
Abstract
Recombinant human interleukin-1 receptor antagonist (rhIL-1ra) markedly protects against focal cerebral ischaemia in the rat, implicating endogenous IL-1 in the events leading to cerebral infarction. The present experiments investigated the effect of intracerebroventricular (i.c.v.) administration of IL-1β or rhIL-1ra on ischaemia damage and physiological parameters after permanent middle cerebral artery occlusion in the rat. IL-1β (5 ng, i.c.v.) markedly (92%) enhanced infarct volume and caused a significant rise in body temperature, but rhIL-1ra (10 μg, i.c.v.) significantly reduced infarct volume and did not significantly affect heart rate, blood pressure, or body temperature. rhIL-1ra administered 30 min before, or at the time of ischaemia significantly reduced infarct volume in cortex (55 and 60%, respectively) and striatum (52 and 41%, respectively). rhIL-1ra administered 30 min after ischaemia significantly reduced total and cortical infarct volume (26 and 29%, respectively), but did not significantly protect striatal tissue. The effects of rhIL-1ra were still evident in both cortex and striatum 7 days after ischaemia. These results support the role of IL-1 in ischaemic brain damage, revealing potent, sustained, neuroprotective effects of rhIL-1ra in the cortex and striatum, which cannot be attributed directly to changes in physiological parameters.
Research on the mechanisms underlying neuronal damage caused by cerebral ischaemia has revealed the importance of immune or inflammatory molecules and cells in the neuronal death that results from stroke. Several recent studies have implicated cytokines such as interleukin-1 (IL-1) in experimentally induced neurodegeneration (Minami et al., 1992a; 1992b; Relton and Rothwell, 1992, Yamaski et al., 1992; Wang et al., 1995; Fuerstein et al., 1995).
IL-1 was originally identified as a mediator of peripheral immune function, but both IL-1 agonists (IL-1α and IL-1β) and the endogenous receptor antagonist (rhIL-1ra) are synthesised in the brain (Breder et al., 1988; Lechan et al., 1990; Licinio et al., 1991). Constitutive expression of IL-1 in normal brain is low, but IL-1β mRNA is induced rapidly after experimentally induced cerebral ischaemia in the rat (Minami et al., 1992a; Liu et al., 1993; Buttini et al., 1994; Yabuuchi et al., 1994; Wang et al., 1995), and increased expression of the protein has been reported after brain injury in rodents and humans (McClain et al., 1987; Woodroofe et al., 1991; Yan et al., 1992; Taupin et al., 1993). IL-1 exerts diverse actions in the brain on both neuronal and glial function, some of which have potentially beneficial effects on neuronal survival after brain insults (Giulian et al., 1988; Miller et al., 1990; Spranger et al., 1990; Plato-Salaman and Ffrench-Mullen, 1992). However, we have proposed that brain IL-1 participates directly in ischaemic and traumatic brain injury. This hypothesis is based on our observation that intracerebroventricular (i.c.v.) administration of rhIL-1ra markedly (>50%) inhibits neuronal damage induced by focal cerebral ischaemia (middle cerebral artery occlusion) (Relton and Rothwell, 1992) or fluid percussion injury in the rat (Toulmond and Rothwell, 1995).
These data suggest that endogenous IL-1 mediates ischaemic brain damage, and are supported by the observations of Minami et al. (1992b) and Yamasaki et al. (1992, 1995), who reported that intracerebroventricular (i.c.v.) administration of IL-1β significantly exacerbates neuronal loss after global ischaemia in the gerbil, and oedema after temporary focal ischaemia. However, IL-1 is a potent pyrogen, and because these forms of damage are highly susceptible to even modest changes in body temperature, the data may reflect an indirect effect of IL-1 on neuronal survival. Thus, it is important to quantify the effect of IL-1 and rhIL-1ra on body temperature and other physiological parameters because these might influence ischaemic damage. The potent effects of rhIL-1ra on neurodegeneration imply that pharmacological manipulation of IL-1 synthesis or action may be beneficial in the treatment of stroke. The time course of action of rhIL-1ra and effects on specific brain regions are important considerations in assessing therapeutic value of this and related treatments (Hunter et al., 1995).
The objectives of the present study were to investigate in more detail effects of IL-1 and rhIL-1ra on neuronal damage induced by focal cerebral ischaemia [middle cerebral artery occlusion (MCAo)] in the rat, to analyse the impact of these treatments on body temperature, and to investigate the time course and tissue distribution of the neuroprotective actions of rhIL-1ra.
MATERIALS AND METHODS
All experiments were performed on male, Sprague-Dawley rats (Charles River, UK), weighing 230–270 g. Animals were housed at 21°C with free access to food and water.
Surgical procedures
Guide cannulae were stereotaxically implanted into the lateral ventricle of the brain of rats under sodium pentobarbitone anaesthesia (60 mg/kg) 7 days before induction of ischaemia to permit administration of substances into the brain. At this time, radiotransmitters (Data Sciences, U.S.A.) were also implanted into the peritoneum, to allow subsequent recording of body temperature by remote radiotelemetry in conscious animals.
Cerebral ischaemia was induced by permanent unilateral occlusion of the middle cerebral artery, using an adaptation (Bederson et al., 1986b) of the method described by Tamura et al. (1981). Anaesthesia was induced by inhalation of 4% halothane in oxygen, and maintained by 2–3% halothane throughout surgery. A 2-cm incision was made midway between the lateral margin of the left orbit and the auditory canal. The temporalis muscle was elevated to expose the subtemporal fossa, and a craniotomy was made using a dental drill. The dura was pierced and the left MCA was exposed and occluded proximally below the lenticulostriate branch, and again, at the level of the inferior cerebral vein, using a bipolar coagulator (Downs Surgical, UK). The artery was then cut between these two points to prevent recannalisation. After surgery, soft tissues were replaced and the wound was stitched. Animals were maintained on a heated mat until they regained consciousness.
Temperature recording
Body temperature was recorded in conscious, free-moving animals by remote radiotelemetry at an ambient temperature of 21°C, immediately after recovery from surgery. To avoid the complication of circadian variation, body temperature was recorded, for 24 h before and the 24 h after MCAo to allow calculation of the change in body temperature due to ischaemia. Data are therefore presented as the difference in temperature after ischaemia relative to the previous day shown at 30-min intervals.
Blood pressure and heart rate recordings
In separate groups of rats, maintained under halothane anaesthesia, mean arterial blood pressure and heart rate were measured by pressure transduction and arterial pulse, respectively, via an indwelling cannula inserted in the left femoral artery. Parameters were measured for 30 min before MCAo to obtain baseline levels, then for 45 min after MCAo. Data are presented as change from mean baseline values.
Measurement of infarct size
In most experiments, delineation of the lesion was determined on fresh tissue. Sequential, 500-μm coronal brain sections were incubated in 2% triphenyl tetrazolium chloride (TTC; Sigma Chemical Co., UK), a stain for mitochondrial viability, until a clear delineation of brain infarction could be observed. The lesion area was charted onto stereotaxic maps (thus correcting for any swelling due to brain oedema), and subsequently measured by automated SeeScan image analysis. Lesion volume was calculated by integration of the areas of infarct for each animal.
For more detailed histological analysis, or analysis at time points later than 24 h, brains were frozen in isopentaneat −30°C, and stored at −70°C. Coronal sections, 15 μm each, were cut on a cryostat at −21°C at 500-μm intervals through the whole brain. Sections were thaw-mounted onto chromium-gelatin-coated microscope slides, dried, and stored at −70°C before staining with haematoxylin and eosin for light microscopic analysis. Infarct volume was calculated as already described. All analysis was performed “blind.”
Experimental procedures
In the first study, effects of i.e.v. injection of human recombinant IL-1β (rhIL-1β, Dupont, U.S.A., specific activity 200 IU/ng, endotoxin content 2.5 EU/mg protein) on ischaemic damage and core temperature after MCAo was investigated. IL-1β (2.5 ng in 4 μl) was infused (over 2 min) into the right lateral ventricle of conscious, lightly restrained rats at two times (30 min and 2 h) after MCAo (total dose 5 ng). Extensive dose-response studies, conducted in our laboratory, on the pyrogenic properties of IL-1β have revealed this dose to be optimal for induction of fever (Rothwell and Hopkins, 1995). Control animals were infused with an appropriate volume of sterile, 0.9% saline. Body temperature was recorded for 24 h before and 24 h after MCAo by radiotelemetry. Ischaemic damage was assessed 24 h after MCAo by TTC staining.
In a separate experiment, the effects of i.e.v. injection of rhIL-1ra on ischaemic brain damage was compared with that of the noncompetitive N-methyl-D-aspartate (NMDA) receptor antagonist, MK-801. An optimum dose (10 μg in 4 μl) of rhIL-1ra (donated by Synergen, U.S.A.) or vehicle (4 μl, sterile saline) was infused into the right lateral cerebral ventricle (as already described) 30 min before and 10 min after induction of ischaemia (total dose 20 ng). MK-801 (4 mg/kg, dizocilpine maleate, MSD, U.K.) or vehicle was administered intraperitoneally 30 min before MCAo. Damage was assessed 24 h after MCAo by TTC staining.
To investigate the possibility that rhIL-1ra protects against ischaemic damage by modifying physiological parameters, core temperature, blood pressure, and heart rate were measured in animals subjected to MCAo and treated with rhIL-1ra or saline (administered as described in the second experiment).
The time course of action of rhIL-1ra was assessed in separate groups of animals injected i.e.v. with a single dose of 10 jig rhIL-1ra or vehicle either 30 min before (–30 min), at the time of ischaemia (0 min), or 30 min after MCAo (+30 min). Animals were killed 24 h after MCAo, and damage was assessed by TTC staining. To determine whether the neuroprotective effects of rhIL-1ra were sustained, rhIL-1ra (10 ng) or vehicle was infused at the time of MCAo (0 h), and groups of animals were killed either 1 or 7 days after MCAo. These brains were frozen and subsequently stained using haematoxylin and eosin for analysis by light microscopy.
Statistical analysis
All data are expressed as mean ± standard deviation. The number of animals in each group are presented in the figure legends (see following section). Statistical differences between lesion volume were assessed using the Students' unpaired t test. Temperature, blood pressure, and heart rate data were analysed by multiple analysis of variance. Probabilities are two-tailed, assuming a probability of <5% for statistical significance.
RESULTS
All animals subjected to MCAo sustained focal brain damage throughout the left cortex and basal ganglia when observed 24 h after surgery. An example of the typical distribution of damage as observed in control animals is shown in Fig. 1A. Damage was always seen in the frontal cortex and lateral striatum, and usually in the sensorimotor and auditory areas of the cortex, and the medial area of the striatum. Very low mortality (<5%) was associated with surgery and injection of either vehicle, rhIL-1ra or IL-1β, and no neuronal damage was observed in sham-operated rats (artery exposed, but not occluded—data not shown). All animals that underwent MCAo demonstrated splaying of the contralateral extremities, and circling; some animals also exhibited “barrel rolling” (ipsilateral turning).
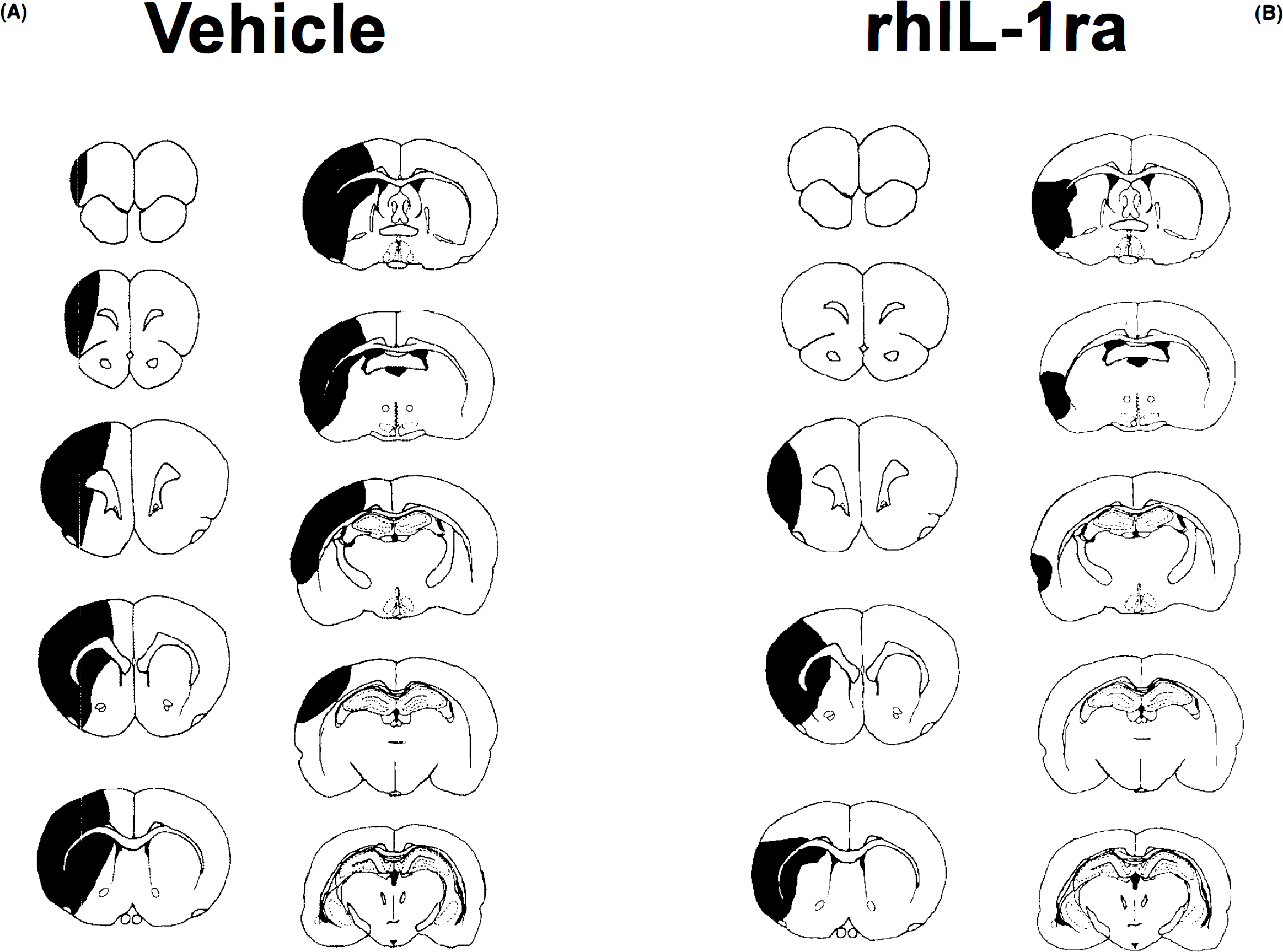
An example of the typical distribution of damage in cortex and striatum at 10 stereotactic levels, measured 24 h after middle cerebral artery occlusion (MCAo) in rats injected i.e.v. with either saline
Experiment 1
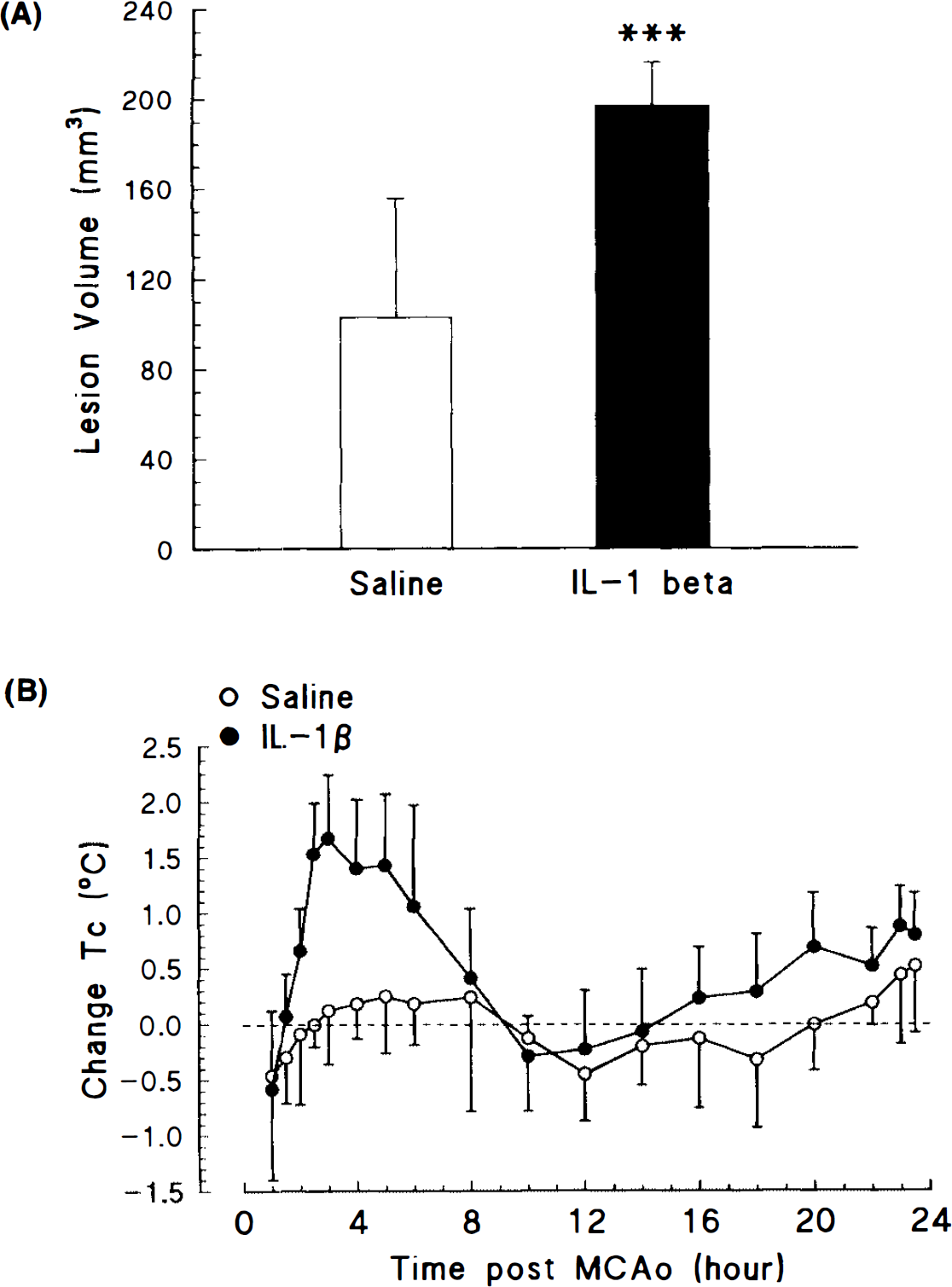
Central administration of IL-1β (2.5 ng, i.e.v., 30 min and 2 h after MCAo) caused a marked exacerbation of ischaemic damage at all brain levels (Fig. 2A), resulting in a large (92%), and statistically significant (p < 0.001) enhancement of lesion volume, compared with animals injected with vehicle. The effect of infusion IL-1β or vehicle on body temperature after MCAo is shown in Fig. 2B. Basal (presurgery) temperatures were similar in groups injected with saline (37.3 ± 0.1°C, n = 7) or IL-1β (37.4 ± 0.2°C, n = 6). Treatment with IL-1β resulted in a rapid increase in core temperature, which began soon after recovery from anaesthesia, and reached a maximal value, of ∼38.9°C, between 3 and 4 h later. This increase in temperature was significantly greater (1.5°C, p < 0.001) than that of vehicle-treated animals. Body temperature had returned to presurgery levels by 8 h after ischaemia. In saline-injected rats, core temperature returned to values similar to those recorded on the previous day within 2 h after surgery, and thereafter remained relatively constant.
Experiment 2
The data presented in Fig. 3 demonstrate that injection of rhIL-1ra (i.c.v.) or MK-801 (i.p.) significantly reduced, by a similar extent, infarct volume measured 24 h after MCAo. Peripheral administration of MK-801 (4 mg/kg, i.p., 30 min before MCAo) caused a marked (67%) and statistically significant (p < 0.01) reduction in lesion volume (vehicle 85.8 ± 45.0 mm3, n = 8, MK-801 27.9 ± 7.0 mm3). This reduction in damage was evident through all levels of the brain. Central administration of rhIL-1ra (10 μg, i.e.v., 30 min before and 10 min after MCAo) also resulted in a marked (72%) and statistically significant (p < 0.01) reduction in lesion volume (control 99.1 ± 43.4, n = 7, rhIL-1ra 27.8 ± 7.8,n = 8). The residual volume of damage was remarkably similar in animals treated with rhIL-1ra (27.8 mm3) or MK-801 (27.8 mm3).
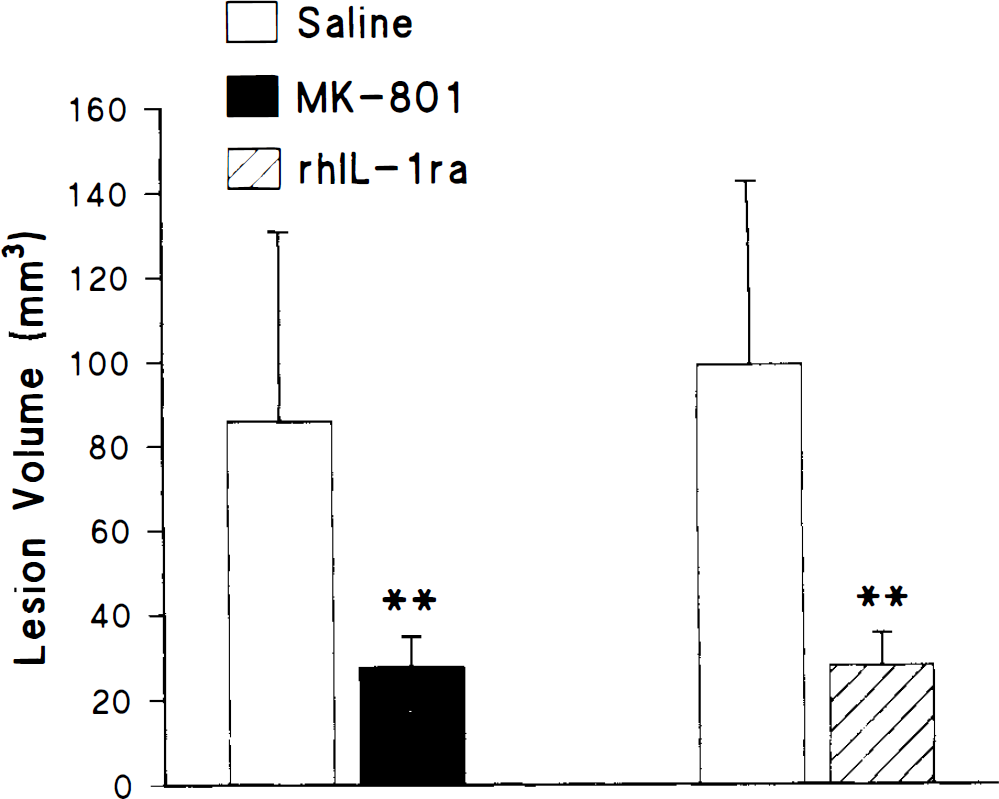
Infarct volume measured 24 h after middle cerebral artery occlusion (MCAo) in rats injected i.e.v. with either saline or rhIL-1ra (10 μg) 30 min before and 10 min after MCAo, or i.p. with either saline or MK-801 (4 mg/kg) 30 min before MCAo. Values denote mean of eight animals and bars indicate SD. ** p < 0.01 versus saline-treated group.
Experiment 3
Changes in heart rate, and mean arterial blood pressure over the 45 min after MCAo are presented in Table 1. Resting heart rate and mean arterial blood pressure were similar and stable in all animals before surgery (320 ± 16 beats/min and 89 ± 3 mm Hg, respectively). Central injection (10 μg, i.e. v.) of rhIL-1ra 30 min before surgery had no significant effect on either parameter. In vehicle-treated rats, MCAo caused modest and transient increases in blood pressure (29%) and heart rate (13%), which were maximal 20 min after surgery. rhIL-1ra slightly, but not significantly attenuated these changes (Table 1). Blood pressure and heart rate were monitored for longer periods (up to 2 h) in a small number of animals, but no differences between vehicle- or rhIL-1ra-treated animals were observed.
Effects of rhIL-1ra on blood pressure (BP) and heart rate (HR) after MCAo
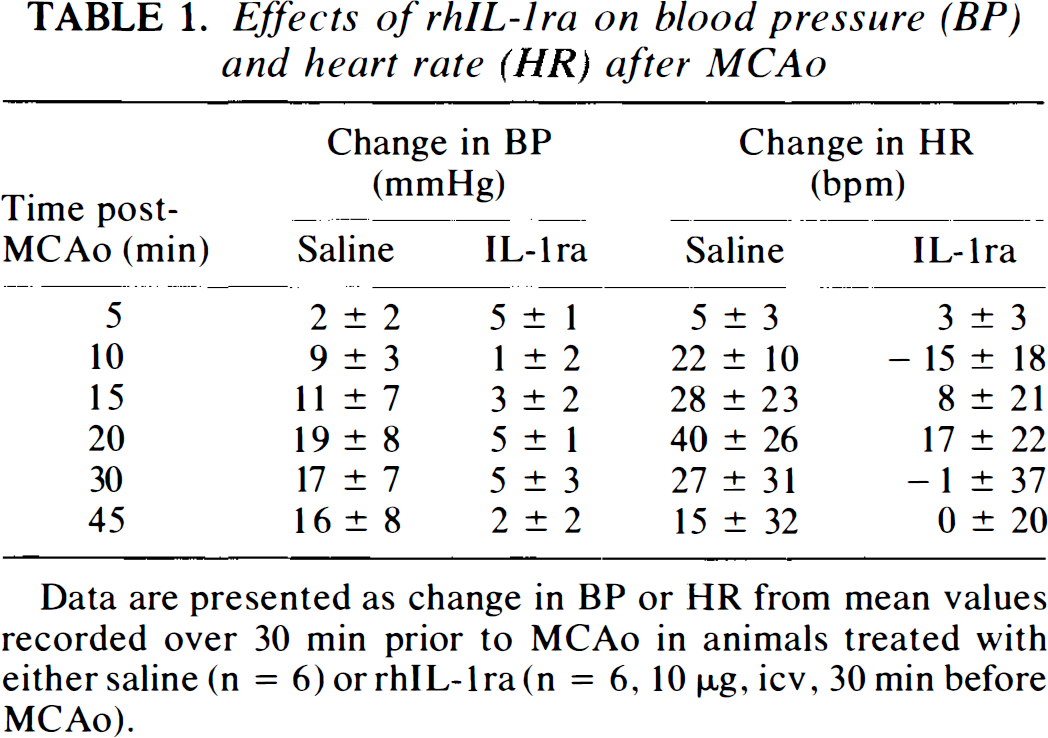
Data are presented as change in BP or HR from mean values recorded over 30 min prior to MCAo in animals treated with either saline (n = 6) or rhIL-1ra (n = 6, 10 μg, icv, 30 min before MCAo).
Changes in body temperature observed during the 24 h after MCAo are presented in Table 2. Basal body temperatures were similar in MCAo rats treated with saline (37.2 ± 0.2°C, n = 7) or rhIL-1ra (37.4 ± 0.3°C, n = 8). After an initial, transient hypothermia (∼1°C), presumably due to the affects of anaesthesia, body temperature remained stable in animals treated with rhIL-1ra, which had no statistically significant effect on the change in temperature observed after MCAo.
Effects of rhIL-1ra on body (peritoneal) temperature (Tc)
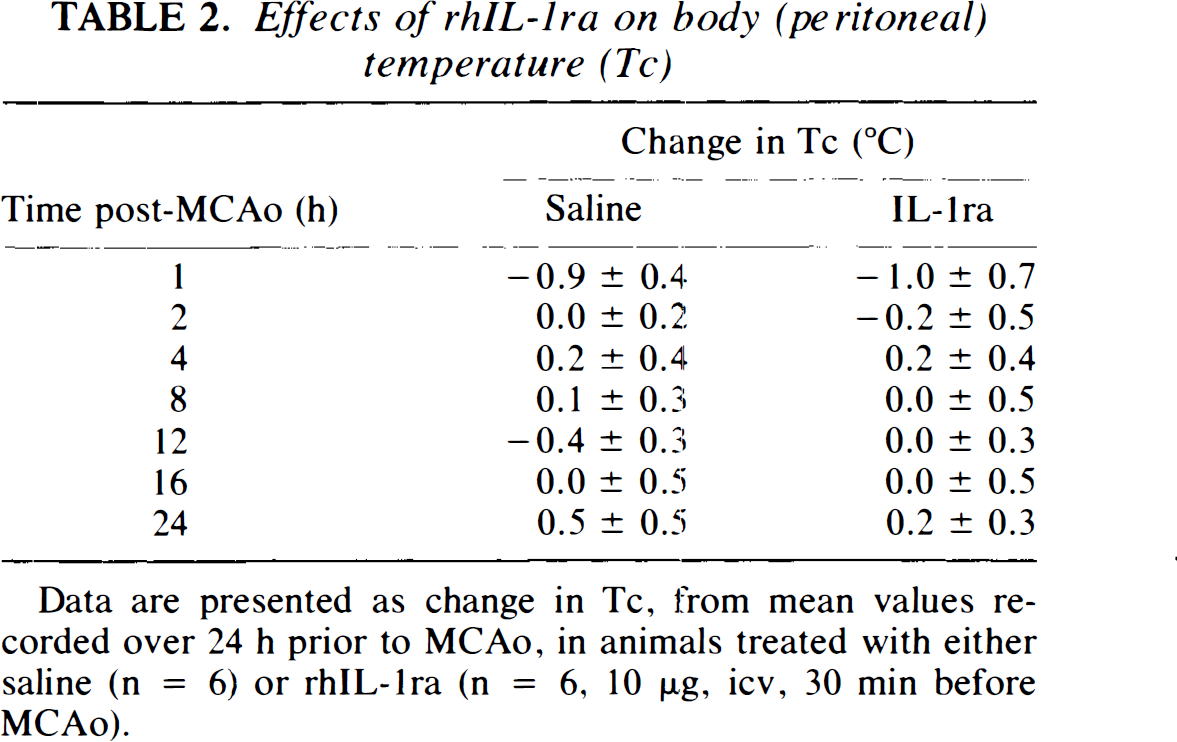
Data are presented as change in Tc, from mean values recorded over 24 h prior to MCAo, in animals treated with either saline (n = 6) or rhIL-1ra (n = 6, 10 ng, icv, 30 min before MCAo).
Experiment 4
Results presented in Fig. 4 illustrate the effect of delayed treatment with rhIL-1ra on infarct volume caused by MCAo. Pretreatment with rhIL-1ra (10 (jug, i.c.v.) 30 min before MCAo resulted in a marked and significant reduction of ischaemic damage throughout both cortex (54.9 ± 6.3%, p < 0.001) and striatum (52.1 ± 7.7%, p < 0.001, n = 7). A typical example of the distribution of damage seen after this treatment is shown in Fig. 2B. A single injection of rhIL-1ra (10 μg, i.c.v.) at the time of MCAo inhibited cortical damage to a similar extent (59.7 ± 3.3%, p < 0.001), but caused a slightly reduced level of striatal protection (41.1 ± 6.8%, p < 0.001, n = 8). When administration of rhIL-1ra was delayed until 30 min after MCAo, a smaller but nonetheless significant inhibition (28.8 ± 9.7%, n = 13) of cortical damage was observed, but no striatal protection was evident.
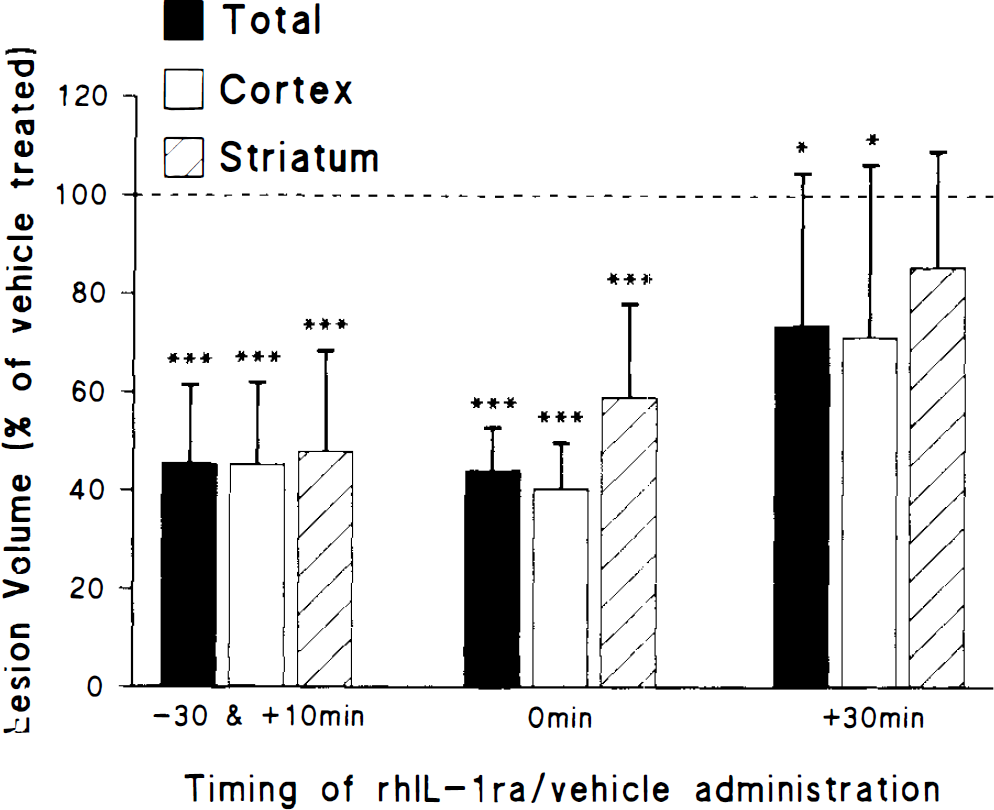
Infarct volume measured 24 h after middle cerebral artery occlusion (MCAo) in rats injected i.e.v. with either saline or rhIL-1ra (10 μg) 30 min before and 10 min after MCAo, or 0 h after or 30 min after MCAo. Values are presented as a percentage of the mean lesion size of the respective vehicle-treated group, and denote the mean of eight animals; bars indicate SD. ***p < 0.001 versus saline-treated group; *p < 0.05 versus saline-treated group.
Data shown in Fig. 5 illustrate that protection with rhIL-1ra (10 μg, i.e.v., 0 h) is still apparent 7 days after ischaemia. The inhibition of infarct volume measured 7 days after MCAo was similar to that observed at 24 h, throughout both the cortex (55.3 ± 5.3%) and striatum (36.7 ± 7.6%), and in both regions infarct volume was significantly lower than that of vehicle-treated rats, p < 0.001 for cortical and p < 0.01 for striatal damage.
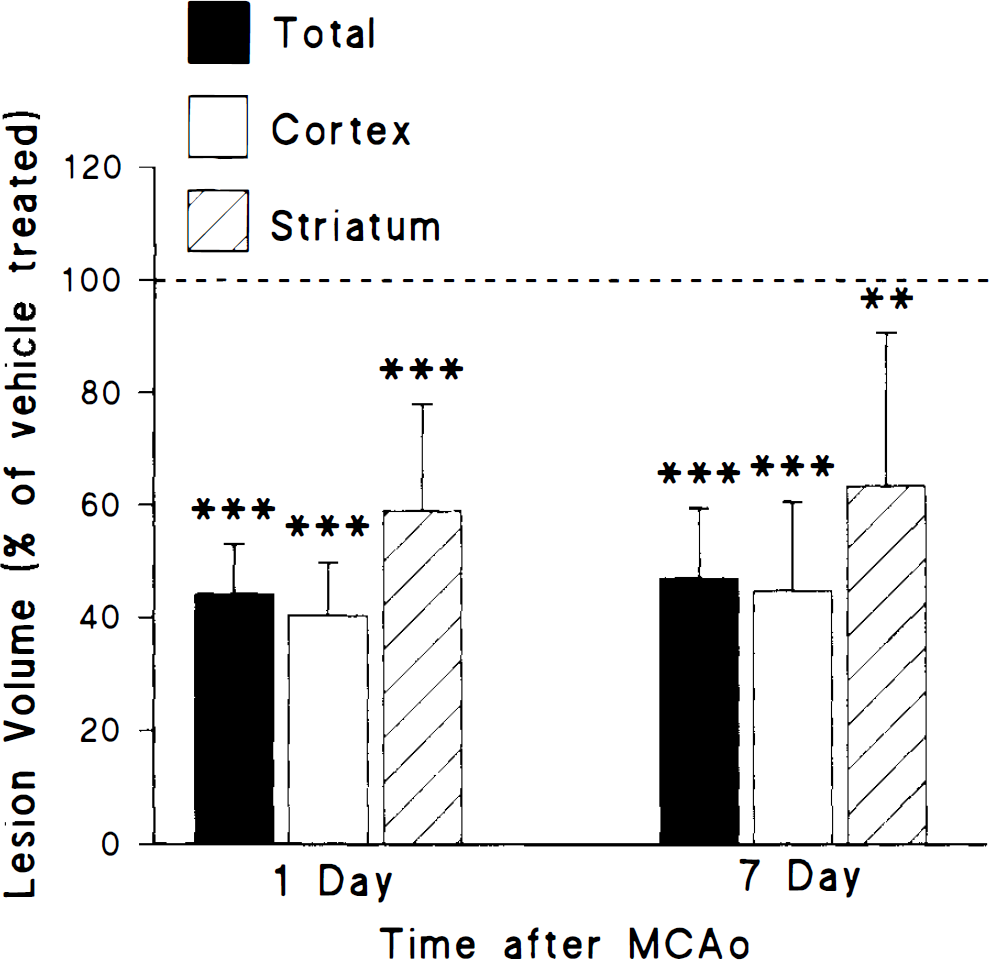
Infarct volume measured 1 or 7 days after middle cerebral artery occlusion (MCAo) in rats injected i.c.v. with either saline or rhIL-1ra (10 μg) 0 h after MCAo. Values are presented as a percentage of the mean lesion size of the respective vehicle-treated group, and denote the mean of eight animals; bars indicate SD. ***p < 0.001 versus saline-treated group; **p < 0.01 versus saline-treated group.
DISCUSSION
The results of these studies confirm and extend our previous findings, and show that rhIL-1ra is a potent inhibitor of experimentally induced ischaemic brain damage. In seven separate experiments we have observed inhibition of ischaemic brain damage in the rat by rhIL-1ra of up to 78%. A similar neuroprotective effect of rhIL-1ra has now been observed in focal ischaemia in the mouse (N. J. Rothwell, L. Rothwell, and C. Davies, unpublished data), traumatic brain damage in the rat (Toulmond and Rothwell, 1995) and perinatal hypoxic brain damage in the rat (Martin et al., 1995).
A number of studies have reported increased expression of IL-1 mRNA or protein after experimental ischaemic, traumatic, or excitotoxic brain damage (e.g., McClain et al., 1987; Minami et al., 1990; Woodroofe et al., 1991; Minami et al., 1992a; Yan et al., 1992; Liu et al., 1993; Taupin et al., 1993; Buttini et al., 1994; Yabuuchi et al., 1994; Wang et al., 1995). These observations implicate IL-1 in the processes of neuronal death and/or repair and recovery. Experiments on cultured neurons in vitro have revealed that IL-1 protects cultured neurons from excitotoxic insults (Strijbos and Rothwell, 1995) and this, together with the fact that IL-1 induces expression of nerve growth factor (Gadient et al., 1990; Spranger et al., 1990), implies that IL-1 acts to limit neurodegeneration. However, the results of the present study are not consistent with this hypothesis, i.e.v. injection of IL-1β massively exacerbated neuronal damage caused by MCAo, but did not cause measurable neuronal damage when infused i.e.v. or into brain tissue in normal rats (data not shown). It is difficult to equate the dose of IL-1β injected (5 ng, i.e., <1 pmol) with brain concentrations after ischaemia, and the quantity administered may have been supramaximal, although this dose is optimal for induction of several other responses to IL-1 such as fever, behavioural changes, and hypothalamus pituitary adrenal activation (Rothwell and Hopkins, 1995). Nevertheless, these results are not consistent with protective actions of IL-1. The mechanisms by which exogenous IL-1 exacerbates ischaemic damage are unknown, but may relate to its effect on body temperature (Fig. 2B). IL-1 exacerbates brain damage caused by global ischaemia in the gerbil (Minami et al., 1992b) and reperfusion injury in the rat (Yamasaki et al., 1992, 1995), but these forms of damage are highly susceptible to changes in body temperature (Ginsberg et al., 1992; Ridenour et al., 1992). Damage resulting from focal ischaemia (MCAo) is considerably less sensitive to hyperthermia (Ridenour et al., 1992; Morikawa et al., 1992), although the extent of fever (>1.5°C) is likely to have exerted some influence on neuronal variability. In an attempt to determine the contribution of the raised body temperature to the exacerbation of ischaemic damage, experiments to block the pyrogenic response to IL-1β were considered. However, treatments that effectively block IL-1β fever (e.g., glucocorticoids, cycloxygenase inhibitors, corticotrophin-releasing factor antagonist, lipocortin, etc.) all affect directly ischaemic damage due to MCAo, independently of effects on body temperature. rhIL-1ra can also inhibit the pyrogenic effects of IL-1β, but because of its potent effects on MCAo, experiments to test whether rhIL-1ra inhibits the potentiation of damage by IL-1β would be difficult to interpret. However, i.c.v. injection of IL-6 at a dose (50 ng) that causes comparable fever inhibited rather than enhanced ischaemic damage (S. A. Loddick and N. J. Rothwell, unpublished data), indicating that fever is not the primary reason why IL-1 increases ischaemic damage.
Experiments using rhIL-1ra provide more compelling evidence that endogenous IL-1 mediates ischaemic brain damage. After testing a range of doses (1–50 μg, i.e.v., data not shown), 10 μg rhIL-1ra was found to cause maximal inhibition of damage, and was used in all subsequent experiments. The rate of diffusion of rhIL-1ra into the brain from CSF and its half-life in the CNS are not known accurately, although our preliminary data indicate a half-life of ∼60 min after i.e.v. injection in the rat (X. Luheshi, X. Miller, X. Poole, and N. J. Rothwell, unpublished data). Thus, sustained infusion or overexpression of rhIL-1ra may have caused greater protection.
Damage caused by permanent occlusion of the MCA was reproducibly observed in areas of the cortex and striatum, consistent with the original studies describing this protocol (Tamura et al., 1981; Bederson et al., 1986b). Tetrazolium chloride staining was employed to assess neuronal damage in all acute (up to 24 h) studies, because it enables infarct size to be rapidly calculated. This method is now used widely to assess ischaemic damage, and has been favourably compared with the more traditional method of haematoxylin and eosin staining (Bederson et al., 1986a; Lin et al., 1993), and in the present study almost identical patterns of damage and effects of rhIL-1ra were observed using tetrazolium or haematoxylin and eosin to assess damage at 24 h (data not shown). In addition, an almost identical pattern of protection from rhIL-1ra was observed when damage was assessed 7 days after MCAo, indicating that rhIL-1ra did not simply delay neurodegeneration (Fig. 5).
rhIL-1ra is a selective, competitive receptor antagonist at the IL-1 receptor (Dinarello and Thompson, 1991) that is present in normal brain (Licinio et al., 1991), and as yet no actions other than inhibition of IL-1 receptors have been demonstrated. The protective effects of rhIL-1ra were comparable to that of the noncompetitive NMDA receptor antagonist, MK-801 (Fig. 2), and the residual damage seen after injection of rhIL-1ra or MK-801 was almost equal. Neuroprotective effects of MK-801 have been studied extensively in several forms of neurodegeneration including focal ischaemia (Albers et al., 1989, 1992), but behavioural side effects mean that it is unlikely to have a clinical application (Olney et al., 1989; Muir and Lees, 1995).
rhIL-1ra did not significantly affect body (peritoneal) temperature, mean arterial blood pressure, or heart rate in sham-operated or MCAo rats (Tables 1 and 2). The possibility that rhIL-1ra affected brain temperature independently of core temperature cannot be excluded, although this seems unlikely as a primary mechanism of action. The modest effect of rhIL-1ra on the small rise in blood pressure after MCAo may reflect an indirect action due to the marked reduction in brain damage in rhIL-1ra-treated animals. These data suggest that rhIL-1ra protects against cerebral ischaemia by modifying the neurochemical events leading to neuronal death, rather than affecting physiological parameters. This proposal is supported by our observations that rhIL-1ra also markedly inhibits neuronal damage induced by pharmacological overactivation of NMDA or AMPA receptors in vivo (Relton and Rothwell, 1992; Lawrence and Rothwell, 1994).
The marked inhibition of ischaemic damage by rhIL-1ra reflected protection against striatal as well as cortical damage (Fig. 1B and Fig. 3). Relatively few agents have been reported to limit striatal damage after MCAo (Muir and Lees, 1995), because this is the primary area of infarct after occlusion of the MCA (Shigeno et al., 1985; Garcia et al., 1995). rhIL-1ra also inhibited damage to a similar extent in both brain regions when administered at the time of ischaemia. However, delayed injection of rhIL-1ra (30 min after MCAo) protected cortical but not striatal tissue, presumably due to the earlier progression of damage in the striatum. Because rhIL-1ra may diffuse slowly from the ventricles into brain tissue (particularly when blood flow is reduced), it is likely that direct application of rhIL-1ra to the site of brain damage may be effective some time after the ischaemic event.
The mechanism of these neuroprotective effects of rhIL-1ra are unknown. Several hypotheses to explain the actions of rhIL-1ra or IL-1 have been described (Relton and Rothwell, 1992), which include modification of oedema, blood–brain barrier damage, release of arachidonic acid or its products, or corticotropin-releasing factor and nitric oxide synthesis. The magnitude of inhibition of damage by rhIL-1ra and its effects on excitotoxic, ischaemic, and traumatic brain damage suggest either a site of action common to diverse forms of insult, or effects at multiple points in the cascade of neurodegeneration.
rhIL-1ra is a large protein, and therefore does not cross the blood–brain barrier readily. However, neuroprotective effects of systemically injected rhIL-1ra have been reported (Relton et al., 1993). Overall, these data indicate that pharmacological strategies to inhibit the synthesis or actions of brain IL-1 may offer considerable benefit in ischaemic and related forms of degeneration.
Footnotes
Acknowledgment:
This work was supported by Biotechnology and Biology Research Council and Medical Research Council. We thank Synergen for the gift of rhIL-1ra, Dupont for IL-1, and Anthea Hardwick for her technical assistance.