
Abstract
Classically, compartmentation of glutamate metabolism in the brain is associated with the fact that neurons and glia exhibit distinct differences with regard to metabolism of this amino acid. The recent use of 13C-labeled compounds to study this metabolism in conjunction with the availability of cell type-specific tissue culture modes has led to the notion that such compartmentation may even be present in individual cell types, neurons as well as glia. To better understand and explain this, it is proposed that mitochondrial heterogeneity may exist resulting in tricarboxylic acid cycles with different properties regarding cycling rates and ratio as well as coupling to amino acid biosynthesis, primarily involving glutamate and aspartate. These hypotheses are evaluated in the light of current knowledge about mitochondrial structure and function.
Metabolic pathways have classically been elucidated using radioactively labeled precursors, the metabolic fate of which may be followed by monitoring the appearance of radioactivity in the products. Alternatively, stable isotopes can be utilized. A specific radioactivity of the product remaining lower than that of the precursor would imply a simple product-precursor relationship, but when the specific radioactivity of the product rapidly exceeds that of the precursor, a compartmentation of the metabolic pathway is likely to exist. Any organ with different cell types, each having a characteristic set of enzymes pertinent to the metabolic pathway in question, could potentially exhibit metabolic compartmentation. Early studies of glutamate metabolism in the brain (e.g., Berl and Clarke, 1969, 1983) clearly suggested compartmentation since the product, glutamine, had much higher specific activity than its precursor, glutamate, after labeling with, e.g., [14C]glucose. More recent studies of glutamate metabolism in either neurons or astrocytes have suggested (e.g., Schousboe et al., 1993) that the concept of metabolic compartmentation may be extended to apply even for individual cell types. To discuss this in detail, it will be necessary to evaluate the possibility of the existence of different types of mitochondria within the same cell. This so-called heteroplasmy is a well known phenomenon in patients with defects of the mitochondrial genome (Taylor et al., 1997). The present review will discuss evidence for the existence of such intracellular mitochondrial heterogeneity in nonpathological brain cells. For a better comprehension of the biochemical differences, some background information will be given on mitochondrial location and ultrastructure.
Characterization of mitochondria was pioneered by Lehninger (1965) using mostly liver preparations as a model system. During the following 20 years, extensive work on mitochondrial characterization took place. For excellent reviews of this research on brain mitochondria, see Abood (1969) and Clark and Nicklas (1984). The latter authors predicted an expansion in the area of mitochondrial research related to aging and senile dementia, and time has proven this prediction to be correct. Neurodegenerative disorders in general are characterized by a gradually progressing neuronal death, frequently accompanied by gliosis. A selective loss is taking place of defined groups of neurons that might be related either anatomically or physiologically. These diseases are exemplified by Alzheimer's, Parkinson's, and Huntington's diseases and cerebellar degenerations such as olivopontocerebellar atrophy. An increasing number of observations suggest that defects in energy metabolism are important pathophysiological mechanisms in neurodegenerative diseases (Plaitakis and Berl, 1988; Beal et al., 1993), although it is at present not possible to ascertain if the observed changes are secondary effects of neuronal loss and astrogliosis. In Parkinson's disease, a decrease in activity of complex I (NADH dehydrogenase, ubiquinone) of the mitochondrial energy metabolism has been implicated in the pathogenesis of the illness (Schapira et al., 1990). Alzheimer's disease has been linked to reduction of pyruvate and 2-oxoglutarate dehydrogenase activities, enzymes necessary for the tricarboxylic acid (TCA) cycle activity and thus oxidative production of NADH (Sheu et al., 1994; Heroux et al., 1996). Central nervous system involvement has also been found in patients with mitochondrial myopathy. Hepatic encephalopathy is a major neuropsychiatric complication of acute and chronic liver disease (Norenberg et al., 1992). An increased brain ammonia concentration, which is the result of liver cirrhosis, has been shown to lead to almost complete inhibition of 2-oxoglutarate dehydrogenase, a rate-limiting enzyme of the TCA cycle (Cooper and Plum, 1987). The above-mentioned conditions lead either to a decreased production of NADH, which in turn results in a decreased activity of complex I, or, as in Parkinson's disease, to decreased activity of complex I directly (Schapira et al., 1990).
It is well known that some neurons are more vulnerable than others to different kinds of toxins, i.e., dopaminergic cells to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and certain areas of the hippocampus to lack of oxygen and increased extracellular glutamate concentrations. Thus, compartmentation of vulnerability involving mitochondria is well established. It is, however, also conceivable that subpopulations of mitochondria within the same cell could exhibit different degrees of vulnerability, but experimental evidence for this is at present still lacking.
MITOCHONDRIA
Mitochondrial location and ultrastructure
Mitochondria are distributed with varying densities throughout the CNS, with the more vascular parts containing most mitochondria (Friede and Pax, 1961). They are the cytoplasmic granules containing the enzymes involved in oxidative metabolism. Since it is known that the brain is responsible for ~25% of the total body oxygen and glucose consumption, it must contain a high concentration of mitochondria. These have their greatest concentration in neurons (Abood, 1969) and are present throughout the perikaryon, cytoplasm, dendrites, axons, and nerve endings. Regions of particular abundance are nerve endings, the axon hillock, and nodes of Ranvier (Abood, 1969). They can be seen as rods (1 × 2.5 μm) or rounded granules (4 μm) in living neurons (McIlwain and Grinyer, 1950). Mitochondrial heterogeneity has been demonstrated in the brain by separation of mitochondria of synaptic and peripheral origin, which showed slight differences in enzyme distribution (Lai et al., 1977). Mitochondria are found in oligodendroglia and Schwann cells, but they are not abundant in astrocytes and microglia.
Mitochondrial origin
Mitochondria originate by fission of preexisting mitochondria. In an evolutionary context, it should be noted that according to the endosymbiont hypothesis, the mitochondria arose by internalization of bacteria by ancient eukaryotic cells (De Duve, 1991; Margulis, 1993; Taylor et al., 1997). This hypothesis is at the present time broadly accepted and supported by a host of observations: The mitochondria have their own DNA, in the form of a circular piece of DNA that codes for a few endogenous proteins that are still synthesized by the organelle itself. The genetic code in the mitochondria shows distinct differences from the more universal code used by the nuclear DNA. The mitochondria replicate at times independently of their host cell in the cell cycle. The majority of the enzymes found in the mitochondria are, however, coded for by the genome in the host cell nucleus, produced by the host cell and subsequently transported to the mitochondria. Thus, all essential enzymes involved in the TCA cycle are synthesized by this mechanism.
METABOLIC COMPARTMENTATION
Metabolic compartmentation in the brain has during the last three decades been studied extensively using radioactive substrates such as glucose, pyruvate, and lactate, which label a large pool of glutamate or substrates such as acetate, 2-oxoglutarate, citrate, and bicarbonate, which label a small pool of glutamate, which is further metabolized to glutamine (Berl and Clarke, 1969; Shank and Campbell, 1983; Hassel et al., 1992). Subsequent elucidation of the cellular localization of glutamine synthetase in astrocytes (Norenberg and Martinez-Hernandez, 1979) confirmed the originally proposed two-compartment model (Balazs et al., 1972) for glutamate metabolism with a small glial pool having a high turnover and a larger neuronal pool with a slow turnover.
As mentioned above, studies in the intact brain or in slices have shown that acetate metabolism takes place in the small glial compartment (Berl and Clarke, 1969; Badar-Goffer et al., 1990; Hassel et al., 1992), a notion subsequently confirmed in cell culture studies using astrocytes (Sonnewald et al., 1993c). Cell culture techniques have also demonstrated that glucose metabolism proceeds efficiently in both astrocytes and neurons (Sonnewald et al., 1991, 1993c; Peng et al., 1994). With [1-13C]glucose as the substrate, glycolysis will produce labeled pyruvate with half of the molecules containing 13C in the C-3 position. Through the action of pyruvate dehydrogenase, which is present in both astrocytes and neurons, acetyl CoA will enter the TCA cycle and subsequently give rise to labeling predominantly in C-4 of glutamate, glutamine, and citrate and C-2 or C-3 in aspartate. As the metabolites remain in the TCA cycle during a second turn, there will also be labeling in C-2 and C-3 of glutamate, glutamine, and citrate. [2-13C]Acetate, which enters the TCA cycle after conversion to acetyl CoA by the mitochondrial enzyme acetyl CoA synthetase, will give rise to the same labeling patterns in glutamate, glutamine, and citrate as [1-13C]glucose (Fig. 1).
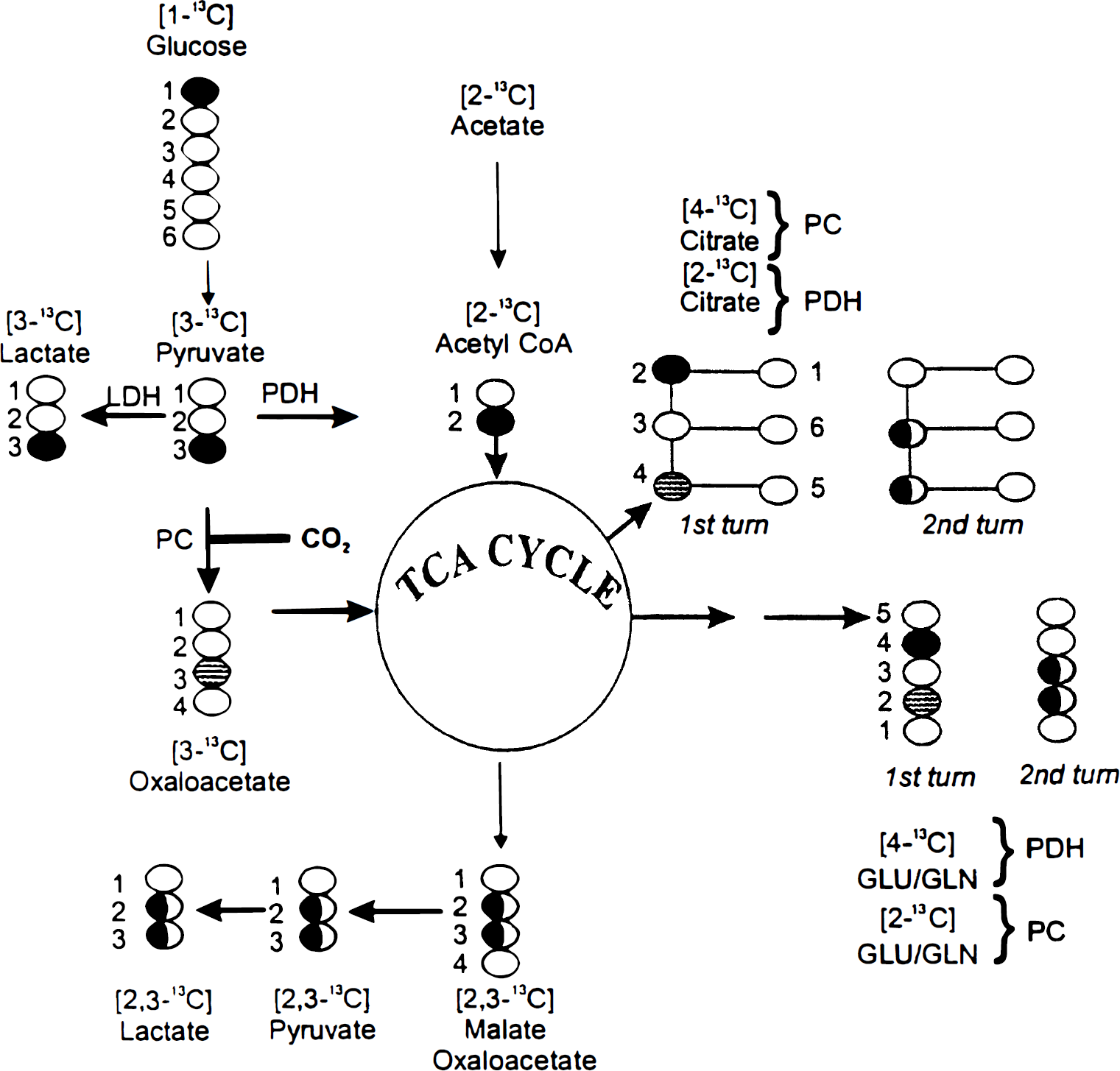
A simplified scheme of the tricarboxylic acid (TCA) cycle showing the conversion of [1-13C]glucose and [2-13C]acetate to pyruvate and acetyl CoA and further metabolism through the first and second turn of the TCA cycle. The labeling patterns of Glu, Gln, and citrate when synthesized through the pyruvate carboxylase (PC) or pyruvate dehydrogenase (PDH) pathways are indicated. Labeling with 13C is indicated by black or shaded circles.
In contrast to neurons, astrocytes express pyruvate carboxylase, a mitochondrial ATP-dependent enzyme (Yu et al., 1983; Shank et al., 1985). This will additionally allow entry of pyruvate into the TCA cycle by carboxylation to [3-13C]oxaloacetate, which gives rise to labeling in C-2 of glutamate, glutamine, and citrate and in C-3 of aspartate (Fig. 1).
Compartmentation of astrocyte metabolism
While compartmentation of glutamate metabolism in the brain has long been an accepted concept (Berl and Clarke, 1969), such compartmentation within one cell type has only recently been reported. Mitochondrial heterogeneity within astrocytes was already 20 years ago suggested by Van den Berg et al. (1975) in an elegant review. More recent studies of metabolism of glutamate and glutamine in astrocytes have also suggested that such a compartmentation of glutamate metabolism may exist. It was shown that extracellular radioactive glutamine gave rise to a higher specific labeling of intracellular glutamate than glutamine and that aspartate formation was dramatically different when exogenous glutamate or glutamine was used as the substrate in astrocytes (Schousboe et al., 1993). Compartmentation of malate metabolism has also been reported, as metabolism of malate at different concentrations was affected differently by certain metabolites and enzyme inhibitors (McKenna et al., 1990). By nuclear magnetic resonance analysis of 13C incorporation into different positions in glutamine and citrate, information has been obtained that gives strong support to the concept of compartmentation of glutamate metabolism and hence of mitochondrial heterogeneity. Thus, using [2-13C]acetate together with unlabeled glucose or [1-13C]glucose together with unlabeled acetate, it could be demonstrated that released glutamine and citrate did not originate from the same TCA cycle in astrocytes (Sonnewald et al., 1993a).
With use of [U-13C]glutamate as the major substrate, it was shown that astrocytes metabolize glutamate extensively with cellular label appearing predominantly in aspartate and malate. Additionally, the incubation medium contained labeled glutamine, lactate, alanine, citrate, and glutamate (Sonnewald et al., 1993b). The labeling of lactate is particularly noteworthy since it indicated that lactate, which must be formed from pyruvate, can be produced from precursors derived from the TCA cycle (Sonnewald et al., 1993b). A similar conclusion has been reported by Bachelard et al. (1994). Thus, citrate leaving the mitochondria may give rise to oxaloacetate in the cytoplasm and subsequently to pyruvate via the activity of cytosolic malic enzyme (McKenna et al., 1990; Kurz et al., 1993) or by the concerted action of phosphoenolpyruvate carboxykinase and pyruvate kinase and then to lactate. Additionally, mitochondrial malic enzyme might be operative. The cystolic and mitochondrial pools of pyruvate did not mix freely as evidenced by the differential labeling of alanine and lactate. This is based on the fact that lactate dehydrogenase resides only in the cytosol, while alanine aminotransferase is located both in the cytosol and in the mitochondria. Uniformly labeled lactate but only monolabeled alanine was detected (McKenna et al., 1996; Sonnewald et al., 1996). Thus, uniformly labeled pyruvate was available in the cytosol for conversion by lactate dehydrogenase but not by alanine aminotransferase. The absence of uniformly labeled and the presence of monolabeled alanine indicated that this amino acid, in contrast to lactate, was derived from a TCA cycle that is associated with pyruvate carboxylation and/or a cycle in which 2-oxoglutarate remains for more than one turn, a strong indication of mitochondrial heterogeneity (Sonnewald et al., 1996).
Compartmentation of neuronal metabolism
Indications that neuronal mitochondrial metabolism of glutamate may be compartmentalized, suggesting mitochondrial heterogeneity, have come from studies of metabolism of 13C-labeled glutamate, glutamine, and lactate in cultured cerebral cortical neurons, the vast majority of which are GABAergic (Yu et al., 1984; Drejer et al., 1987). These neurons metabolized [U-13C]glutamate extensively, with label appearing predominantly in aspartate and γ-aminobutyric acid (GABA) (Westergaard et al., 1995). Label from [U-13C]glutamine appeared in the same molecules but with different labeling ratios. Label incorporation into GABA after 3 hours was similar from glutamate and glutamine, 13 and 15%, respectively, implicating the existence of a large unlabeled GABA pool, due either to a slow turnover of GABA in cell cultures or the preference of synthesis from other precursors (Westergaard et al., 1995). Even though incorporation of label was similar from the two precursors, the labeling patterns were different. Approximately 50% of GABA formed from labeled glutamine was derived from the TCA cycle, whereas only 30% of GABA formed from glutamate originated from this cycle (Westergaard et al., 1995). TCA cycle involvement in the synthesis of GABA, aspartate, glutamate, and glutamine could be determined by analysis of doublets present in the nuclear magnetic resonance spectra. The 13C enrichment in aspartate was almost twice as high from glutamate (37%) as from glutamine (20%). In addition, aspartate was more highly labeled than GABA. When glutamine was the precursor, aspartate was also more highly labeled than glutamate (Westergaard et al., 1995). Also, in the intact brain, there is evidence to suggest that synthesis of GABA from glutamine and glutamate may be complex. Thus, enrichment of 13C in analogous C atoms in these amino acids after infusion of rabbits with [U-13C]glucose showed the same values for glutamate and GABA, while that in glutamine was different (Lapidot and Gopher, 1994).
When cerebral cortical neurons were incubated for 4 hours with [U-13C]lactate, an extensive incorporation of 13C into the cellular pool of the amino acids aspartate, glutamate, and GABA was observed, although net synthesis of these amino acids from lactate is not possible. This finding suggests that lactate is metabolized in a TCA cycle that also gives rise to aspartate, glutamate, and GABA. A reduction of enrichment in 13C atoms was seen in aspartate and glutamate but not in GABA when nonlabeled glutamine was added to the incubation medium, suggesting the existence of at least two glutamate pools, of which one serves as a precursor for the synthesis of GABA and the other for synthesis of aspartate. A decrease in cycling ratios for the TCA cycle precursors, in the presence of glutamine, points in the same direction, since cycling for the TCA cycle constituents serving as precursors for GABA decreased more than the corresponding cycling for glutamate and aspartate precursors. It could be argued that the presence of a small number of astrocytes in the neuronal cultures (Larsson et al., 1985) could contribute to the difference in glutamate/aspartate and GABA labeling. It should, however, be noted that glutamine was only marginally labeled (U. Sonnewald, unpublished data) and that astrocytes utilize lactate poorly compared with neurons (H. S. Waagepetersen et al., submitted).
Surprisingly, alanine did not exhibit labeling from [U-13C]lactate in cerebral cortical neurons. Since the equilibrium constant for the alanine aminotransferase reaction is close to unity (Krebs, 1953), this observation is most likely explained by compartmentation of pyruvate metabolism or by rapid formation of mitochondrial acetyl CoA from pyruvate (Fitzpatrick et al., 1988). Since alanine is labeled in astrocytes under these conditions (Sonnewald et al., 1991), this is a further indication that the presence of a small number of astrocytes in the cultures could not explain the differential labeling patterns in glutamate/aspartate and GABA.
These results are best explained postulating that lactate enters at least two TCA cycles (Fig. 2). In the absence of added glutamine, one TCA cycle, which has a rapid turnover, is in exchange with the aspartate pool and one glutamate pool. The other cycle in which intermediates stay for only a few turns is in contact with the GABA pool. Glutamine added exogenously gains access to the aspartate-forming TCA cycle as indicated by an increase in the aspartate concentration (Waagepetersen et al., 1998) but has no direct access to the TCA cycle from which intermediates serve as precursors for GABA synthesis (Fig. 2). The model suggests that aspartate flows between the two TCA cycles located in different mitochondria, but it should be noted that other C-4 units such as malate could substitute for aspartate.
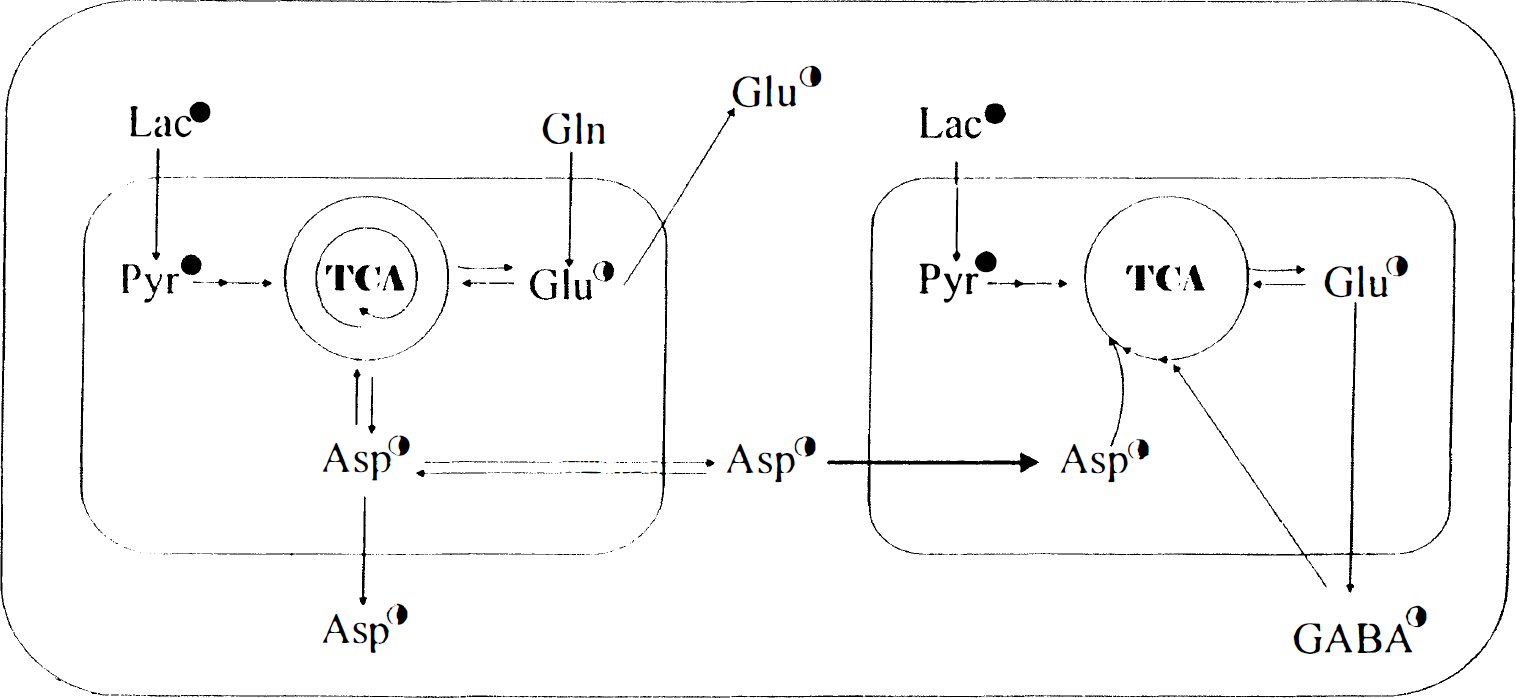
An illustration of a two-compartment model, explaining the metabolism of [U-13C]lactate in the presence of 0.5 mmol/L glutamine in cultured cerebral cortical neurons. The left part represents the Asp- and Glu-generating tricarboxylic acid (TCA) cycle and the right part the corresponding 7-aminobutyric acid-generating cycle. The number of circular arrows in the TCA cycle indicates in a semiquantitative manner the number of turns that metabolites remain in the cycle; i.e., it reflects cycling ratios. 13C labeling is indicated by filled or half-filled (less enrichment) circles. Lac, lactate; Pyr, pyruvate. (Modified from Waagepetersen et al., 1998).
MITOCHONDRIAL HETEROGENEITY
Involvement in metabolic compartmentation
Biochemical (Gallo et al., 1987), physiological (Trimmer et al., 1986), and developmental (Raff, 1989) heterogeneity of mitochondria has been described in the literature. Although heterogeneity of cell cultures is possible, a growing amount of evidence seems to point strongly toward intracellular compartmentation of metabolism in individual cells as well. In the following paragraphs, some models are presented that may explain such compartmentation.
Intercellular heterogeneity in astrocytes has been shown by Oh et al. (1991) by visualizing oxidative metabolism in cultured glial cells by indirect fluorescence labeling of mitochondrial malate dehydrogenase. In process-bearing astrocytes and oligodendroglia, mitochondrial malate dehydrogenase was reported to be uniformly distributed, whereas in polygonal astrocytes that contained relatively similar numbers of mitochondria, immunoreactivity to mitochondrial malate dehydrogenase varied among cells from very weakly positive to intensely positive.
Several possibilities exist for cellular compartmentation of the TCA cycle metabolism. There could be different types of mitochondria within the same cell, or different TCA cycles might be operative and separated within the same mitochondrion. Support for the latter notion is obtained from a nuclear magnetic resonance study of mitochondrial viscosity by Lopez-Beltran et al. (1996), which showed that free diffusion of metabolites within mitochondria is unlikely.
Heterogeneous microenvironments are also indicated by the observations of Jones (1986), who reported that mitochondria appeared to be clustered in sites of high ATP demand and concluded: “Thus, microheterogeneity of metabolite concentrations can occur in cells without membranal compartmentation and may be important in determining the rates of various high flux processes.”
Origin of mitochondrial heterogeneity
The observations mentioned above may be interpreted in favor of the existence of at least two compartments with differently functioning TCA cycles. This is unlikely to be caused by two genetically discrete types of mitochondria in the same cell. The mitochondrial genome has been completely mapped and is of a very small size. More likely, therefore, this compartmentation is a consequence of the development of distinctly different environments for mitochondria in astrocytes and neurons. Differential transport of proteins coded for by the cell nucleus to the different mitochondria will result in different environments and thereby facilitate modulation of basic mitochondrial functions. Several reports on differential transport of mRNA within neurons might support this view. Thus, Landry et al. (1994) observed that mRNA encoding glutamate decarboxylase-67, an enzyme associated with membranes (Dirkx et al., 1995), was confined to cell somas. Furthermore, Chun et al. (1996) reported differential transport of mRNA species into axonal domains of the squid giant axon. One has to bear in mind, however, that relative abundance of mRNA not always correlates with the amount of the corresponding proteins.
Mitochondrial heterogeneity in neurons would appear to be the rule sooner than the exception since these cells are highly polar, being composed of dendrites, cell bodies, and axons, i.e., compartments with widely differing needs. Because of the lack of the machinery for protein synthesis in axons, materials required in axons and synapses have to be transported down the axon after synthesis in the cell body. Furthermore, fast anterograde transport conveys mitochondria to their destinations (Kondo et al., 1994). It is not known if such mitochondria contain a complete set of enzymes already from the start or, as appears more likely, collect the enzymes required for the type of work that needs to be carried out at their destination.
Footnotes
Abbreviations used
Acknowledgment
The authors thank Ms. Hanne Danø for her expert secretarial assistance.