
Abstract
Herpes simplex virus vectors bearing a glucose transporter (GT) gene and a marker gene were found to protect neurons against a 1-h focal ischemic insult. Rats receiving the GT vector vα22βgalα4GT exhibited a 67.4 ± 35.3% survival of virally targeted neurons in the ischemic hemisphere compared with the contralateral control (n = 7), whereas rats receiving a control vector exhibited only 32.8 ± 17.9% survival (n = 9). This significant improvement in survival (105%, p = 0.022) suggests that energy failure is an important contributor to the neuropathology of ischemic damage in the striatum, and that it can be alleviated by gene transfer. This is the first demonstration of protection against ischemic cerebral injury by the direct transfer of GT genes to neurons.
Neurons are highly susceptible to the hypoxia–ischemia that occurs during stroke. Due to their nonreplaceable post-mitotic nature, it is essential that effective protective interventions be developed to prevent neuron death. Because of these same properties, however, the development of such interventions demands novel therapeutic approaches. With the recent advent of herpes simplex virus (HSV) and other viral vectors that are capable of overexpressing exogenous genes in mature neurons, the possibility of achieving neuroprotection by direct gene transfer has emerged. HSV vectors afford the advantage of being both neurotropic and capable of packaging large amounts of DNA for delivery to nondividing CNS neurons (Glorioso et al., 1992). Employing such vectors, we have chosen as a principal target the energetic deficits intrinsic to many neurologic crises. An HSV vector, designated vα22βgalα4GT, was generated bearing the glut-1 gene encoding the GLUT-1 isoform of the rat brain glucose transporter (GT) and the Escherichia coli lacZ marker gene. We have previously demonstrated that such GT vectors increase glucose uptake in neurons both in vitro and in vivo (Ho et al., 1993), and that this enhanced uptake protects a variety of cultured neurons against an array of metabolic and excitotoxic insults (Ho et al., 1995; Lawrence et al., 1995), and against seizure-induced (Lawrence et al., 1995) and antimetabolite-induced neuronal damage in vivo (R. Dash et al., 1995).
With a cessation of blood flow during stroke, there is a failure of glucose delivery and shift to less efficient anaerobic metabolism. Thus, stroke represents a significant energetic crisis to the affected neuron populations (Auer and Siesjo, 1988; Beal et al., 1991; Sapolsky, 1992). In this report we have investigated the potential neuroprotective effects of overexpressing the glucose transporter in neurons within the striatum before a transient focal ischemia induced by a 1-h middle cerebral artery occlusion (MCAO).
MATERIALS AND METHODS
HSV vectors were generated by harnessing the natural packaging properties of the virus (Spaete and Frenkel, 1982; Ho et al., 1993). As previously described (Lawrence et al., 1995), the plasmid pα22βgalα4GT was constructed containing the HSV origin of replication and packaging sequences and the rat brain glut-1 gene and the Escherichia coli lacZ gene under the control of the HSV α4 and α22 promoters, respectively. This plasmid was transfected into E5 cells, which are stably transfected with the HSV immediate early gene α4. Superinfection of the E5 cells with the HSV deletion mutant d120, lacking the α4 gene, gave rise to vectors, designated pα22βgalα4GT, bearing concatemers of the plasmid and replication-incompetent helper virus particles at a ratio of 1:2 (Lawrence et al., 1995). Previous investigations had demonstrated that such vector/helper virus stocks were not cytopathic when delivered to the striatum at the titers employed (Ho et al., 1994) and that they significantly enhanced glucose uptake in targeted neurons as assessed by 2-deoxy[14C]glucose uptake (Ho et al., 1993). A control vector, designated vα22βgalα4gts, was generated bearing the glut-1 gene with the insertion of a stop codon so as to produce a truncated dysfunctional gene product (Lawrence et al., 1995).
After microconcentration of viral stocks 5–10-fold in Microcon 100-kDa microfiltration devices (Amicon), 6 μl of either vα22βgalα4GT or vα22βgalα4gts was delivered bilaterally to the striatum of 350–400-g Sprague–Dawley rats (from bregma: AP = 0, ML = 3.5, with two injection sites of 3 μl each at DV = 4.5 and 3.5 mm; vector titer of 1 × 106 infectious particles/ml, helper virus titer of 2 × 106 plaque forming units/ml). Six hours later ischemia was induced by the insertion of an intraluminal 3–0 nylon suture through the common carotid artery to the branch point of the MCA and the anterior cerebral artery, resulting in the occlusion of the MCA and the reduction of collateral blood flow from the anterior and posterior cerebral arteries (Longa et al., 1989; Memezawa et al., 1992; Yoon and Steinberg, 1994). To confirm in vivo coexpression of the glut-1 and lacZ genes, one rat from each treatment group was killed 24 h after viral infusion without being subjected to MCAO. Brains were frozen in 2-methylbutane (−20°C) and 5 μm coronal sections were taken at the site of injection. Sections were then labeled with anti-β-galactosidase mouse monoclonal antibody (1:20; Sigma), and anti-GT rabbit serum (1:100; East Acres Biologicals), followed by goat anti-rabbit antibody conjugated to rhodamine (1:100; Vector Laboratories) and goat anti-mouse antibody conjugated to fluorescein (1:20; Vector Laboratories). GT and β-gal signal was detected and photomicrographs were taken on a fluorescent microscope with a 100× objective.
Brain sections from the occluded animals were prepared as previously described (Lawrence et al., 1995). Briefly, animals were killed and perfused with 4% paraformaldehyde 48 h after the occlusion. After postfixing, 25-n,m frozen sections were taken at 50-μm increments 1 mm anterior and posterior to the infusion site. Slices were stained with 3-bromo-4-chloro-5-indoyl-galacto-pyranoside (X-gal) and cresyl violet to allow the identification of healthy, intact, virally targeted neurons and to assess the extent of stroke-induced damage. The number of positive-staining blue neurons were counted at 40× magnification in successive sections within each striatum by a researcher blind to the identity of the experimental groups. Relative survivorship of vector targeted neurons for each brain were expressed as the ratio of the total number of positive neurons in the occluded hemisphere versus the number in the unoccluded control hemisphere. Infarct damage within the striatum was assigned a degree of severity on a scale of 0–5, in which 0 represented no visible damage in the occluded hemisphere under 20× magnification and 5 represented complete ablation of the striatum and surrounding cortex. Those brains exhibiting 0 damage were not included in the analysis and a comparison of damage between GT and control groups was made using a χ2 test. A comparison of lacZ-positive neuron survivorship between GT and control groups was performed by analysis of variance, and a comparison of the number of surviving neurons in the occluded hemispheres of each brain to their own contralateral controls within each group was made using a paired Student's t test.
RESULTS AND DISCUSSION
Modeling stroke by the temporary occlusion of the MCA results in lesions within the medial and lateral striatum, and overlying cerebral cortex (Longa et al., 1989; Memezawa et al., 1992; Yoon and Steinberg, 1994). Delivery of both vα22βgalα4GT and vα22βgalα4gts before the occlusion gave rise to the, expression of lacZ in striatal neurons, some of which lay within the lesioned cell field (Fig. 1A–F). Delivery of the vectors vα22βgalα4GT, but not vα22βgalα4gts, gave rise to coexpression of Glut-1 and lacZ as assessed by double immunofluorescence (Fig. 1G and H). We have previously demonstrated by immunofluorescence that the expression of lacZ in cultured Vero cells infected with vα22βgalα4GT is virtually always accompanied by the strong coexpression of GT (Lawrence et al., 1995), and now finding that the same is true in striatal neurons in vivo, we interpret positive striatal neuron lacZ staining to indicate overexpression of GT within these cells. Likewise, the morphological characteristics of lacZ-positive cells, and previous demonstrations of the strong propensity of HSV vectors to infect neurons rather than glia in cell culture and in the intact brain (Ho et al., 1993; Lawrence et al., 1995), led us to conclude that the cells included in our analysis were striatal neurons. A comparison of the titer of the infused vector stock and an estimate of the total number of successfully targeted neurons in unoccluded control cell fields indicated that there was variability in the number of neurons successfully infected as result of each microinfusion, but the overall efficiency of infection was ∼10%.
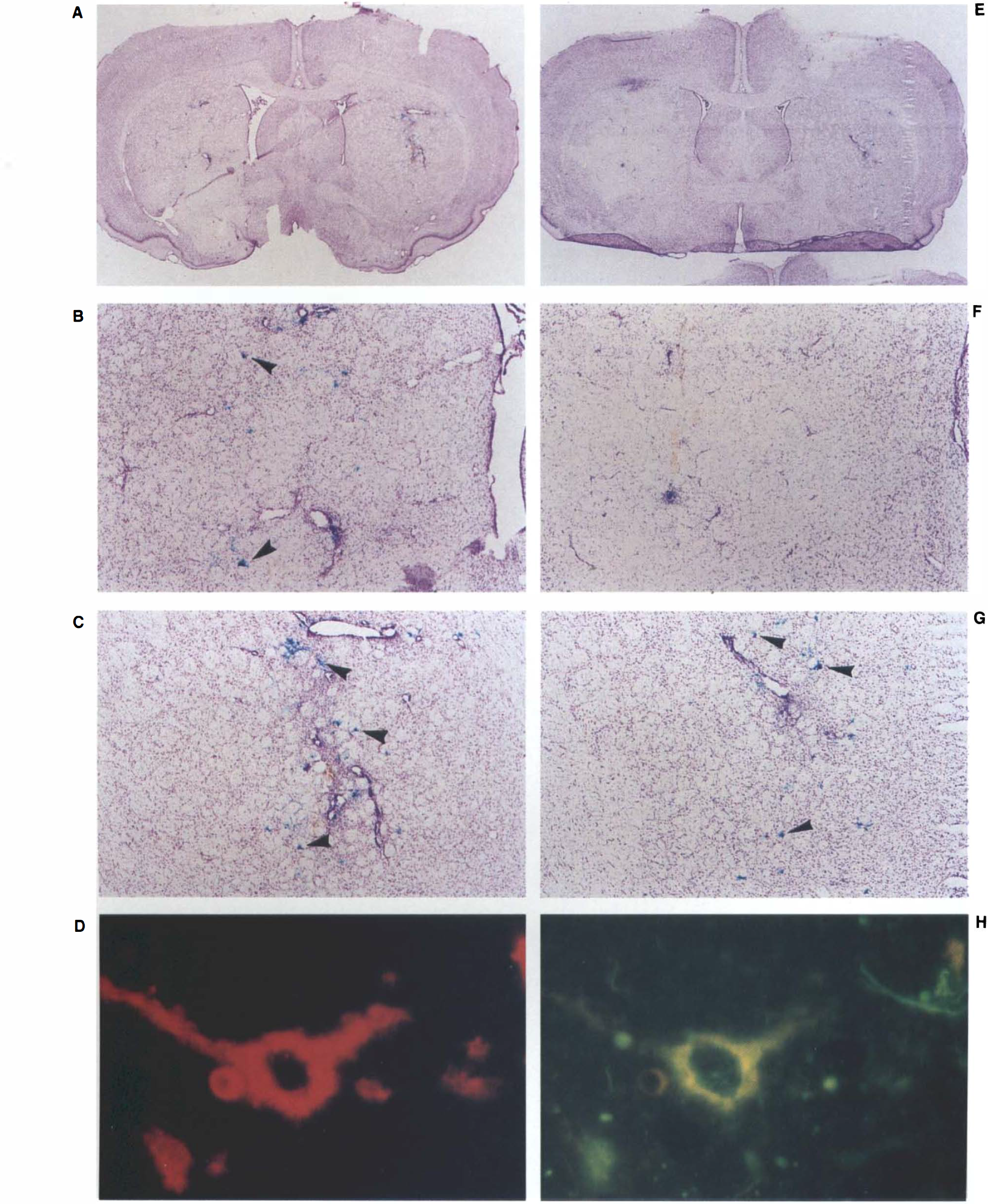
Photomicrographs of a striatal section targeted with vα22βgalα4GT reveal the extent of middle cerebral artery occlusion damage in the left hemisphere
Although both the GT and control animals exhibited reduced survivorship of lacZ positive neurons within the occluded hemisphere (p < 0.05 and p < 0.001, respectively), the number of such neurons surviving, expressed as a percent of the number of positive neurons in the contralateral control hemisphere, was significantly greater in animals receiving the GT vector (67.4 ± 35.3%) than in animals receiving the control vector (32.8 ± 17.9%; p = 0.022) (Fig. 2). Brains in GT and control groups exhibited no significant difference in the amount of total damage within the region of infusion (GT damage = 3.43 ± 1.13; control damage = 3.44 ± 1.01; χ2 = 0.237, n. s.); hence, the differential survivorship cannot be attributed to differing degrees of hypoxia–ischemia endured. The results suggest, instead, that enhanced glucose uptake can protect striatal neurons in the face of hypoxic–ischemic insults. The possibility that the GT overexpression merely acts to slow neuron death, and hence, contribute to a greater number of surviving neurons at the 48-h time point, cannot be excluded. Most cellular degradation and neuronal death that occurs after focal ischemia, however, is detectable within 48 h (Chen et al., 1993). Thus, the intact cell membranes and processes we observed in the lacZ positive neurons suggest that they survived the ischemic event. Whether damage to the neighboring neurons, glia, and microvasculature will ultimately contribute to the death of these neurons cannot be determined from the present results.
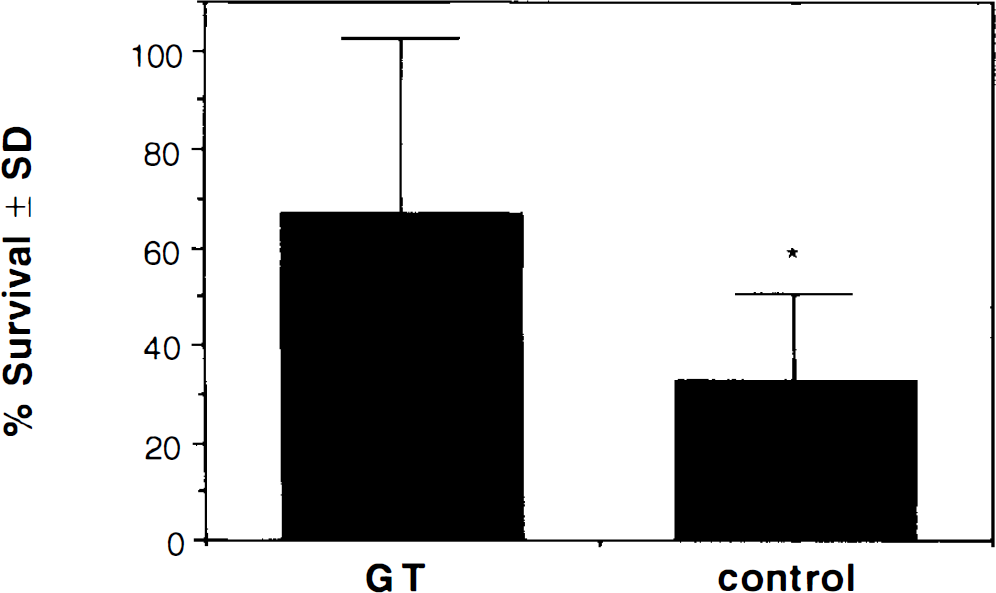
Survivorship of vα22βgalα4GT and vα22βgalα4gts-infected striatal neurons 48 h after middle cerebral artery surgery. Data are represented as the mean percent ± SD of lacZ-expressing neurons remaining in the occluded hemisphere versus the contralateral unoccluded hemisphere. Differences in survivorship between occluded and nonoccluded hemispheres within each group were significant [i.e., significantly different from 100%; p < 0.05 for glucose transporter (GT) and p < 0.001 for control] and differences between GT (n = 7) and control groups (n = 9) were significant (*p = 0.022).
Genes placed under the control of the HSV α4 promoter express within 4 h of infection and peak within 8 h, maintaining high levels of expression for 48–72 h thereafter. Thus, targeting striatal neurons with the vector 6 h before a 1-h occlusion of the MCA results in GT overexpression both during the occlusion and the subsequent reperfusion period. Previous studies have shown that supplementing neuronal energy supplies by increasing circulating glucose levels at the time of a global hypoxic–ischemic insult can be damaging, perhaps by contributing to the increased production of lactic acid (Ginsberg et al., 1980; Pulsinelli et al., 1982). Others, however, have shown that energy supplementation before a focal ischemic insult or an in vivo hypoxic insult can be protective (Schurr et al., 1987; Zasslow et al., 1989; Kraft et al., 1990), a disparity that may reflect the relative importance of acidosis in the different models studied (Tombaugh and Sapolsky, 1990). It is clear, however, that energy supplementation after a hypoxic–ischemic event is protective (Farias et al., 1990). Thus, the protective effect derived from the overexpression of the glucose transporter may arise principally from enhancement of glucose uptake in the aftermath of the occlusion. Testing such a possibility by delivery of viral vectors simultaneous to or in the immediate aftermath of MCAO was prevented by the rigor of the surgeries involved. The plausibility of such a postinsult effect, however, is supported by our previous investigations in which neuroprotection was achieved in the hippocampus by the delivery of the vector 1 h after the infusion of the excitotoxin kainate (Lawrence et al., 1995) or the antimetabolite 3-acetylpyridine (R. Dash et al., 1995). Such findings are consistent with the delayed nature of necrotic neuron death and the slow cumulative effects of the energy loss that ultimately leads to this fate. The resequestration of glutamate, restoration of ion and proton imbalances, and repair of oxidative damage place continued energetic demands on the neuron long after the hypoxic–ischemic event itself (Auer and Siesjo, 1988; Beal et al., 1991).
Under normal circumstances the transport of glucose from the blood to the brain exceeds the rate of neuronal glucose use by two- to fourfold (Betz et al., 1976; Gjedde, 1980). During ischemia, however, this unidirectional transport may be reduced to the point of becoming a rate-limiting determinant of neuronal energy states (Betz et al., 1983). The importance of rectifying the shortfall in glucose delivery that occurs during ischemia is supported by the finding that ischemia induces the expression of endogenous GT genes in neurons, astrocytes, and microvessels (Lee and Bondy, 1993). Thus, the enhancement of glucose transport by gene transfer may strengthen a natural adaptive response that allows the restoration energy balances during the reperfusion period. In the absence of reperfusion, extracellular glucose levels remain depressed and the number of transporters becomes less relevant. Under these circumstances it is unlikely that overexpression of either an endogenous or exogenous GT would be protective.
In this experiment only a small percentage of the striatal neuron population was targeted to overexpress GT, and consequently there was no reduction on the overall extent of the MCAO lesion. Furthermore, there was not complete protection of all of those neurons that were targeted. The enhanced survival of individual neurons, however, is noteworthy. Mechanistically, these results suggest that metabolic distress is an important contributor to the neuropathology of hypoxic–ischemic damage in the striatum. From a broader therapeutic perspective, they suggest that HSV vectors and other evolving viral vector strategies offer a promising means of delivering novel neuroprotective genes to the striatum and elsewhere in the CNS.
Footnotes
Acknowledgment:
This study was supported by a Howard Hughes predoctoral fellowship (M.S.L.), a Howard Hughes undergraduate research fellowship (R.D.), the American Paralysis Association and the Adler Foundation (R.M.S.), and National Institutes of Health National Institute of Neurological Diseases and Stroke grant ROl NS27292-01A2 (G.K.S.).