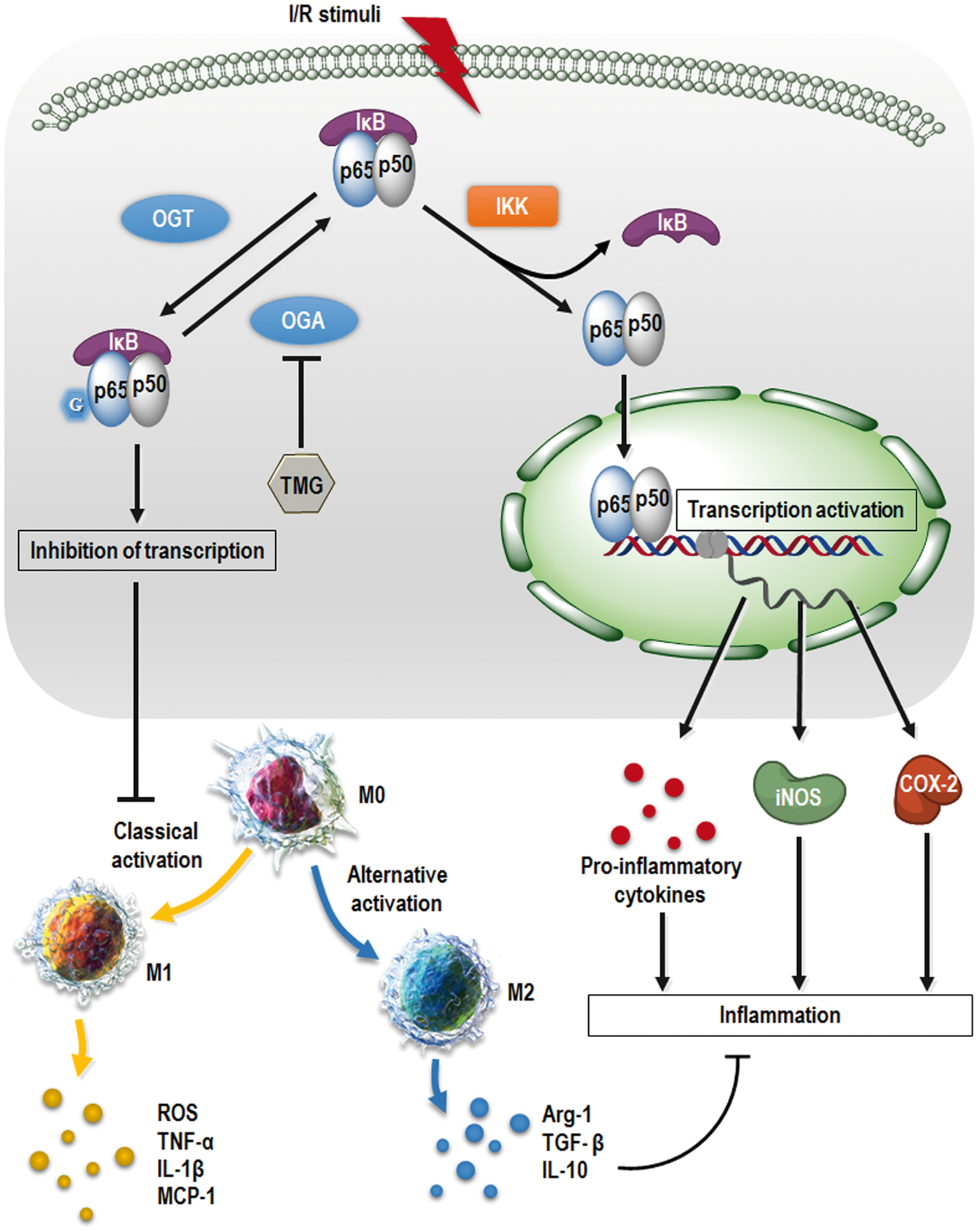
As is well known, OGT and OGA are highly expressed in the brain, and O-GlcNAcylations accumulates in neurons and Purkinje cells.29,30 In fact, the involvement of O-GlcNAc modification in neurological degeneration diseases, such as Alzheimer and Parkinson, has been reported. 31 Recent studies also revealed a dramatic increase of O-GlcNAc modification in the cortex after stroke.16,32 Supposedly, an elevation of O-GlcNAc modification is associated with neuronal apoptosis in the early stage of stroke and the extent of recovery after ischemic injury. Moreover, Hwang et al. 17 provided evidence that treatment with GlcN could inhibit the production of inflammatory cytokines and activation of microglia by elevating the O-GlcNAcylation level.
In the present work, we used TMG to prompt the accumulation of O-GlcNAcylations in mice with ischemia-damaged brains (Supplemental Figures 2 and 3). As a selective inhibitor of OGA, TMG is also a blood–brain barrier-permeable agent and suitable for animal experiments. Compared to the neurologic damage from mice in our PBS-treated MCAO group, TMG provided a dramatically different performance as estimated in mice rated by mNSS, adhesive-removal test, foot-fault test, and inclined plane test. The mNSS combines sensory, motor, reflex, and balance tests and is graded from 0 to 18 (normal score 0; maximal deficit score 18). 33 The adhesive-removal test can be used at the early stage of somatosensory recovery, but outcomes do not change significantly after a one-month interval. 34 Forelimb placement dysfunction was assessed by the foot-fault test. 35 The results of these tests strongly supported the protective effect in TMG-treated groups. When we also calculated infarct volumes by TTC staining of brain sections after MCAO, the TMG-treated groups manifested sharply reduced infarct volumes, in accordance with the study of Hwang et al. 17 We also evaluated the inflammatory microenvironment in the mouse brain after MCAO. In multiple ELISAs and qRT-PCRs, even as the content of several pro-inflammatory cytokines diminished, the anti-inflammatory cytokine IL-10 clearly increased in TMG-treated groups. However, only a slight decrease of TNF proteins was observed, whereas TNF transcripts were dramatically reduced. We proposed that the expression of TNF proteins might be elevated at an early stage after MCAO, 36 so that the total amount of TNF proteins changed slightly when TNF mRNA expression was suppressed by TMG treatment. A change of hemopoietic growth factors was also observed. That is, the expression of G-CSF was reduced in both TMG-treated groups, whereas GM-CSF expression was upregulated only in the treatment group. This may result when G-CSF enhances an inflammatory response after stroke by mobilizing macrophage infiltration and promoting microglial proliferation.37,38 One could then infer that a decrease of G-CSF is associated with the reduction of Iba-1+ cells in the ischemic brains of mice. Also, GM-CSF is believed to improve functional recovery after stroke by downregulating neuronal apoptosis as a result of suppressing apoptosis-related gene expression. 39 Additionally, we found that the neuroprotective effect of TMG treatment after the occurrence of stroke was greater than that of pretreatment with TMG. Since neuroprotection induced by TMG, as previously proposed, is mediated by an increase of O-GlcNAcylation, we attempted to identify a change of O-GlcNAcylation in mouse brains. However, the O-GlcNAcylation level turned out to decrease with time after pretreatment with TMG, although this action indeed upregulated O-GlcNAc modification in the brains of these mice. To recapitulate, an increase of O-GlcNAc modification provided neuroprotection that forestalled the otherwise destructive effects of ischemic stroke. Moreover, treatment with TMG after MCAO strengthened that neuroprotective effect.
Acute I/R injury and inflammation are closely allied, but precisely how inflammatory cells and molecules react remains elusive. Since the activation of microglia/macrophages might cause neuronal death in the ischemic penumbra, 40 we analyzed the cells by immunofluorescent staining. Microglia/macrophages have critical functions as immune-competent and phagocytic cells by acting as scavengers in the event of infection, inflammation, trauma, ischemia, and neurodegeneration.41–44 In the present study, expression of the microglia marker Iba-1 decreased in mice of the TMG-treated groups compared to those in the MCAO group. We also found that TMG could inhibit the production of pro-inflammatory cytokines in LPS-stimulated microglia in vitro. This suggests that an increase of O-GlcNAc modification decreases activated microglia/macrophages and acts as an important protective mechanism that negates immune response injury.
We observed a phenotype shift of microglia/macrophages in the ischemic brain; that is, levels of the M2 anti-inflammatory cytokines (Arg-1, CD206, TGF-β, IL-10, and YM-1) significantly increased, whereas gene expression of the M1 inflammatory cytokines (IL-1β, TNF-a, MCP-1, CD16, and CD32) decreased compared to those levels in the MCAO group. Experiments performed in vitro indicated that TMG suppressed classical activation of microglia/macrophages, rather than promoting alternative activation of these lymphocyte subsets, as shown in Figure 7. Immunoblots also hinted at a potential link between suppression of the NF-κB pathway and inhibition of M1 polarization. However, only further investigation will adequately illustrate the underlying mechanisms.
In a rodent trauma-hemorrhage model, increased O-GlcNAc modification improved cardiac function and perfusion of peripheral organs. 17 The levels of IL-6 and TNF-α were also suppressed. 11 Some studies demonstrated that increased O-GlcNAc modification reduced phosphorylation of IκB and the NF-κB pathway. Elsewhere, O-GlcNAcylation was effective in exerting vasoprotective and anti-inflammatory effects in a model of acute vascular injury.14,15 In other words, O-GlcNAc modification of p65 decreased TNF-α-induced phosphorylation on Ser536, resulting in an increased association of NF-κB with IκB, thus lowering NF-κB activity and inhibiting the production of pro-inflammatory mediators. 45 Another report stated that the administration of GlcN at 3 h post-MCAO sharply suppressed the induction of the pro-inflammatory markers, IL-1β, TNF-α, iNOS, and COX-2 in ischemic hemispheres as measured by qRT-PCR. 17 In the present study, we used TMG to define the relationship between enhanced O-GlcNAc modification and its role as an inhibiter of NF-κB activity before or after I/R injury. We demonstrated that O-GlcNAc modification is significantly elevated by TMG coincident with the decreased nuclear translocation of p65. Thus, down-regulation of iNOS and COX-2 provides a molecular basis for the neuroprotective effects of TMG in I/R injury (Figure 7). On examining O-GlcNAcylation of total protein, we documented a significant difference between TMG-treated and MCAO groups. Nuclei of ischemic cortex cells from TMG-treated groups contained less p65 than found in our MCAO group, although the cells’ cytoplasmic content was not significantly different. The mRNA expression of iNOS and COX-2 concurred with the result obtained by immunofluorescence using cell counting methods, strongly supporting the conclusion that increasing O-GlcNAc modification provides a neuroprotective role in acute vascular injury of the brain by suppressing the activation of NF-κB and the expression of iNOS and COX-2.
In conclusion, we found that TMG treatment suppressed the inflammatory response in mice used as an experimental model of stroke. Our findings also supported the concept that TMG modulated the phenotype shifting of microglia/macrophages and inhibited NF-κB p65 signaling to suppress the immune response after ischemic injury. Therefore, TMG offers considerable potential as a therapeutic agent to alleviate the neurologic damage from stroke.
Footnotes
Funding
This work was supported by the National Natural Science Foundation of China (81571600, 81322018, and 81273287 to JWH, and 81401361 to XFM), and the Youth Top-notch Talent Support Program (to JWH).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
JWH, XFM, and YTH designed the research studies, and YTH, XFM, and DJL conducted the experiments and prepared figures. YTH, XFM, and DJL analyzed the data and wrote the manuscript. JWH and XFM reviewed and revised the manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.