
Abstract
Mitochondrial function is critical to maintain high rates of oxidative metabolism supporting energy demands of both spontaneous and evoked neuronal activity in the brain. Mitochondria not only regulate energy metabolism, but also influence neuronal signaling. Regulation of “energy metabolism” and “neuronal signaling” (i.e. neurometabolic coupling), which are coupled rather than independent can be understood through mitochondria’s integrative functions of calcium ion (Ca2+) uptake and cycling. While mitochondrial Ca2+ do not affect hemodynamics directly, neuronal activity changes are mechanistically linked to functional hyperemic responses (i.e. neurovascular coupling). Early in vitro studies lay the foundation of mitochondrial Ca2+ homeostasis and its functional roles within cells. However, recent in vivo approaches indicate mitochondrial Ca2+ homeostasis as maintained by the role of mitochondrial Ca2+ uniporter (mCU) influences system-level brain activity as measured by a variety of techniques. Based on earlier evidence of subcellular cytoplasmic Ca2+ microdomains and cellular bioenergetic states, a mechanistic model of Ca2+ mobilization is presented to understand systems-level neurovascular and neurometabolic coupling. This integrated view from molecular and cellular to the systems level, where mCU plays a major role in mitochondrial and cellular Ca2+ homeostasis, may explain the wide range of activation-induced coupling across neuronal activity, hemodynamic, and metabolic responses.
Introduction
How brain integrates neural activity (cell signaling events) and oxidative metabolic demand (enzymatic activity) in vivo is not completely understood, but may have a common link to mitochondrial function. In vitro cellular and molecular studies have established the integrative role of mitochondrial Ca2+ influx and cycling and its influence on cellular function.1–15 These foundational studies characterized subcellular mitochondrial Ca2+-dependent physiological processes, including maintenance of intracellular Ca2+ ion homeostasis, synaptic activity, vesicle exocytosis, energy metabolism, and hormonal secretion.1–15 In recent years, normal and pathophysiological brain function is increasingly studied using neuroimaging such as functional magnetic resonance imaging (fMRI) and optical imaging, which measure cerebrovascular and metabolic correlates of neural activity.16–21 Building upon earlier cellular and molecular studies, recent in vivo systems-level approaches have determined the neurophysiological impact of mitochondrial Ca2+ uptake and cycling on neural signaling8,19,22,23 and energy metabolism.10,19,22,23 Both “neural signaling” and “energy metabolism” being fundamental to cerebral blood flow (CBF) and blood oxygenation changes, correlate to a certain extent with tissue level Ca2+ responses. 24 Hence local mitochondrial Ca2+ homeostatic pathways in brain tissue can be hypothesized to impact hemodynamic-related neuroimaging markers, including fMRI and optical spectroscopic measures of cerebral blood oxygenation, CBF, and cerebral blood volume (CBV). In vivo studies on resting state fluctuations and task-evoked changes using laser Doppler-CBF and fMRI-blood oxygen level dependent (BOLD) signals indicate that both intrinsic and evoked brain activity are impacted by interventions altering mitochondrial Ca2+ influx.22,23,25 Other independent systems-level studies of the cerebellum 19 and olfactory epithelium 8 also indicate similar effects. Considering recent systems-level and earlier in vitro studies, a larger integrative role of mitochondrial Ca2+ uptake emerges with a strong ability to influence neuronal network activity, neurovascular, and neurometabolic coupling.
Introducing cellular diversity of mitochondria and their ion homeostatic functions in various tissues, energy metabolic, ion homeostatic, and cellular signaling roles of mitochondria are reviewed with particular emphasis on the central nervous system (CNS). An integrated view of mitochondrial impact across spatial scales and cellular types is presented in order to explain the diverse evoked neuronal activity and hemodynamic responses observed in the brain at the systemic level. This is crucial to better interpret experimental results from emerging approaches including those that simultaneously map in vivo Ca2+ and activation-induced hemodynamic functional responses in the working mammalian brain.24,26
Subcellular spatial distribution of mitochondria
Mammalian mitochondria are distributed with varying densities across the CNS, where regions with larger vascular density contain relatively more mitochondria. 27 Mitochondrial localization and heterogeneity in the brain is well studied, 28 with the largest concentration of mitochondria in neurons, 29 followed by glial and endothelial cells, respectively. Within CNS cells, mitochondria differ in their spatial distribution. For example, neuronal mitochondria are present throughout the perikaryon, cytoplasm, dendrites, axons, and nerve endings with relatively greater abundance in presynaptic terminals30,31 and dendrites. 29 High concentrations of neuronal mitochondria enable the intense oxidative metabolic capacity for energy-dependent restoration of ionic balance after firing of action potentials. Astrocytic mitochondria are important to support energy needs for glutamate–glutamine shuttle activity and ion homeostasis at the tripartite-synapse. 32 Within astrocytes, mitochondria are abundant in the processes, including the fine astrocytic processes.33,34 Large populations of astrocytic mitochondria are also found to co-localize with glutamate transporters to dynamically regulate excitatory signaling and brain energetics. 35 Mitochondrial populations are relatively lesser in endothelial cells compared to neurons and glia. Though mitochondrial contribution to endothelial oxidative metabolism is insignificant, they play a major in intracellular Ca2+ homeostasis in close association with the endoplasmic reticulum (ER). 36
Mitochondria are dynamic and move within cells by their interactions with cytoskeleton (i.e. intermediate filaments and microtubules). Miro1, a mitochondrial Rho-GTPase protein has been identified as the mitochondrial trafficking and activity-driven positioning of mitochondria in neuronal synapses. 37 However, recently Miro 1 has been shown to have a role in mitochondrial dynamics and positioning in astrocytes too. 34 It is not very clear if functional properties of individual mitochondrion change when they move to different locations within cells, but several brain studies demonstrate mitochondrial functional heterogeneity with synaptic and peripheral organelles differing in their enzymatic distribution. 38 These differences in proteins, based on the spatial location of mitochondria, support the hypothesis that they maybe functionally specialized. Such heterogeneous localization maybe important for the source specificity of mitochondrial Ca2+ uptake whether near plasma membrane Ca2+ channels or ER Ca2+ release channels. Mitochondrial numbers per cell depend on the particular cell’s functional specialization such as its signaling and metabolic roles. For example, larger numbers of mitochondria in neurons correlate with their high oxidative metabolic capacity and ion homeostatic activity, whereas mitochondrial numbers are relatively lesser in astrocytes with further differences in their Ca2+ uptake 39 and metabolic properties. 40
Calcium ions (Ca2+) and neural signaling in the brain
Synaptic communication in the brain is enabled by Ca2+ movement within several tissue compartments. Free Ca2+ ions are typically maintained at low levels in the cytoplasm (100–400 nM) compared to the extracellular space (1–2 mM) (Figure 1(a)). Such a large difference in Ca2+ concentration leads to substantial intracellular–extracellular electrochemical gradient enabling Ca2+ to enter cells through various Ca2+ channels during signaling events such as release of neurotransmitters, action potentials, vasomotion, hormone secretion, and gene expression. Of these processes, release of neurotransmitters, action potentials, and vasomotion are likely to affect neuronal activity and its related hemodynamic responses in time-scales of most neuroimaging experiments. During task-related brain activity, action potentials in presynaptic neurons depolarize their plasma membrane, releasing neurotransmitters into the synapse in a cytoplasmic Ca2+-dependent manner.
1
The released neurotransmitters bind postsynaptic receptors transferring Ca2+ from extracellular space into the cytoplasm41–45 or release Ca2+ from ER stores into the cytoplasm of postsynaptic neurons.46,47 The ensuing membrane depolarization opens voltage sensitive ion channels that generate action potentials in postsynaptic neurons, enabling neuronal communication. However, neural cytoplasmic Ca2+ homeostasis needs to be maintained through efficient buffering and extrusion for normal brain function and this consumes energy from hydrolysis of adenosine triphosphate (ATP) molecules. During neuronal activity, transient cytoplasmic Ca2+ increase leads to mitochondrial Ca2+ uptake via the mitochondrial calcium uniporter (mCU) channel complex, thereby increasing mitochondrial matrix Ca2+ level.9,48–51 During moderate to high Ca2+ loads (0.5 to 2.0 mM range), Ca2+ uptake into mitochondrial matrix is catalyzed by the mCU, driven by the membrane potential constituent of the respiratory proton motive gradient, Δψm (about −180 mV).
52
mCU allows Ca2+ entry aided by its electrochemical gradient maintained across the mitochondrial inner membrane without any ATP hydrolysis or co-transport of other ions. Influx of Ca2+ into mitochondria during neural activity not only stimulates dehydrogenase enzymes in a Ca2+-dependent manner to accelerate ATP production,
14
but also influence neural signaling by modulating the amplitude and duration of cytoplasmic Ca2+ transients.11,53 Uptake of Ca2+ by mitochondria generally occurs via microdomains of high Ca2+ located around ER channels, plasma membrane, and nucleus (reviewed by Rizzuto and Pozzan
54
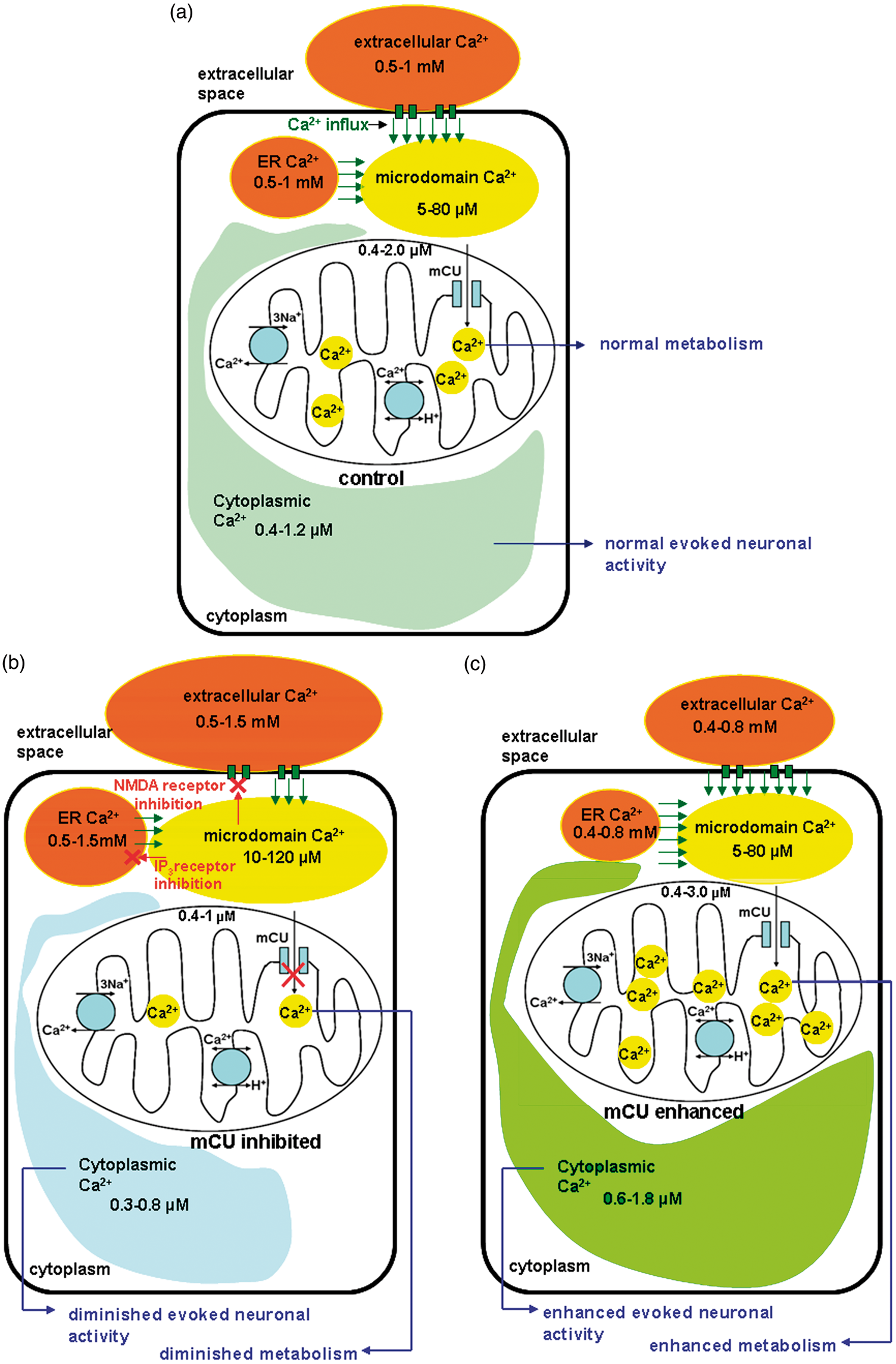
). These Ca2+ microdomains, in their interactions with mitochondria, have the ability to alter inositol 1,4,5 tris-phosphate (IP3) channel openings to affect IP3 receptor activation through a positive feedback of local Ca2+ concentration.55,56 Additionally, Ca2+ microdomains proximal to plasma membrane also interact with N-methyl D-aspartate (NMDA) receptor channels to modulate their activity through a negative feedback.11,57,58 From extensive in vitro evidence on cellular signaling and its integration by mitochondrial Ca2+ uptake, neuronal activity, and neurometabolic regulation can be hypothesized to be controlled through mitochondrial Ca2+ uptake and its consequence on the net cytoplasmic Ca2+ distribution (Figure 1(a)). Systems level studies that perturb mitochondrial Ca2+ uptake indicate that decreased or increased mitochondrial Ca2+ uptake capacity impact both intrinsic23,59 and evoked brain activity in the form of altered neuronal network activity and neurovascular coupling.19,22 These scenarios of decreased or increased mitochondrial Ca2+ uptake on the resulting activity-induced intracellular Ca2+ ranges and their cellular consequences are depicted in Figure 1(b) and (c), respectively.
Multi-compartment distribution of free Ca2+ in the brain in vivo during evoked neural activity conditions based on in vitro literature Ca2+ measurements from neurons, glia, and brain slices (a) during normal physiological conditions, evoked brain activity decreases extracellular and endoplasmic reticular Ca2+ and increases cytoplasmic and mitochondria Ca2+ (b) during mCU inhibition, evoked neural activity leads to a relatively smaller decrease in extracellular and endoplasmic reticular (ER) Ca2+ and smaller increases in cytoplasmic and mitochondrial Ca2+. This is due to accumulating microdomain Ca2+ which leads to a feedback inhibition of plasma membrane glutamate receptors and ER Ca2+ release. Hence diminished mitochondrial Ca2+ influx capacity reduces neocortical excitability, metabolism, and hemodynamic response (c) during mCU enhancement evoked neural activity leads to relatively larger decreases in extracellular and ER Ca2+ and relatively larger increases in cytoplasmic and mitochondrial Ca2+. Clearance of Ca2+ in the microdomains by augmented mitochondrial Ca2+ uptake and cycling delays N-methyl-D-aspartate (NMDA) receptor desensitization, resulting in larger cytoplasmic Ca2+. Hence an enhanced mitochondrial Ca2+ influx state increases neocortical excitability, metabolism, and hemodynamic response. Reproduced with modifications from Sanganahalli et al.
22
Mitochondrial Ca2+ uptake and cycling
During periods of increased cellular activity, transient cytoplasmic Ca2+ increases are actively sequestered by mitochondria via the mCU channel complex leading to increases in mitochondrial matrix Ca2+ levels.9,48–51 Ca2+ enters mitochondria through the electrochemical gradient provided by the mitochondrial membrane potential (Δψm). Excess mitochondrial Ca2+ is extruded through the mitochondrial Na+-Ca2+ (mNCX) and H+-Ca2+ exchangers (mHCX) in a relatively slower manner into the cytoplasm. Mitochondria via the influx and cycling process act as integrators of intracellular Ca2+ signals.
15
At the molecular level, rates of Ca2+ influx via the mCU channel complex maybe influenced by certain regulating domains, MICU1, outside the transmembrane domain of the mCU channel complex
60
(Figure 2). These domains are regulated by intracellular Ca2+ itself, the increase of which tends to increase mitochondrial Ca2+ influx rates, resulting in non-linear rates of Ca2+ uptake.
61
Recent studies using Förster resonance energy transfer (FRET) have characterized a novel protein MCUb, which is a subunit of the oligomer mCU.
At the molecular level, the mitochondrial calcium uniporter (mCU) complex is a transmembrane protein complex situated on the inner mitochondrial membrane. mCU complex is made up of four subunits formed by different combinations of mCU and mCUb. Outside the transmembrane regions are certain regulating domains such as EMRE, MICU1, and MCUR1, all of which regulate Ca2+ influx through the transmembrane pore formed by the four subunits. (a) Neuronal and glial baseline intracellular Ca2+ is typically in the range of 100–200 nM and the mCU channel is mostly in the closed state. (b) During evoked neuronal and glial activity, intracellular Ca2+ rises above 500 nM and the mCU channel is in the open state, activated by intracellular Ca2+ rising above the set level of 500 nM. In the open configuration, cytoplasmic Ca2+ within microdomains close to mitochondria enter the matrix via the mCU channel pore driven by the electrochemical gradient provided by negative mitochondrial membrane potential (Δψm).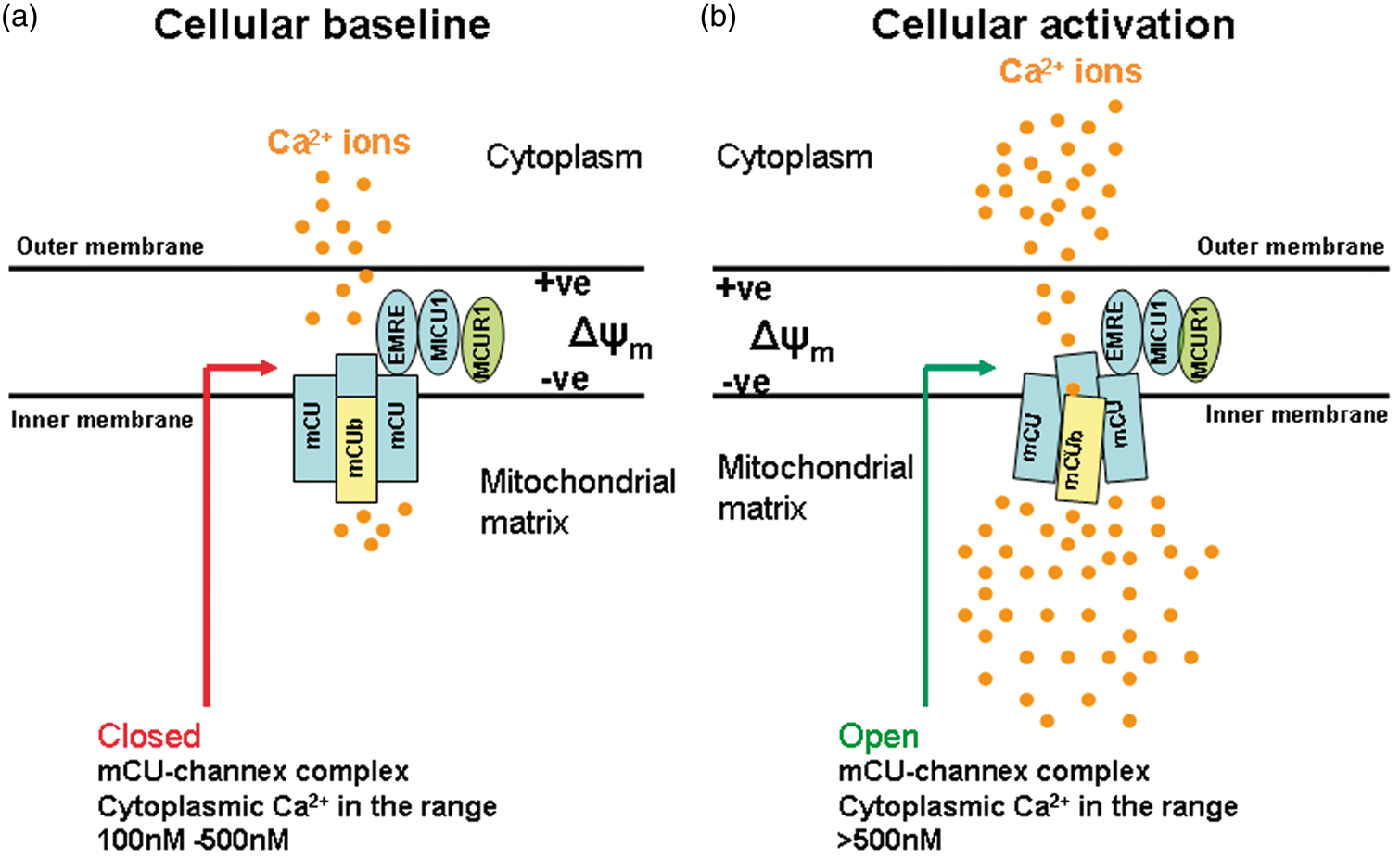
Computational modeling and experimental studies of MCUb reveal critical amino-acid substitutions in the pore region that do not form a Ca2+-permeable channel in planar lipid bilayers. 62 This dominant negative pore forming subunit when selectively expressed has been suggested to offer protection against excitotoxicity. Additionally, mCU expression can also be repressed in a nuclear Ca2+-dependent manner leading to protective effects against NMDA-receptor dependent excitotoxicity. 63 mCU and mCUb activities are regulated by additional subunits MICU1, MICU2, MCUR1, and EMRE. 64 Hence mCU channel population numbers and their regulatory subunit configurations that efficiently conduct Ca2+ and other isoforms that do not conduct Ca2+ during cellular activity may become important factors determining the diversity of mitochondrial function in different brain regions during normal and pathological states 59 (Figure 2).
Properties of the mCU channel complex may vary in different types of cells depending on their subunit and regulating domain configuration. A critical analysis of mitochondrial Ca2+ influx from electrophysiological studies of reconstituted mCU protein in lipid bilayer and mitoplasts indicated larger mCU-mediated current amplitudes in skeletal muscle mitochondria compared to cardiac mitochondria. 61 Since there is a relatively larger density of mitochondria and hence mCU channel populations in cardiac myocytes compared to skeletal muscle cells, larger mitochondrial density (and hence more mCU population) in cardiac myocytes, may adequately compensate for the relatively lesser mCU-related Ca2+ current amplitudes compared to skeletal muscle cells. Channel numbers versus flux rate compensations result in a overall equivalent mitochondrial Ca2+ uptake in cells from the two different tissues (cardiac vs. skeletal) despite differences in current amplitudes per mCU channel. 61 If similar variations of mCU-mediated Ca2+ currents are assumed across different cell types in the brain (e.g. neurons vs. astrocytes), neurons containing the largest mitochondrial density, and hence more mCU channels, can be expected to have a relatively smaller rate of mitochondrial Ca2+ uptake per mCU channel relative to astrocytes to compensate for equivalent mitochondrial Ca2+ uptake across the cell types. While the primary mechanism of mitochondrial Ca2+ entry is through the mCU channel complex, this process is relatively smaller in scale compared to alternative Ca2+ buffering process such as the sarcoplasmic reticulum Ca2+–ATPase (SERCA) and plasma membrane Na+–Ca2+ exchanger (pmNCX) activities. Hence mitochondria are unlikely to control cytoplasmic Ca2+ levels only through uptake and buffering of Ca2+ given its small magnitude compared to alternative and stronger clearance mechanisms. 61
A re-evaluation of mitochondrial Ca2+ uptake and cellular Ca2+ homeostasis
Two decades ago, critical research using cerebellar granule cells called for the re-evaluation of mitochondrial Ca2+ uptake and homeostasis during cellular activity.4,65 These studies provided evidence of decreased mitochondrial Ca2+ transients when mitochondrial Ca2+ uptake was inhibited. The results contradicted majority of the existing neuronal cell culture level studies, which showed increases in cytplasmic Ca2+ transients during mitochondrial Ca2+ uptake blockade. An important caveat in most of the in vitro neuronal studies probing cellular effects of mitochondrial Ca2+ uptake was the use of mitochondrial uncouplers in their approach, which lead to bioenergetic stress (i.e. decreased ATP levels). Mitochondrial uncouplers not only depolarize mitochondria by drastically reducing mitochondrial membrane potential (ΔΨm), but also consequently reverse the F0F1 ATPase enzyme activity. This leads to uncontrolled hydrolysis of ATP and a compromised cellular bioenergetic state. 4 With sub-optimal energy-dependent Ca2+ clearance from the cytoplasm (by SERCA and pmNCX activities), which are the larger scale Ca2+ clearance mechanisms, higher cytoplasmic Ca2+ responses to stimulation are imminent.66–68 However, in studies that did not compromise neuronal bioenergetics during depolarization of ΔΨm in order to inhibit mitochondrial Ca2+ uptake, stimulation with excitatory amino acids attenuated cytoplasmic Ca2+ responses. 4 In subsequent studies in CNS neurons and brain slices, a similar trend of attenuated Ca2+ responses were reproduced after inhibition of mitochondrial Ca2+ uptake by various methods.11,53,65 Recently in peripheral neurons in vivo and non-CNS cells from the mCU knockout mice, no significant changes were observed in cytoplasmic Ca2+ transients with and without mitochondrial Ca2+ uptake inhibition.8,69 Overall, when cellular bioenergetics is not compromised, in vitro, ex vivo and in vivo measures suggest a minimal impact of mitochondrial Ca2+ uptake on cytoplasmic Ca2+ transients in non-CNS cells and a significant reduction of cytoplasmic Ca2+ transients in CNS cells and tissue. Hence, both cellular and mitochondrial bioenergetic states should be carefully considered for accurate determination of mitochondrial Ca2+ uptake functions and its impact on CNS cytoplasmic Ca2+ levels. Based on the overall evidence, the re-evaluated scheme of inhibited mitochondrial Ca2+ uptake better explains recent in vivo neuronal activity and neurovascular coupling responses (Figure 1(b)). In this revised view, under physiological Ca2+ loads, mitochondrial Ca2+ uptake is more likely to dynamically influence cytoplasmic Ca2+ levels through its mCU-dependent feedback inhibition of plasma membrane and ER receptor activities. Hence mitochondria should be considered as active controllers and integrators of neural Ca2+ signals as opposed to just passive buffers of cytoplasmic Ca2+. 22
Average cytoplasmic Ca2+ responses also need to be interpreted cautiously. Based on results at the cellular and systemic levels, there are indicators that mitochondrial Ca2+ uptake process may dynamically control cellular signaling and oxidative metabolism through interactions with Ca2+ microdomains.4,70–72 Hence mitochondrial interactions with cytoplasmic Ca2+ ions are spatially compartmentalized 73 and may not exactly reflect average cytoplasmic Ca2+ levels typically measured by wide-field fluorescence measurements of Ca2+.19,26 Mitochondrial Ca2+ uptake directly influence Ca2+ influx through the NMDA subtype of glutamate receptors, thereby reducing its activity during mitochondrial Ca2+ uptake inhibition shown by Mn2+ quench experiments. 11 Such reduced cytoplasmic Ca2+ influx may occur through a feed back inhibition of NMDA channels by high concentrations of accumulating Ca2+ in microdomains between mitochondria and plasma membrane locations of NMDA channels.57,58 Hence, mitochondria may not only influence the overall intracellular Ca2+ levels by active uptake via the mCU channel complex, but additionally control cytoplasmic Ca2+ mobilization through feedback activity on plasma membrane receptors 11 and ER stores. Hence mitochondrial Ca2+ uptake capacity may determine the achievable level of brain electrical and metabolic activity. 22 In this active role, the maintenance of cytoplasmic Ca2+ homeostasis through mitochondrial Ca2+ uptake and buffering is no more passive but rather dynamic through its capacity to influence plasma membrane, ER receptor activity and hence tissue level neurophysiological responses (Figure 1(a), (b) and (c)).
Mitochondrial Ca2+-dependent regulation of cellular oxidative energy metabolism
Energy demand in the brain is mainly oxidative 74 with the brain consuming approximately 20% of the body’s oxygen supply. Since mitochondria are the main source of cellular oxidative metabolism, it is a critical subcellular organelle in the brain. 75 Neural energy demand increases during both spontaneous and evoked activities necessitating increases in oxidative energy metabolism.10,74 Mitochondria not only respond to context-dependent fluctuations in energy by accelerating or decelerating mitochondrial dehydrogenase activity in a Ca2+-dependent manner, 14 but also maintain baseline metabolic states of the brain in a dynamic neural activity-coupled manner.76–79 Association of mitochondria to ER and plasma membrane Ca2+ channels enable rapid and efficient transfer of Ca2+ into mitochondria to stimulate oxidative metabolism during increased ATP demand, thus maintaining brain energy equilibrium. 80 mCU channel complex-mediated rapid Ca2+ uptake is the primary mechanism known by which mitochondrial oxidative metabolism is regulated via the activity of several mitochondrial dehydrogenases in a matrix Ca2+-dependent manner.6,14 As the mCU channel complex and its regulatory domains render it as a low affinity Ca2+ uptake channel, efficient transfer of Ca2+ into the mitochondrial matrix mostly occur through interactions of mCU channel with cytoplasmic Ca2+ microdomains near specific plasma membrane Ca2+ influx channels and ER channels where the Ca2+ concentrations can be many fold higher than net cytoplasmic Ca2+ levels. In situations where moderate Ca2+ rises occur within the cytoplasm, a complementary mechanism of mitochondrial Ca2+ uptake exists through the activity of aralar, a mitochondrial aspartate–glutamate carrier protein.81,82 Aralar is present in most principal neurons with the most abundance in neurons of the brain stem and spinal cord. 83 Though some studies indicate that astrocytes and inter-neurons do not express aralar, 83 others indicate its presence,84,85 and there is currently no consensus. Alternate pathways of mitochondrial Ca2+ uptake have been recently discovered with studies showing the presence of canonical transient receptor potential-3 (TRPC3) channels in mitochondrial fractions isolated from liver and brain. 86 However the exact physiological roles of TRPC3 channels related to mitochondrial function need to be established in further detail. Hence, at present only the mCU channel can be certainly considered as a dominant channel protein involved in the rapid influx of Ca2+ into mitochondria.
Mechanisms of neurovascular, neurometabolic coupling, and mitochondrial Ca2+ integration
During normal physiological states, CBF changes due to evoked brain activity correlate linearly with oxygen consumption,20,87 however oxygen or energy consumption alone are considered insufficient to drive cerebral hyperemia.
88
For example, in humans an increased lactate/pyruvate ratio can augment evoked activation-induced CBF responses.
88
Thus, brain baseline states and the extent of its perturbation from the normal baseline are needed to accurately interpret evoked hemodynamic responses.
89
Baseline differences also confound ex-vivo brain slice studies wherein dilation and constriction of vasculature compete against each other with dilations dominating at lower pO2.
90
Depending on baseline pO2 levels, opposing effects of vasoconstriction or vasodilation have been obtained in ex-vivo preparations that lack blood hemodynamics and vascular pressures.91,92 While this suggests that a transient lowering of pO2 may drive much of the CBF response during activated states, it is very unlikely that pO2 mechanisms may operate effectively in the time scale of seconds during cerebral hyperemia.
93
Hence in vivo, activation-induced hemodynamic measures such as CBF, BOLD, and CBV may be considered more reliable indicators of neurovascular coupling and have also been used to model cerebral oxidative metabolism (CMRO2).
94
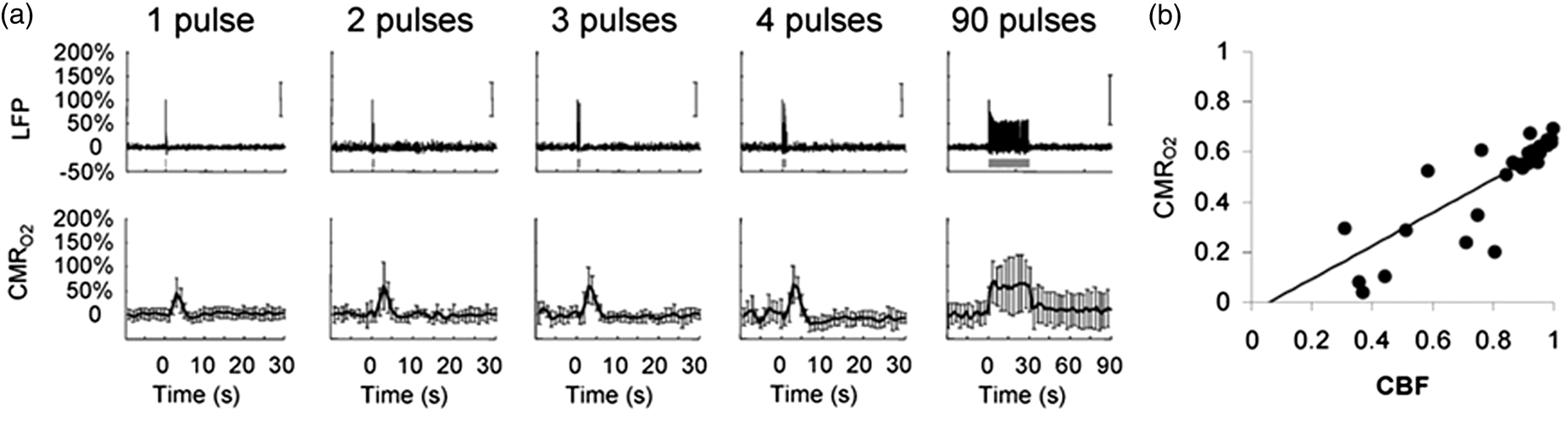
These in vivo results show a linear correlation between neuronal electrical activity and oxidative metabolic demand (ΔCMRO2) during both steady state and transient conditions in the neocortex20,95 (Figure 3(a) and (b)). Similar electrical activity and oxidative metabolic demand relationship has been observed in the cerebellum.
19
Such in vivo measures obtainable non-invasively in preclinical animal models or humans, combined with neuronal electrical measures, may have the potential to delve deeper into mechanisms of neurovascular and neurometabolic coupling and its relationship with mitochondrial Ca2+ uptake and cycling. Based on a stable neurometabolic coupling relationship, earlier brain studies considered spontaneous neuronal activity as a sensitive measure of baseline oxidative metabolism and mitochondrial redox state.
96
Stable activation-induced neurometabolic coupling can exist when mitochondrial Ca2+ uptake capacities vary in either direction (inhibited or enhanced). Supporting this, recent systems-level studies indicate that mitochondrial Ca2+ uptake inhibition decreased resting-state spontaneous neuronal activity and oxidative metabolism to a lower baseline, whereas enhanced mitochondrial Ca2+ uptake increased neuronal activity and oxidative metabolism to a higher baseline.
22
Such a tight coupling between oxidative metabolic changes that parallel evoked synaptic activity is possible through the mitochondrial Ca2+ uptake process. This way, mitochondrial Ca2+ uptake and cycling via the mCU may actively control spontaneous and evoked neuronal activities and their related metabolic responses. These interacting hot spots of Ca2+ can be highly compartmentalized in space and may not be accurately measured by conventional fluorescent dye measurements of intracellular Ca2+ in most circumstances. Hence a constant neuronal activity to oxidative metabolic demand ratio can be maintained by the brain under widely varying conditions of average cytoplasmic Ca2+ levels, oxidative metabolism, and neuronal activity (Figure 1(b) and (c)).22,23 Hence evoked increases in CMRO2 may not always require proportional neuronal cytoplasmic Ca2+ increases.
19
It is also important to consider that average cytoplasmic Ca2+ distribution measurements may have limited value compared to microdomain Ca2+ which is the actual spatial location of mitochondrial Ca2+ uptake interaction within the cell (Figure 1(a)). Considering different brain regions, activity-dependent local CMRO2 and postsynaptic currents proportionally correlate along with a CBF and tissue PO2 decrease during mitochondrial Ca2+ uptake blockade with Ru360 in the cerebellum,
19
very similar to Ru360-induced baseline BOLD signal decrease in the cerebral cortex.
22
Differences in regional mitochondrial Ca2+ uptake capacities may also exist as indicated by differences in the resting state spontaneous intrinsic activity of the brain as measured by fMRI-BOLD fluctuations during mitochondrial perturbations.
59
Given the constant CMRO2/LFP ratios between cerebrum and cerebellum, a constant activity–metabolic relationship can be expected throughout the brain, despite the possibility of regional differences in intrinsic mCU kinetics and mCU population density across regions.
Neurovascular and neurometabolic coupling. (a) Neocortical electrical activity from neuronal populations (local field potential-LFP) evoke a proportional cerebral metabolic rates of oxygen utilization (CMRO2) during transient to steady-state stimuli. (b) During these transient to steady-state stimuli neural activity-dependent changes in CBF and CMRO2 are linear (slope = 0.661, interception = −0.039, r2 = 0.81). Reproduced with permission from reference Herman et al.
95
Vascular smooth muscle and endothelial mitochondrial Ca2+ uptake on cerebrovascular reactivity
Arterial smooth muscle and endothelial cells bring about vasomotive (vasodilatory and vasoconstrictive) activity in the brain. Mitochondria have been shown to regulate local and global Ca2+ signals in cerebral arterial smooth muscle cells. 97 Inhibition of mitochondrial Ca2+ uptake using Ru360 and release using CGP37157 differentially regulate transient Ca2+ sensitive potassium channel (KCa) currents, suggesting that mitochondrial membrane potential may regulate Ca2+ sparks and transient KCa currents by modulating mitochondrial matrix Ca2+ concentration. 97 Mitochondrial Ca2+ uptake stimulates the production of nitric oxide (NO) in vascular endothelial cells. 98 NO being a modulator of neurovascular coupling,99,100 may exert some mitochondrial Ca2+-dependent effects. Testing for effects of mitochondrial Ca2+ uptake on cerebral endothelial or smooth-muscle cells, we used a transient hypercapnic vasodilatory stimulus and measured hypercapnia-induced CBF responses in the neocortex using Laser Doppler Imaging. 25 No significant difference in the hypercapnia-induced changes in neocortical CBF was observed. Hence there is no compelling evidence yet to support a significant role of endothelial or smooth muscle mitochondrial Ca2+ uptake and cycling on physiological cerebrovascular reactivity responses.
Neuronal and glial Ca2+ change mitochondria and neurovascular coupling
Mechanisms of blood supply regulation in the brain in its baseline and activated state in relation to its energy need is complex involving metabolism and neurotransmission. Hence cellular and molecular level experiments studying activity-induced hemodynamics, systems level functional neuroimaging of hemodynamic control, and theoretical modeling are needed to accurately interpret neurovascular coupling in the brain. Earlier defined mechanism for neurovascular coupling was the metabolic feedback, signaling the demand for oxygen and energy substrates during evoked brain activation, thereby increasing CBF.93,101 Feedback is initiated by consumption of oxygen and energy substrates such as glucose during neural activity via signaling intermediates including carbon-dioxide (CO2), potassium ions (K+) or adenosine
102
(Figure 4). However, manipulation of blood oxygen levels and glucose do not support oxygen or glucose demand alone to control cerebral hyperemia (reviewed by Attwell et al.
103
). Subsequent feedforward mechanisms of neuronal and glial neurotransmission controlling CBF have been proposed.
104
Here, neurons either directly or in interactions with astrocytes via glutamate neurotransmitter signaling, dilate blood vessels in a Ca2+-dependent manner92,105,106 (reviewed by Attwell et al.
103
) (Figure 4). Hence neuronal activity-dependent metabolic feedback and feedforward mechanisms of neurovascular coupling via astrocytic Ca2+ signaling are equally important to completely explain neurovascular coupling. Confounds of vasodilation and vasoconstriction responses in ex vivo brain slice studies91,92,107 and discrepencies related to astrocytic Ca2+ responses were some challenges to the feedforward mechanism. Using a knockout mouse model lacking the inositol 1,4,5-triphosphate (IP3) receptor mediated astrocytic Ca2+ mobilization, intact stimulation-induced vasodilation response was recently observed. But the astrocytic end-feet Ca2+ responses temporally followed vasodilative responses leading to the conclusion that astrocytic Ca2+ may not be involved in the neurovascular coupling cascade.
108
Hence astrocytic Ca2+-mediated control of stimulation-induced cerebral hyperemia generated several new questions (reviewed by Kowianski et al.
109
).
Feedback mechanism of neurovascular coupling controls vasodilation of cerebral blood vessels through neuronal and glial metabolism. Glucose and oxygen (O2) demand during neuronal activation generates signaling molecules such as CO2, nitric oxide (NO) and increased extracellular K+ which dilate cerebral blood vessels. Glucose and oxygen (O2) demand during glial activation due to glutamate transmitter uptake lead to increased extracellular Na+. Increased anaerobic metabolism in glia also lead to higher extracellular lactate. Increased extracellular acidification, combined with higher intracellular Na+ and extracellular K+, dilate cerebral blood vessels. Feedforward mechanism of neurovascular coupling controls vasodilation of cerebral blood vessels through neurotransmitter signaling. During neuronal activity activation of N-methyl-D-aspartate receptors (NMDARs) lead to increased intracellular Ca2+ levels. This leads to activation of neuronal nitric oxide synthase (nNOS) and phospholipase A2 pathway to produce arachidonic acid (AA) and prostaglandins (PG) which lead to vasodilation of cerebral blood vessels. Glial activation by metabotropic glutamate receptors (mGLURs) increase Ca2+ concentration in the end feet of astrocytes lining blood vessels. Increased end feet Ca2+ activates phospholipase A2 pathway to produce AA, epoxyeicosatrienoic acids (EET) via P450 epoxygenase and PG, which dilate blood vessels. Additionally increased end feet Ca2+ in astrocytes can activate Ca2+ gated K+ channels; gk(Ca), releasing K+ which dilates blood vessels. The feedback and feedforward mechanisms are unified by intracellular Ca2+ signals and the mitochondrial Ca2+ uptake process.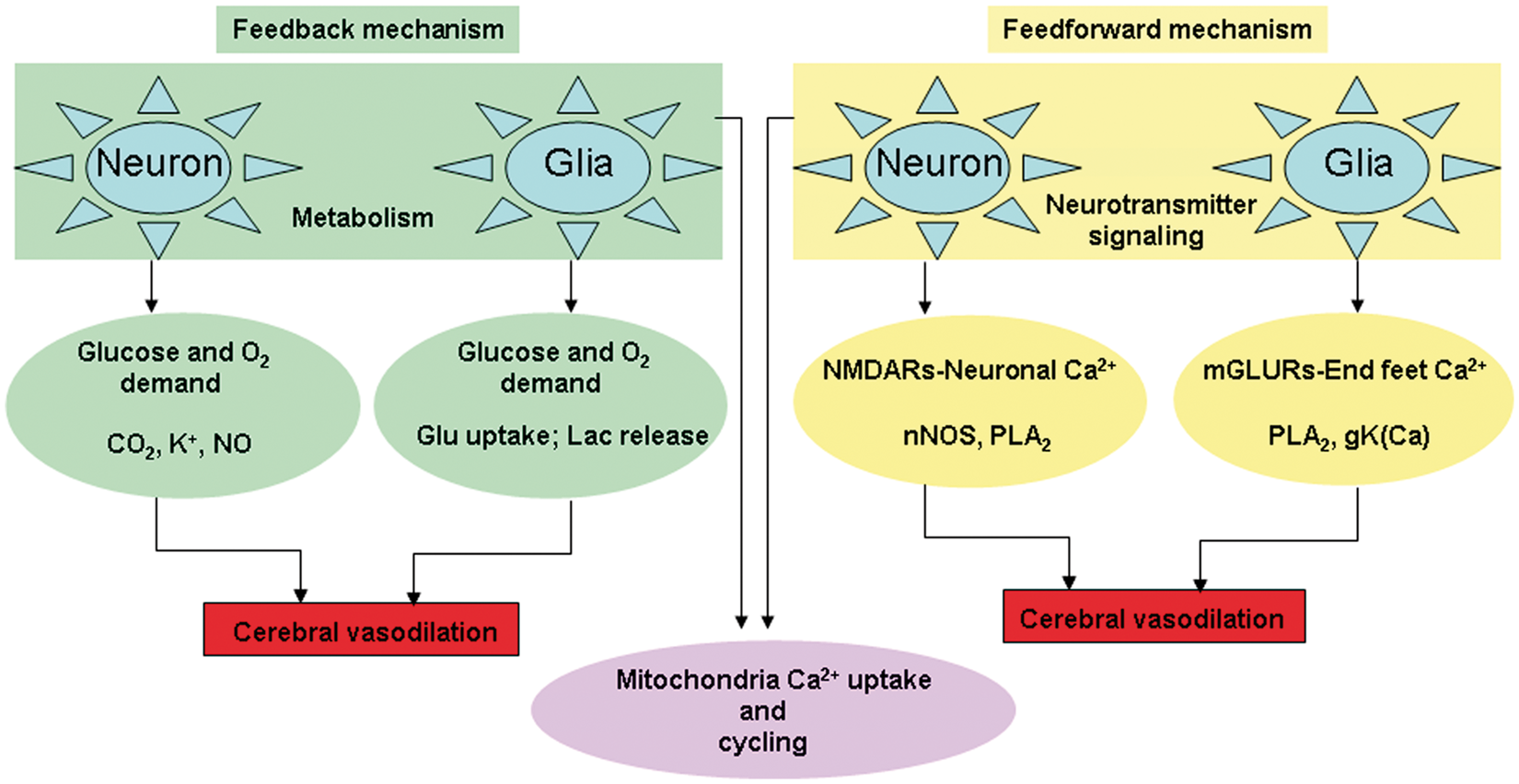
Recent in vivo studies, however, provide clear evidence of astrocyte Ca2+-mediated vasodilation,110,111 thus clearing some of the earlier ex vivo confounds. Measures of rapid onset-short durations of cytoplasmic Ca2+ in the somatosensory cortex suggest that astrocytic soma and end feet Ca2+ transients temporally precede local vasodilatation. 112 These fast Ca2+ transients in the astrocytes also correlate with synaptic activity. Very recently the role of astrocytic Ca2+ in neurovascular coupling was re-examined in the olfactory cortex, in vivo. 110 By selectively expressing a genetically encoded Ca2+ sensor in astrocytes of the olfactory bulb, physiological activation of olfactory sensory neuron (OSN) terminals reliably triggers Ca2+ increases in astrocyte processes but not in somata, which preceeded the onset of functional hyperemia by 1–2 s. Given these strong in vivo correlation of astrocytic end feet Ca2+ responses with cerebral vasodilation 111 and a high localization of mitochondria in these regions, 34 mitochondrial Ca2+ uptake in astrocytes may need to be critically considered to completely understand neurovascular coupling at the systems level. In order to explain why in certain scenarios cytoplasmic Ca2+ in astrocytes may not precede vasodilatation, it is important to differentiate average cytoplasmic Ca2+ with microdomain Ca2+ levels and their mitochondrial interactions. 113 This amounts to limiting the importance of sporadic average distribution of cytoplasmic Ca2+ in a few astrocytes during stimulation-evoked cerebral hyperemia in vivo. Mitochondria and ER are strategically placed and in close proximity in cells of many tissues including the brain that interact through mitochondria-associated membranes (MAMs) in neurons and astrocytes 114 (reviewed by Patergnani et al. 115 ). There is a strong possibility that neuronal and astrocytic excitations through the metabotropic glutamate receptors that lead to IP3 receptor-mediated Ca2+ release from ER maybe rapidly transferred to mitochondria with limited spill over into the bulk cytoplasm and maybe visible in only specialized optical experimental configurations capable of measuring rapid onset Ca2+ transients. 112 While this may occur within the time ranges of activation-induced hyperemia, such a rapid mitochondrial matrix Ca2+-dependent energy metabolic response explains the fast kinetics of the astrocyte-glutamate transporter. 116
Overall, ‘Neuronal and glial signaling’ and ‘oxidative energy metabolism’ seem to be the principal basis defining the feedforward and feedback mechanisms, respectively, and the two processes are coupled by mitochondrial Ca2+ uptake process (Figure 4). This is supported by recent in vivo evidence on the dependency of ‘neuronal signaling’, ‘oxidative energy metabolism’, and ‘neurovascular coupling’ on mitochondrial Ca2+ uptake capacity.8,19,22,23,25,26,59 Furthermore, mathematical modeling approaches to explain neurovascular coupling in vivo suggest that addition of metabolism to the neurotransmitter feedforward model structure better explained biologically plausible experimental neuroimaging outcomes. 117 Hence separating feedforward and feedback mechanisms may be unnecessary since they seem to be coupled via common biochemical pathways. 118
Mitochondrial function and multimodal neuroimaging markers
Evoked neuronal activation in the brain gives rise to changes in CBF, which increases in parallel with cerebral metabolic rate of oxygen utilization (CMRO2) and CBV. Changes in these physiological variables influence deoxyhemoglobin (dHb) concentrations within the brain vasculature. fMRI using the BOLD and CBF contrast has emerged as an efficient and non invasive technology to map brain function in humans.119,120 fMRI relies on hemodynamic changes representative of neural activity in response to a task that is dependent on a complex interplay of neural, vasomotive, and metabolic processes.121–123 While task-related fMRI (T-fMRI) study designs help in determining evoked brain responses, alternate designs that measure resting state spontaneous brain activity are equally important. In humans, resting state-fMRI (R-fMRI) is increasingly used to measure ‘intrinsic’ brain activity. 124 R-fMRI considers the temporal correlations of low frequency (<0.1 Hz) physiological fluctuations in the absence of any task to represent functional connectivity between brain regions. 125 R-fMRI studies are expanding in clinical neuroscience applications due to its patient friendly and simple experimental design not necessitating demanding tasks. R-fMRI studies after removal of physiological and vasomotive noise 126 is an effective way to study the intrinsic modes of brain activity in health124,127,128 and disease.129–133 R-fMRI signal fluctuations are very similar to hemodynamic fluctuations demonstrated by other biophysical methods134,135 and the temporal coherence of intrinsic activity between brain regions has led to the hypothesis of a “default mode of brain function”. 124 Both T-fMRI and R-fMRI signal amplitudes correlate to a great extent, indicating they may have a similar underlying physiological mechanism. 136 Complementary in vivo measures of cerebral hemodynamics and mitochondrial oxidative metabolism through neuroimaging modalities that report changes in CBF and flavoprotein autofluoresence have strengthened interpretations of brain activity and metabolism.137,138 For example, autofluorescence imaging in anesthetized cats have shown metabolic oscillations in the low frequency (<0.1 Hz) similar to BOLD oscillations. Pyridine nucleotide oscillations indicate that the mitochondrial redox state fluctuates, preceding CBF oscillations.134,135,139 CBF oscillations 140 additionally correlate with R-fMRI BOLD fluctuations in intact animal models. 141 These results support the hypothesis that mitochondrial redox state oscillations correlate with cytoplasmic Ca2+ oscillations via mitochondrial Ca2+ uptake and cycling. To this effect, recent in vivo studies that pharmacologically modulate mitochondrial Ca2+ uptake capacity show that mitochondrial Ca2+ uptake capacity indeed influences neocortical excitability during spontaneous and evoked activity states.10,22,23 Perturbations of mitochondrial Ca2+ uptake ability in vivo also modify electrical activity of neuronal populations and systemic variables such as CBF and BOLD dynamics. These recent experimental approaches, at the systemic scale, indicate profound alterations in oxidative metabolism and the collective excitability of neuronal networks during perturbed (either increased or decreased) mitochondrial Ca2+ cycling activity.
Theoretical models that consider cellular Ca2+ changes have not only helped better understand neurovascular coupling, 142 but also support the coupling of feedback and feedforward mechanisms to better align with experimental results. 117 If the impact of mitochondrial Ca2+ cycling in neuronal and glial cells can be included, it may improve theoretical models to a more precise interpretation of neurovascular coupling. For example, there are currently unexplained scenarios where insignificant neuronal activity evoke robust neurovascular responses 143 and vice versa. 144 While baseline bioenergetic state is an important determinant, 144 differences in mitochondrial Ca2+ uptake capacities (either due to inherent mCU channel kinetics or mCU population density across cell types and brain regions) may also impact neuronal and neurovascular coupling differently. 59
Conclusion
Mitochondrial Ca2+ homeostatic functions need to be integrated from the molecular and cellular to the systemic levels in order to better understand neurovascular and neurometabolic coupling. In vivo designs that build upon prior in vitro approaches, preferably in the working brain are presently possible. To this end, net cellular Ca2+ levels in the working brain have already been assessed simultaneously or within same animal subjects with fMRI-BOLD signals and found to covary temporally and spatially during evoked brain activity.24,26 With magnetic resonance imaging and spectroscopic approaches being used to determine mitochondrial metabolic impact on the human brain145,146 and calibrated fMRI methods to simultaneously measure neurovascular and neurometabolic coupling, 147 an integrated view of mitochondrial Ca2+ homeostasis may provide a unified hypothesis to explain neurovascular coupling changes and how it may sometimes not correlate with neurometabolic coupling in the working brain.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: the New Jersey Commission for Brain Injury Research (CBIR15IRG010).
Acknowledgments
The author would like to thank Dr Fahmeed Hyder for initial in depth discussions and critical review during the development of this article.
Declaration of conflicting interests
The author(s) declared nopotential conflicts of interest with respect to the research, authorship, and/or publication of this article.