
Abstract
We investigated the density and distribution of nitric oxide synthase (NOS) binding by quantitative autoradiography using [3H]L-NG-nitroarginine ([3H]L-NNA) after transient focal ischemia or intrastriatal injection of N-methyl-D-aspartate (NMDA) in wild-type (SV-129 and C57black/6) and type I (neuronal) and type III (endothelial) NOS-deficient mice. The middle cerebral artery (MCA) was occluded by an intraluminal filament for 3 h followed by 10 min to 7 days of reperfusion. Specific [3H]L-NNA binding, observed in the wild-type and type III mutant mouse at baseline, increased by 50–250% in the MCA territory during ischemia and the first 3 h of reperfusion. The density of binding sites (Bmax), but not the dissociation constant (Kd), increased significantly during the ischemic period as did type I NOS mRNA as detected by quantitative reverse transcription polymerase chain reaction. [3H]L-NNA binding after intrastriatal NMDA injection also increased by 20–230%. In the type I NOS-deficient mouse, [3H]L-NNA binding was low and only a very small increase was observed after ischemia or excitotoxicity. Under conditions of this study, [3H]L-NNA did not bind to type II NOS as there was no difference in the distribution or density of [3H]L-NNA binding in the rat spleen obtained after lipopolysaccharide treatment despite induction of NOS type II catalytic activity. Our data suggest that an ischemic/excitotoxic insult up-regulates type I NOS gene expression and [3H]L-NNA binding and that this up-regulation may play a pivotal role in the pathogenesis of ischemic/excitotoxic diseases.
Nitric oxide (NO) is a gaseous molecule that has beenimplicated in many physiological and pathological processes. NO is synthesized by nitric oxide synthase (NOS) during the conversion of L-arginine to L-citrulline. At least three isoforms have been identified, including neuronal (type I) (Bredt et al., 1991), endothelial (type III) (Lamas et al., 1992), and inducible (type II) (Curran et al., 1989) NOS. NO production increases after transient middle cerebral artery (MCA) occlusion (Malinski et al., 1993; Kumura et al., 1994; Zhang et al., 1994). Accumulated evidence suggests that enhanced synthesis of NO by type I NOS is deleterious in ischemia. In particular, type I NOS mutant mice were resistant to injury after permanent (Huang et al., 1994) or transient (Hara et al., 1996a) focal ischemia. Type III NOS-deficient mice developed larger lesions after MCA occlusion, and these lesions decreased after L-NG-nitroarginine (L-NNA) treatment (Huang et al., 1996). Excitotoxic lesions following intrastriatal N-methyl-
MATERIALS AND METHODS
Animals
Wild-type (adult male SV-129 and C57black/6; Taconic Farms, Germantown, NY, U.S.A.) and type I (adult male and female) (Huang et al., 1993) and type III (Huang et al., 1995) NOS mutant mice weighing 20–28 g and Sprague–Dawley rats (adult male; Charles River Laboratory, Wilmington, MA, U.S.A.) weighing 350–400 g were allowed free access to food and water. Wild-type (type I NOS+/+), and heterozygote (type I NOS+/–) littermate controls in addition to homozygotes (type I NOS–/–) were used in NMDA excitotoxicity experiments. Pig aorta was obtained from Pel-Freeze Biologicals (Rogers, AR, U.S.A.). LPS (Klebsiella pneumoniae) was purchased from Sigma Chemical Co. (St. Louis, MO, U.S.A.).
Physiology
Animals were initially anesthetized with 1% and maintained by 0.5–1% halothane in 70% N2O/30% O2 (Fluotec 3 vaporizer; Colonial Medical, Amherst, NH, U.S.A.). Regional CBF (rCBF) was determined by laser–Doppler flowmetry (PF2B; Perimed, Stockholm, Sweden) using a flexible 0.5-mm fiberoptic extension to the master probe. The tip of the probe was fixed to the intact skull with cyanoacrylate glue (Aron Alpha, Toa, Tokyo, Japan) over the ischemic cortex (2 mm posterior and 6 mm lateral to bregma). Steady-state baseline values were recorded before MCA occlusion, and rCBF during and after occlusion was expressed as percentage of the baseline values. rCBF, blood pressure, and heart rate were monitored as described (Hara et al., 1996a). Arterial blood pH and oxygen (Pa
Ischemia model
The mouse left MCA was occluded for 3 h and reperfused as described previously with minor modifications (Hara et al., 1996a). The dental resin-coated filament was introduced into the internal carotid artery through the common carotid artery up to the origin of the anterior cerebral artery (ACA) so as to occlude the MCA and posterior communicating artery. Three hours after ischemia, animals were briefly reanesthetized with halothane and the filament withdrawn. [3H]L-NNA binding was studied in SV-129 mice at 15 min, 1 h, and 3 h of MCA occlusion and also after reperfusion for 10 min, 3 h, 24 h, 3 days, and 7 days (n = 4–6 in each group). In addition, [3H]L-NNA binding was examined 3 h after reperfusion in C57black/6, type I, or type III NOS mutant mice.
NMDA-induced neurotoxicity
Mice were anesthetized with halothane (2.5% for induction, 1–1.5% for maintenance) and the head was fixed in a stereotaxic frame (David Kopf, Tujunga, CA, U.S.A.). Three-tenths microliter containing 67 mM NMDA (20 nmol, dissolved in 0.1 M phosphate-buffered saline) was injected by a Hamilton syringe (26 G) at 0.5 mm anterior and 2 mm lateral to bregma and −2.5 mm below the dura over 2 min. The needle was kept in place for an additional 8 min. Mice were killed after 3–48 h and brains were rapidly removed. The area of the lesion (striatum 3.5 mm posterior from olfactory bulb) was measured by an image analysis system (M4; Imaging Research, St. Catharines, Ontario, Canada) and integrated to calculate volume.
Tissue preparation and histopathology
Mice and rats were killed by decapitation under deep halothane anesthesia and brains were rapidly removed. The tissues were immediately frozen in powdered dry ice and stored at −70°C. Coronal sections, 12 (ischemia study) or 20 (neurotoxicity study) μm, were cut in a cryostat at −18°C (Leitz 1720; Leica, Deerfield, IL, U.S.A.) and thaw-mounted onto gelatin-coated slides [1% gelatin and 0.1% CrK(SO4)2].
Adjacent sections were stained with hematoxylin/eosin for evaluation of neuronal damage after ischemia or excitotoxicity.
[3H]L-NNA autoradiographic binding assay
The method for the autoradiographic visualization of [3H]L-NNA binding has been described previously (Hara et al., 1996b). Sections were brought to room temperature and preincubated for 15 min in 50 mM Tris-HCl (pH 7.3), then incubated for 30 min in 50 mM Tris-HCl buffer containing 10 mM CaCl2, 10 μM
Nonspecific binding was defined as that remaining in the presence of an excess of unlabeled L-NNA (10 μM; Sigma). Ten micromolar
To evaluate the specificity of [3H]L-NNA binding, we determined the displacing effects of 7-nitroindazole (7-NI) (100 μM; Tocris Cookson, St. Louis, MO, U.S.A.) and aminoguanidine (100 μM; Sigma).
To evaluate the selectivity of [3H]L-NNA binding, frozen sections from pig aorta (12 μm) were incubated with ligand or with rat spleen tissue sections taken 6 h after LPS injection (5 mg/kg i.p.) as described.
Saturation studies were performed by incubating adjacent sections with increasing concentrations of [3H]L-NNA (5–160 nM) in the presence of 10 μM unlabeled
Since the quenching of tritium is greater in white than in the gray matter (Kuhar and Unnerstall, 1985), an apparent decrease in binding density could result if there were a change in the proportion of gray and white matter in the type I mutant mouse. Histopathological studies, however, revealed no such change. Furthermore, the binding pattern, using the tritiated ligands [3H]PN200-110 (L-type Ca2+ channels) or [3H]phorbol-12,13-dibutyrate (protein kinase C), was similar in wild-type and mutant mouse brain (H. Hara and M. A. Moskowitz, unpublished data).
Densitometric analysis
Films were developed in Kodak D-19 and fixed in Kodak Rapid Fixer, according to the manufacturer's instructions. The optical density of the regions of interest [striatum (caudate putamen), amygdala (amygdalohippocampal area, amygdalopiriform transition, basolateral and basomedial amygdaloid nuclei, posterolateral and posteromedial cortical amygdaloid nuclei), MCA territory (temporal cortex area 3 and perirhinal cortex), and ACA territory (retrosplenial agranular cortex and retrosplenial granular cortex)] was measured by a computer-assisted image analysis system. In the striatum, the sampled area was adjusted to the shape of the lesion. In the other areas (i.e., cortex, hippocampus, amygdala), the shape and site of the regions of interest were kept constant in control animals and animals with lesions. The relationship between optical density and radioactivity was examined with reference to the tritium standards co-exposed with the tissue sections. The optical density of the brain regions measured in the present study was in a range in which the optical density and the radioactivity of the 3H microscale showed a near linear relationship. The densities of NOS binding are expressed in femtomoles bound [3H]L-NNA per milligram tissue using the [3H]L-NNA concentration of 5–160 nM.
NOS catalytic activity
Total NOS catalytic activity was measured by the conversion of [3H]arginine to [3H]citrulline according to the method of Bredt and Snyder (1990) with minor modifications as previously described (Hara et al., 1996a). Selective measurement of calcium-independent NOS (type II NOS) activity was performed by omitting calcium and calmodulin in the reaction mixture and adding the co-factor (6R)-5,6,7,8-tetrahydro-L-biopterin (50 μM; RBI, Natick, MA, U.S.A.).
Quantitative reverse transcription polymerase chain reaction
To determine whether increased [3H]L-NNA binding during ischemia is associated with increased NOS transcription, RNA was isolated from ischemic and contralateral control mouse brain at different time points following MCA occlusion by the guanidinium isothiocyanate method (Chirgwin et al., 1979). RNA quality and average molecular size were determined by formaldehyde agarose gel electrophoresis.
First strand cDNA was synthesized using a fixed amount (5 μg) of brain mRNA in a 20-μl reaction using Superscript II reverse transcriptase (mRNA Preamplification Reagents; Gibco BRL). One-tenth of the cDNA was used as template for polymerase chain reactions (PCRs) to amplify sequences corresponding to type I NOS, type III NOS, type II NOS, and actin.
Type I NOS primers are as follows: B1 primer: 5′CCTTAGAGAGTAAGGAAGGGGGCGGG3′ (26-mer); B2 primer: 5′GGGCCGATCATTGACGGCGAGAATGATG3′ (28-mer). These primers amplify a 404-bp fragment from type I NOS cDNA. Type III NOS primers are as follows: E1 primer: 5′GGGCTCCCTCCTTCCGGCTGCCACC3′ (25-mer); E2 primer: 5′GGATCCCTGGAAAAGGCGGTGAGG3′ (24-mer). These primers amplify a 254-bp fragment from type III NOS cDNA. Type II NOS primers are as follows: I1 primer: 5′ATCAGGAACCTGAAGCCCCAGGAC3′ (24-mer); I2 primer: 5′TGTTGCCAGATTTCTCTGCACGGT3′ (24-mer). These primers amplify a 338-bp fragment from type II NOS cDNA. Each NOS primer is specific to its isoform and does not prime the other NOS isoforms. The actin primers are as follows: A1 primer: 5′ATGGATGACGATATCGCTG3′ (19-mer); A2 primer: 5′ATGAGGTAGTCTGTCAGGT3′ (19-mer). These primers amplify a 600-bp fragment from actin mRNA.
The PCR reactions contained 1.5 mM MgCl2 and 0.2 nmol of each primer. Denaturating was at 94°C for 30 s, annealing at 55°C (actin and type II NOS) or 60°C (type I and III NOS) for 30 s, and polymerizing at 72°C for 60 s. Pilot reactions indicated that 35 cycles was necessary to detect the products. Twenty microliters of each reaction was loaded onto 1% agarose gels in the presence of 0.1 μg/ml ethidium bromide and electrophoresed at 80 V. The HaeIII digest of øX was used as molecular weight markers and as internal calibration controls for densitometry. The gels were photographed and scanned into a Macintosh 7100/80 using Silverscanner and NIH Image 1.60 software, with settings 400 dpi and 256 gray scale.
Image analysis was performed by inverting the image and calibrating for optical density to record relative DNA quantity. The intensity of the molecular weight marker bands was used to confirm that optical density is linearly proportional to the amount of DNA on each gel. The lanes were then analyzed and plotted, and the areas under each band were determined. All results were expressed as relative amounts to the control 10-min sample (defined as 100%).
The validity of the technique was confirmed by using fixed amounts of brain RNA (5 μg) as starting material for reverse transcription (RT). The proportion of type I NOS mutant mRNA to wild-type mRNA was varied, from 100% type I NOS mutant (no wild-type mRNA) to 0% type I NOS mutant (100% wild-type RNA). The total amount of RNA was constant, but the level of type I NOS mRNA varied between 0 and 100%. The amount of PCR product varied according to the amount of type I NOS RNA present.
Statistical analysis
Data are presented as means ± SD. Statistical comparisons were made by one- or two-way analysis of variance (ANOVA) and Dunnett's multiple-range, Turkey's multiple-range, or Student's t test using the software super ANOVA (Abacus Concepts, Berkeley, CA, U.S.A.).
RESULTS
Physiology
The MABPs of wild-type and mutant mice were as reported previously (Huang et al., 1995). Type III NOS mutant mice were hypertensive (Table 1). Immediately after MCA occlusion and during ischemia, rCBF decreased to 10–20% of baseline (Fig. 1). After reperfusion, rCBF immediately returned to preischemic levels in SV-129 and type I NOS mutant mice, whereas recoveries were gradual and incomplete in C57black/6 and type III NOS mutant mice (p < 0.05, C57black/6 vs. SV-129; p < 0.01, C57black/6 vs. type I NOS mutant mice; 5 min after reperfusion) (Fig. 1). There were no significant group differences in heart rate (data not shown), arterial blood gases and pH, or rectal and temporalis muscle temperature before ischemia or after reperfusion (Table 2).
Mean arterial blood pressure during and after ischemia in wild-type and mutant mice
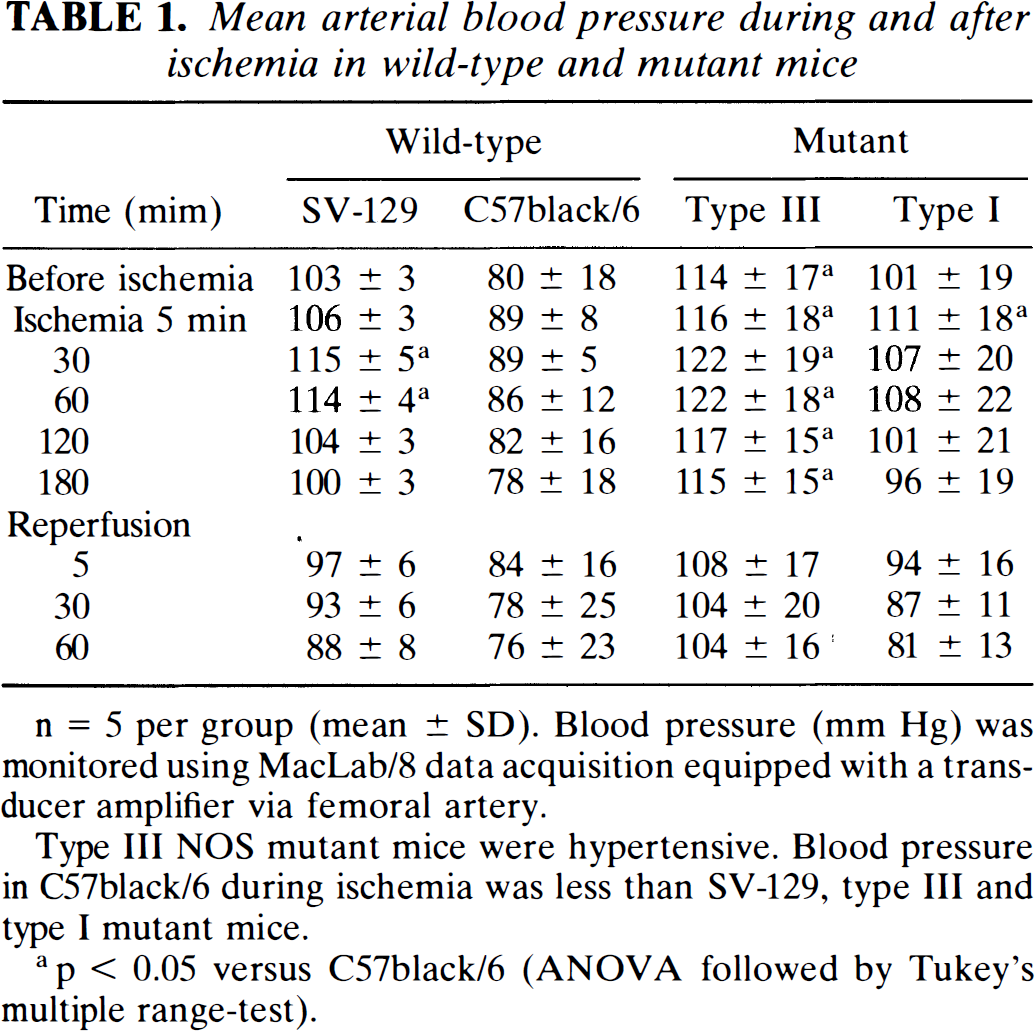
n = 5 per group (mean ± SD). Blood pressure (mm Hg) was monitored using MacLab/8 data acquisition equipped with a transducer amplifier via femoral artery.
Type III NOS mutant mice were hypertensive. Blood pressure in C57black/6 during ischemia was less than SV-129, type III and type I mutant mice.
p < 0.05 versus C57black/6 (ANOVA followed by Tukey's multiple range-test).
Physiological parameters, body temperature, and temporal muscle temperature before and after ischemia in wild-type and mutant mice under halothane anesthesia
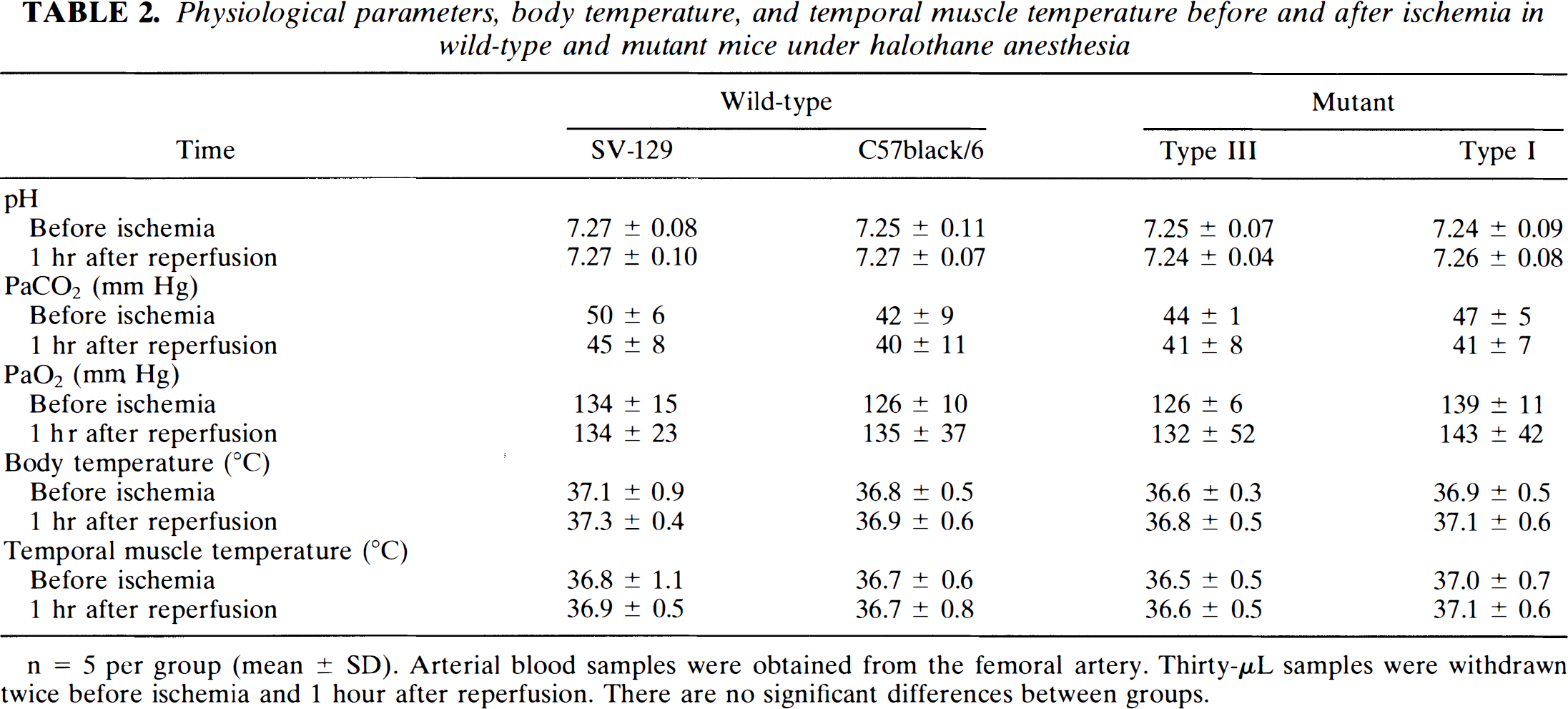
n = 5 per group (mean ± SD). Arterial blood samples were obtained from the femoral artery. Thirty-μL samples were withdrawn twice before ischemia and 1 hour after reperfusion. There are no significant differences between groups.
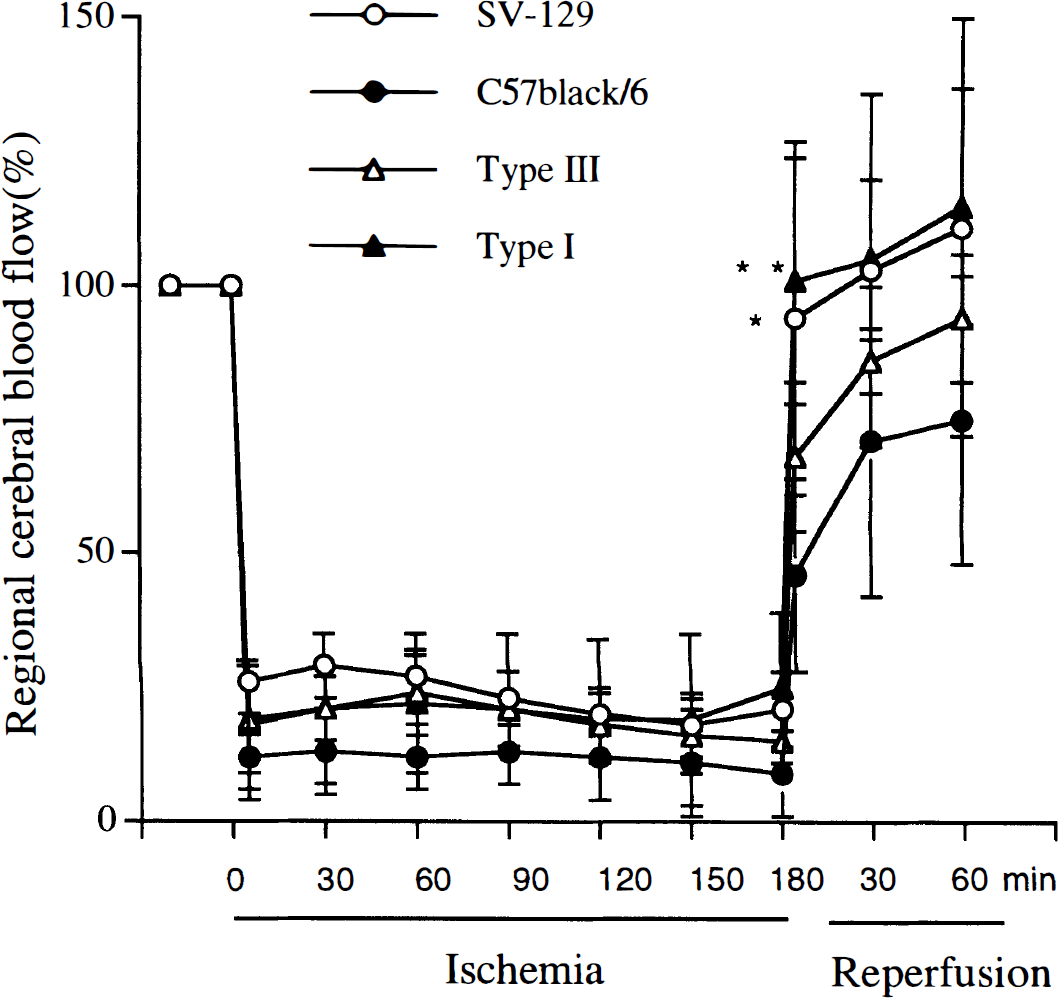
Regional CBF (rCBF) was determined by laser–Doppler flowmetry during 3-h ischemia (middle cerebral artery occlusion) and 1-h reperfusion in wild-type and mutant mice (see Materials and Methods). During ischemia, rCBF decreased to 10–20% of baseline. After reperfusion. rCBF immediately returned to preischemic levels in SV-129 and type I nitric oxide synthase (NOS) mutant mice, whereas recoveries were gradual and incomplete in C57black/6 and type III NOS mutant mice. The tip of the probe was placed on the intact skull over the ischemic cortex (2 mm posterior and 6 mm lateral to bregma). n = 5/group (mean ± SD). *p < 0.05, **p < 0.01 vs. C57black/6 analysis of variance followed by Tukey's multiple-range test).
Transient ischemia
[3H]L-NNA binding in normal brains was consistent with our previous report (Hara et al., 1996b). At baseline, type I NOS mutant mice displayed very low binding, whereas the binding in type III NOS mutant was similar to wild-type. Binding did not change 15 min after MCA occlusion, but began to increase 1 h later (although not statistically significantly). Binding significantly increased in all the ischemic regions (striatum, cortical MCA territory, and amygdala) 3 h after MCA occlusion (no reperfusion) and was still increased 10 min and 3 h after reperfusion. Binding returned to control levels 1 day after reperfusion (Fig. 2). A small decrease below baseline was observed in amygdala and cortex after 7 days (Fig. 2). Radioligand binding in the ACA territory and in contralateral brain did not change during ischemia or reperfusion. A large increase in binding to the ischemic zone (striatum, amygdala, and cortical MCA territory) was found in C57black/6 and type III mutants 3 h after reperfusion. For example, [3H]L-NNA binding values in striatum in SV-129, C57black/6, and type III mutant mice were 9.1 ± 1.7 vs 22.9 ± 10.1 fmol/mg tissue (contralateral vs. ipsilateral, mean ± SD, n = 6, p < 0.01), 9.6 ± 1.8 vs. 34.3 ± 8.5 fmol/mg (n = 5, p < 0.01), and 6.0 ± 2.2 vs. 17.2 ± 7.0 fmol/mg (n = 6, p < 0.01), respectively. However, no increase was observed in type I NOS mutant mice (1.9 ± 1.8 vs. 2.3 ± 2.0 fmol/mg tissue, n = 6) (Fig. 3). A similar pattern was found in amygdala and cortical MCA territory (data not shown). 7-NI (100 μM) completely displaced [3H]L-NNA binding in all brain regions 3 h after reperfusion. Aminoguanidine (100 μM) displaced only ∼30% of bound [3H]L-NNA (not shown). This proportion of displaced ligand was constant in all brain areas, including within the lesion.
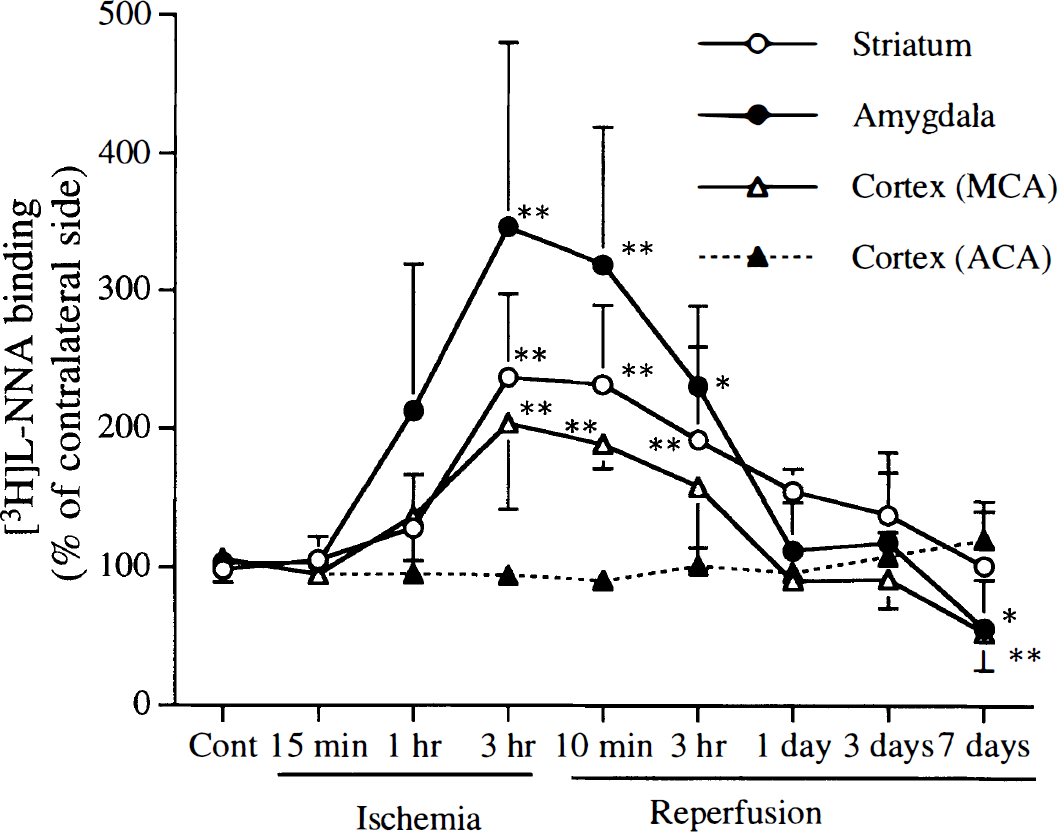
Time course of [3H]L-NG-nitroarginine ([3H]L-NNA) binding after 3 h of filament middle cerebral artery (MCA) occlusion in wild-type (SV-129) mice. [3H]L-NNA binding increased in the ipsilateral striatum, amygdala, and cortical MCA territory at the end of the ischemia (3 h) and during reperfusion, but not in the ipsilateral anterior cerebral artery territory (nonischemic region). After 7 days of reperfusion, [3H]L-NNA binding decreased in the ipsilateral striatum and cortical MCA territory. There were no significant time-dependent binding changes in the contralateral hemisphere. Values are expressed as percentage of contralateral side [mean ± SD, n = 4–6, *p < 0.05, **p < 0.01 vs. Cont (control, no ischemia) (analysis of variance followed by Dunnett's multiple-range test)].
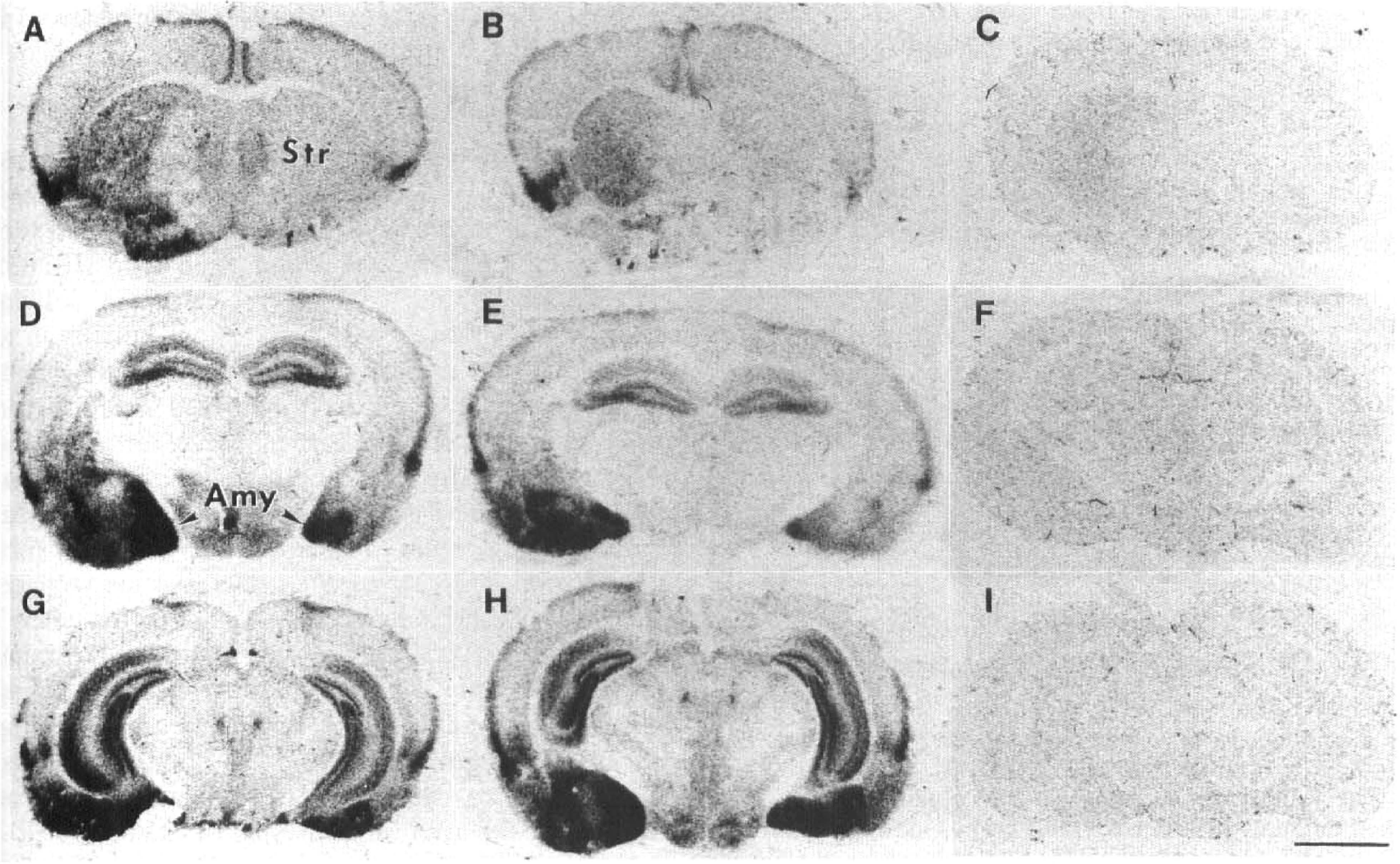
Autoradiographic distribution of [3H]L-NG-nitroarginine [3H]L-NNA) binding after 3 h of left middle cerebral artery occlusion and 3 h of reperfusion in wild-type (SV-129) (
Saturation studies
[3H]L-NNA binding was saturable and of high affinity. A three- to fivefold increase in maximum binding (Bmax) was found in the ischemic regions 3 h after reperfusion. No change in the dissociation constant (Kd) was observed (Fig. 4; Table 3).
Kd and Bmax values of [3H]L-NNA binding at 3 h after reperfusion in wild-type (SV-129) mouse brain
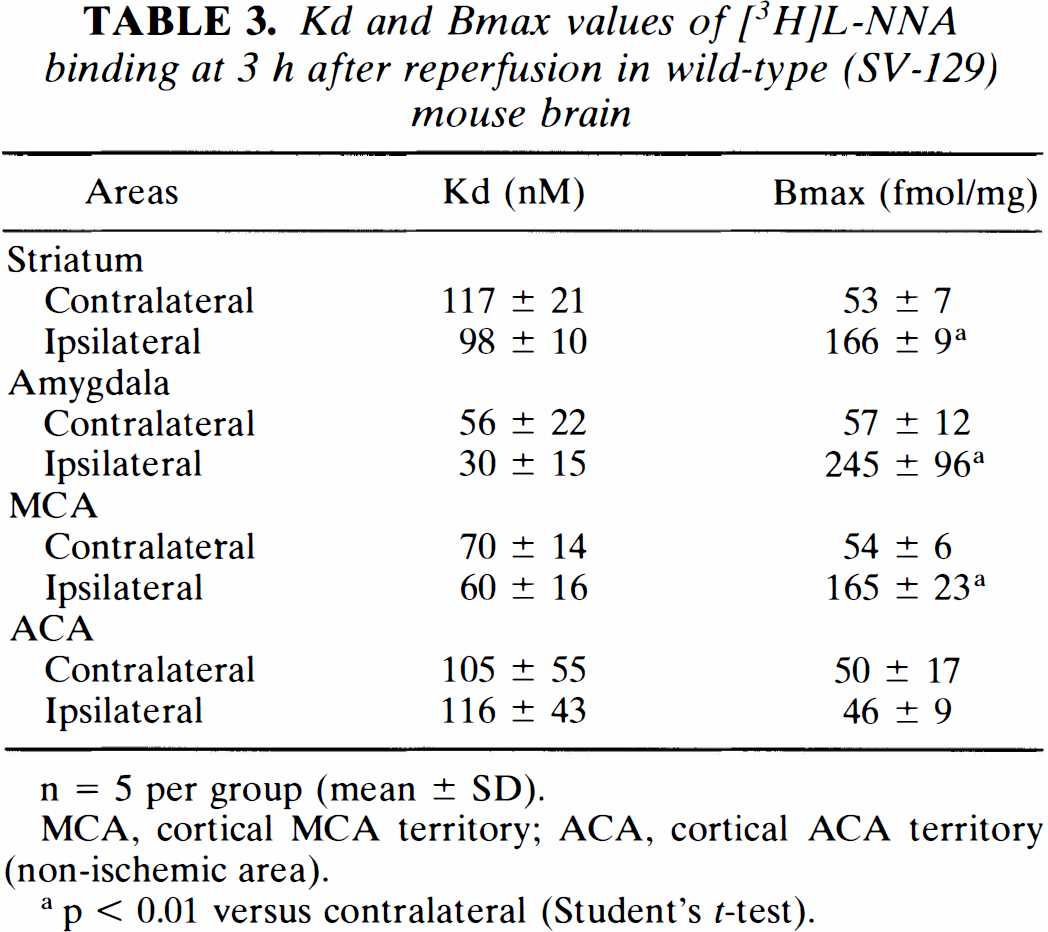
n = 5 per group (mean ± SD).
MCA, cortical MCA territory; ACA, cortical ACA territory (non-ischemic area).
p < 0.01 versus contralateral (Student's t-test).
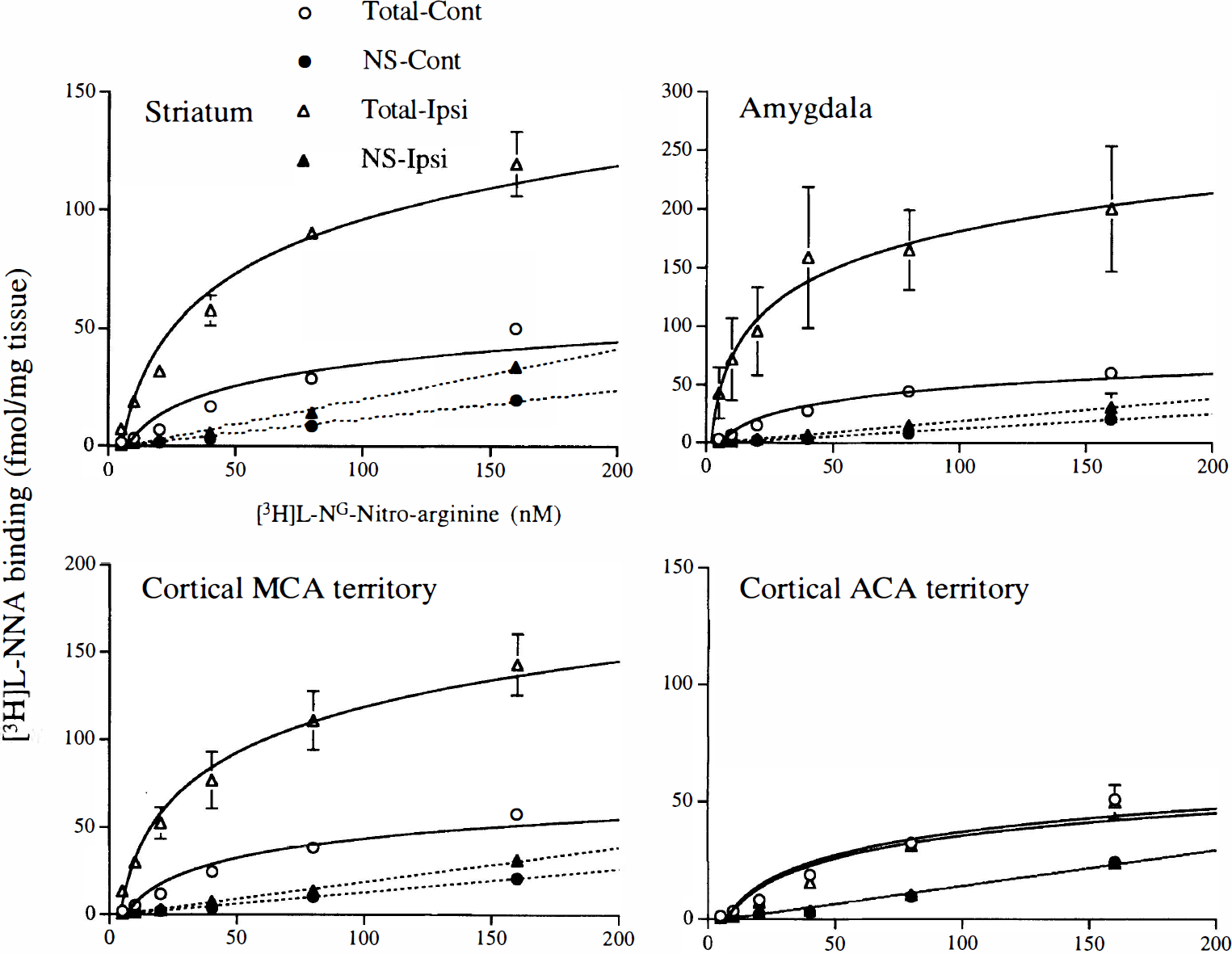
Saturation analysis of [3H]L-NG-nitroarginine ([3H]L-NNA) binding to sections of SV-129 mouse brain at 3 h after middle cerebral artery occlusion (3 h). Sections were incubated with increasing concentrations (5–160 nM) of [3H]L-NNA in the absence (total binding) and presence (nonspecific binding) of 10 μM unlabeled L-NNA for 30 min at room temperature. Each point presents the mean ± SD of five separate brains per group. Total-Cont, total binding in contralateral nonischemic side; NS-Cont, nonspecific binding in contralateral nonischemic side; Total-Ipsi, total binding in ipsilateral ischemic side; NS-Ipsi, nonspecific binding in ipsilateral ischemic side.
Ligand selectivity
A high density of [3H]L-NNA binding was observed in the endothelium of pig aorta (Fig. 5). After LPS injection, type II NOS catalytic activity increased in rat spleen from <0.3 to 21.1 ± 0.5 fmol/mg wet wt/min (n = 3). However, there was no corresponding change in the distribution or density of [3H]L-NNA binding at this time (Fig. 6B). There were no differences in Ca2+-dependent NOS (type I and III) between LPS- and saline-treated animals.
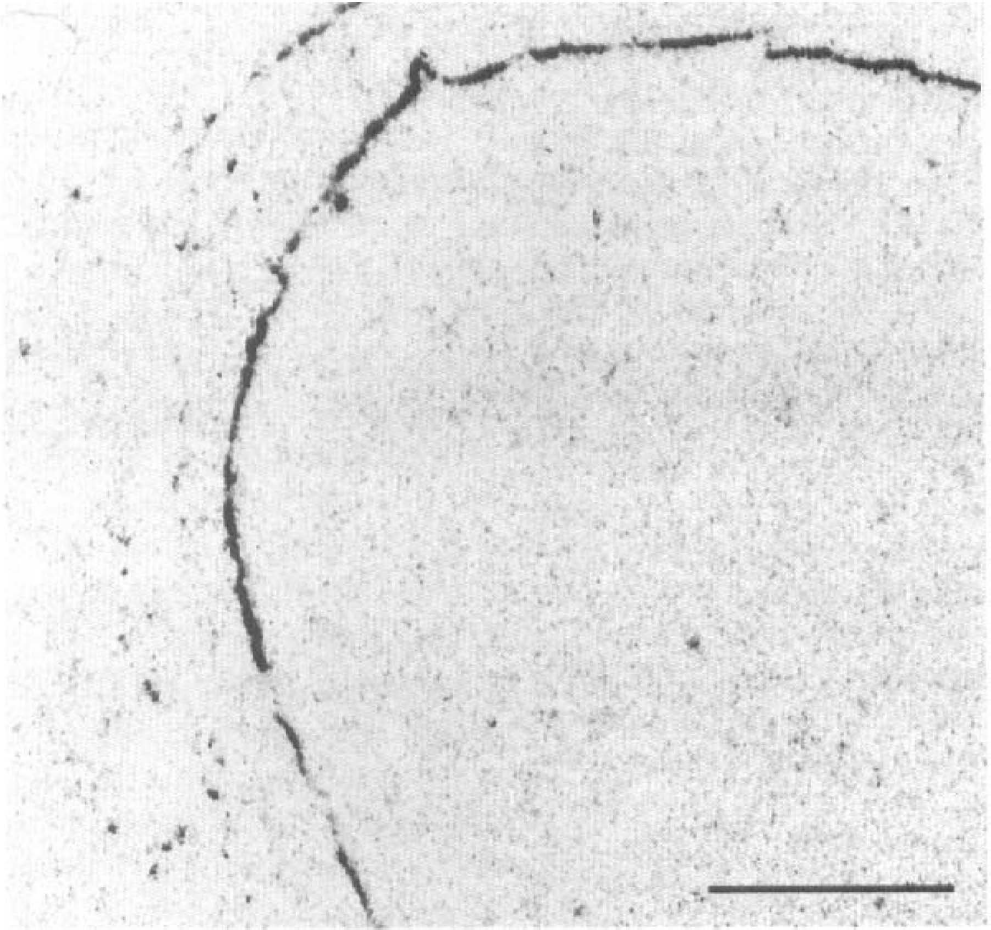
Autoradiographic distribution of [3H]L-NG-nitroarginine ([3H]L-NNA) binding in pig aorta, showing specific binding to the endothelial layer. [3H]L-NNA concentration: 20 nM. Bar = 2 mm.
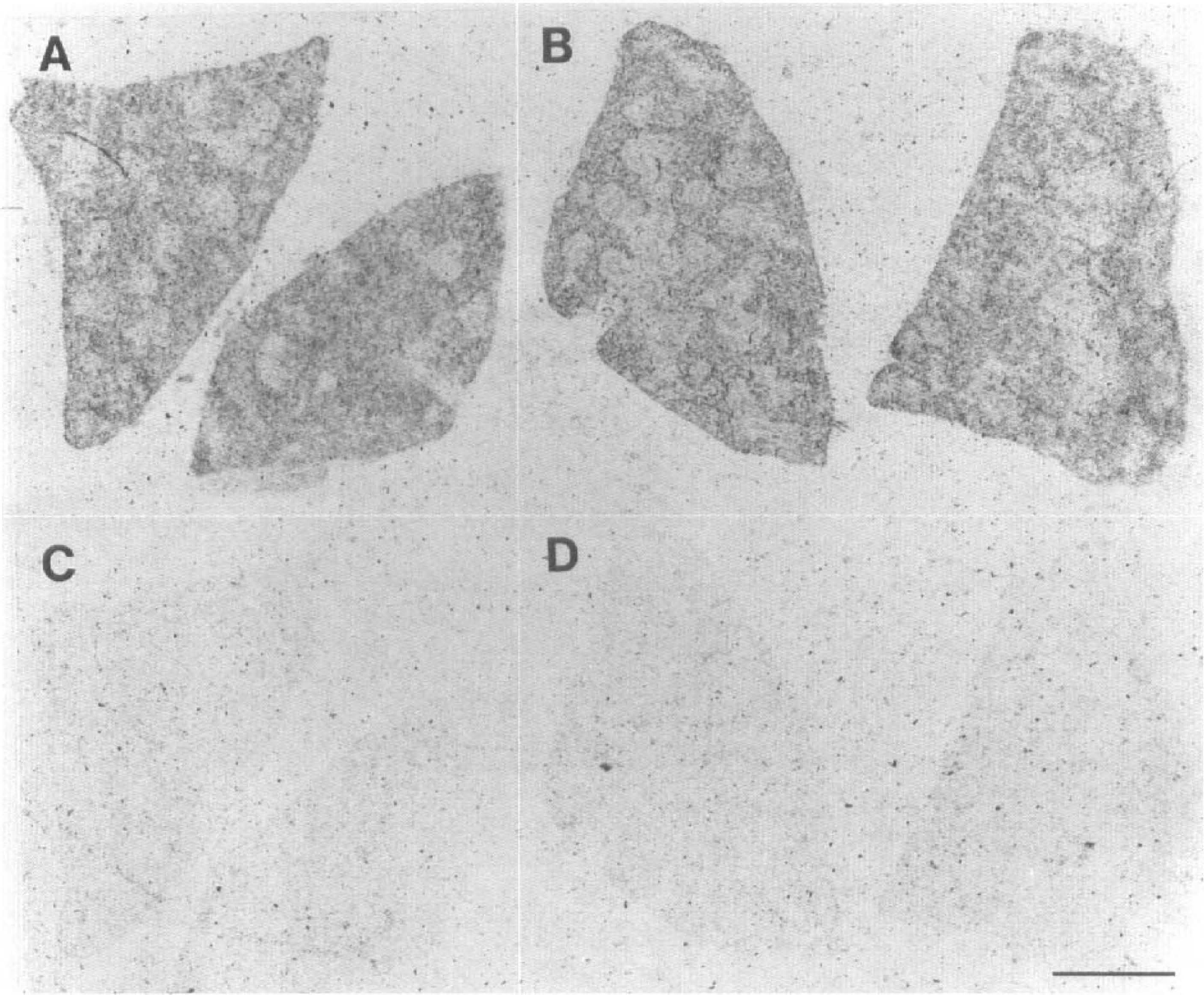
Autoradiographic distribution of [3H]L-NNA binding in rat spleen 6 h after administration of lipopolysaccharide (LPS; 5 mg/kg i.p.). (A and B) Total binding in vehicle (saline) and LPS-treated group. No difference was observed. (
Intrastriatal NMDA injections
NMDA increased binding by 30% starting 6 h following injection and reached a peak (200–230%) at 24–48 h (Fig. 7B–D). Binding in the ipsilateral hippocampus did not change (data not shown). Forty-eight hours after NMDA injection, wild-type littermate controls showed the largest increase, followed by type I NOS heterozygotes. There was only a small elevation in [3H]L-NNA binding in type I NOS mutant mice. [3H]L-NNA binding values in lesion area 48 h after intrastriatal NMDA injection in type I NOS+/+ (sham, no NMDA injection), type I NOS+/+, type I NOS+/–, and type I NOS–/– were 8.9 ± 2.0 vs. 9.1 ± 2.0 (mean ± SD, contralateral vs. ipsilateral, n = 5), 8.4 ± 1.6 vs 19.3 ± 4.9 (n = 5, p < 0.01, ANOVA followed by Student's t test), 6.6 ± 2.2 vs. 13.1 ± 5.4 (n = 6, p < 0.01), and 4.0 ± 0.9 vs. 6.0 ± 1.1 (n = 5, p < 0.01) fmol/mg, respectively. Consistent with previous findings, NMDA lesion size was smallest in type I mutant mice (Fig. 7A and E). [3H]L-NNA binding at 0, 3, 6, 12, 24, and 48 h after NMDA injection in striatum was 100 ± 0 (% of contralateral side, mean ± SD, n = 5), 105 ± 3 (n = 5), 130 ± 3 (n = 5), 163 ± 4 (n = 6, p < 0.01 compared with before injection; time 0 h, ANOVA followed by Dunnett's test), 329 ± 22 (n = 4, p < 0.01, and 295 ± 11 (n = 4, p < 0.01) fmol/mg, respectively.
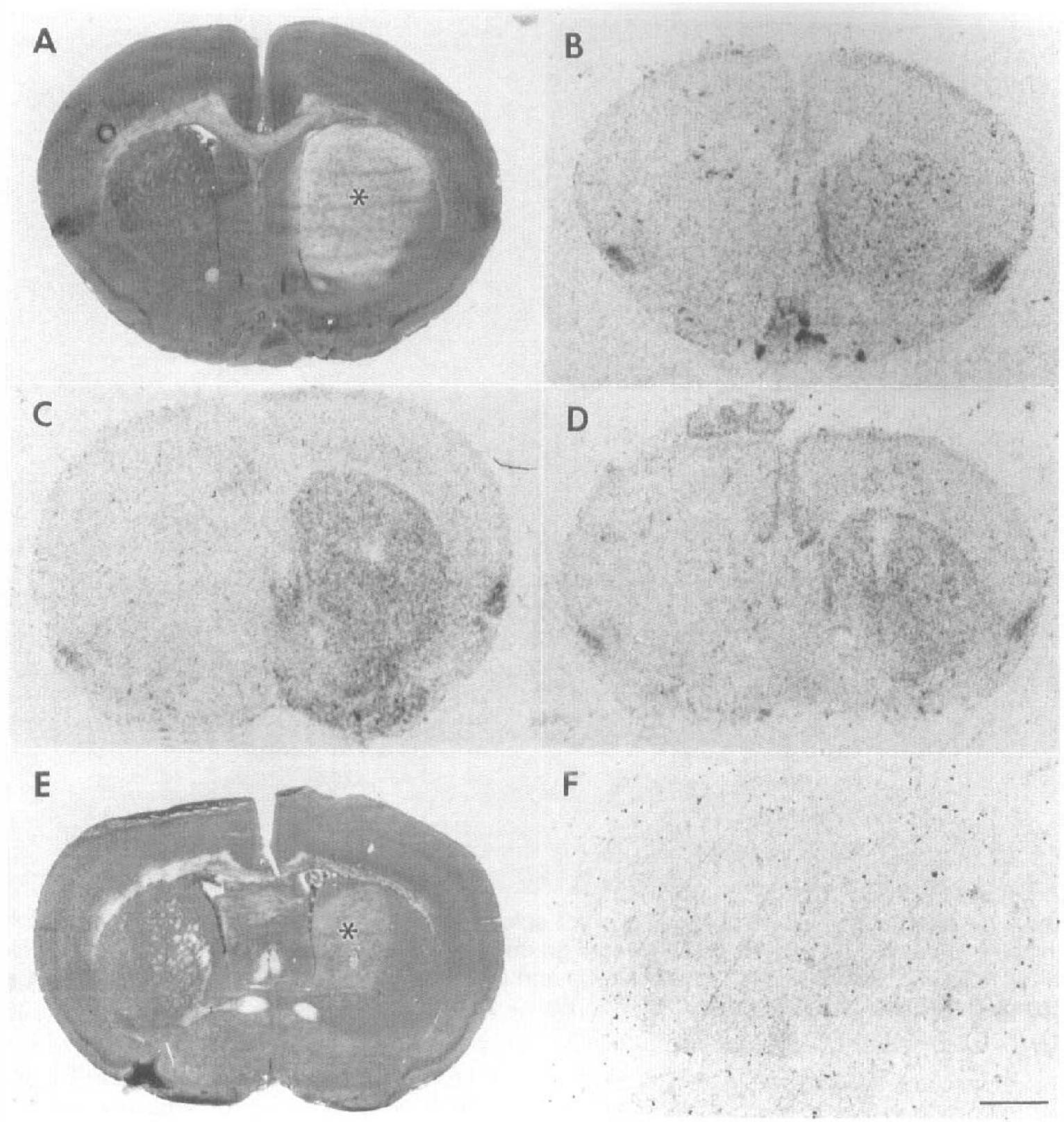
[3H]L-NNA [3H]L-NG-Nitroarginine binding after intrastriatal N-methyl-
Changes in NOS catalytic activity
Twenty-four hours after NMDA injection, total NOS catalytic activity was 7.6 ± 1.1 (mean ± SD, n = 4) and 4.5 ± 1.7 (n = 4) fmol/mg wet wt/min in contralateral and ipsilateral striatum, respectively (p < 0.05, Student's t test). Type II NOS catalytic activity was below the level of assay detection at this time.
RT-PCR following transient ischemia
RT-PCR was used to quantitate changes in mRNA levels for each NOS isoform. First, we confirmed the validity of the technique. Using a fixed total amount of RNA, we varied the level of type I NOS mRNA between 0 and 100% of wild-type levels by combining wild-type brain RNA with type I NOS mutant brain RNA. With use of these mixtures, first strand cDNA was synthesized and used as template for PCR with type I NOS-specific primers. Under the reaction conditions, the amount of PCR product varied according to the amount of type I NOS RNA present in the initial RNA sample (r = 0.97, p < 0.001, χ2-test).
We determined the amount of each NOS isoform during reperfusion for 10 min, 30 min, 60 min, and 3 h. RNA was isolated from ischemic and contralateral control hemispheres. Type I NOS mRNA levels began to increase at 10 min and persisted for 3 h (Figs. 8A and 9). The amount of PCR product was >150% of control values. During this time, the level of type I NOS in the control samples diminished to ∼70% of its original value.
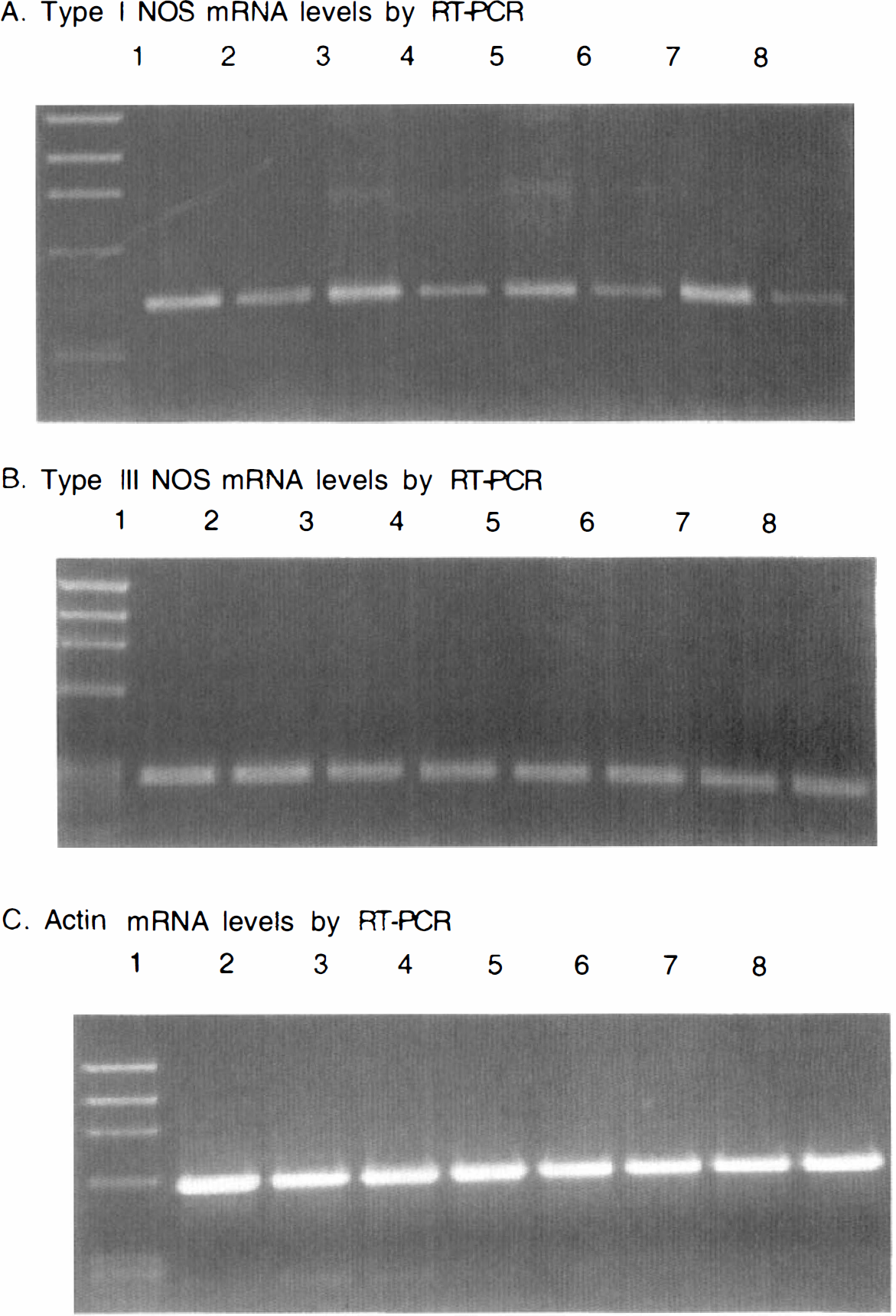
Agarose gel of polymerase chain reaction (PCR) products reflecting mRNA levels of type I nitric oxide synthase (NOS) (
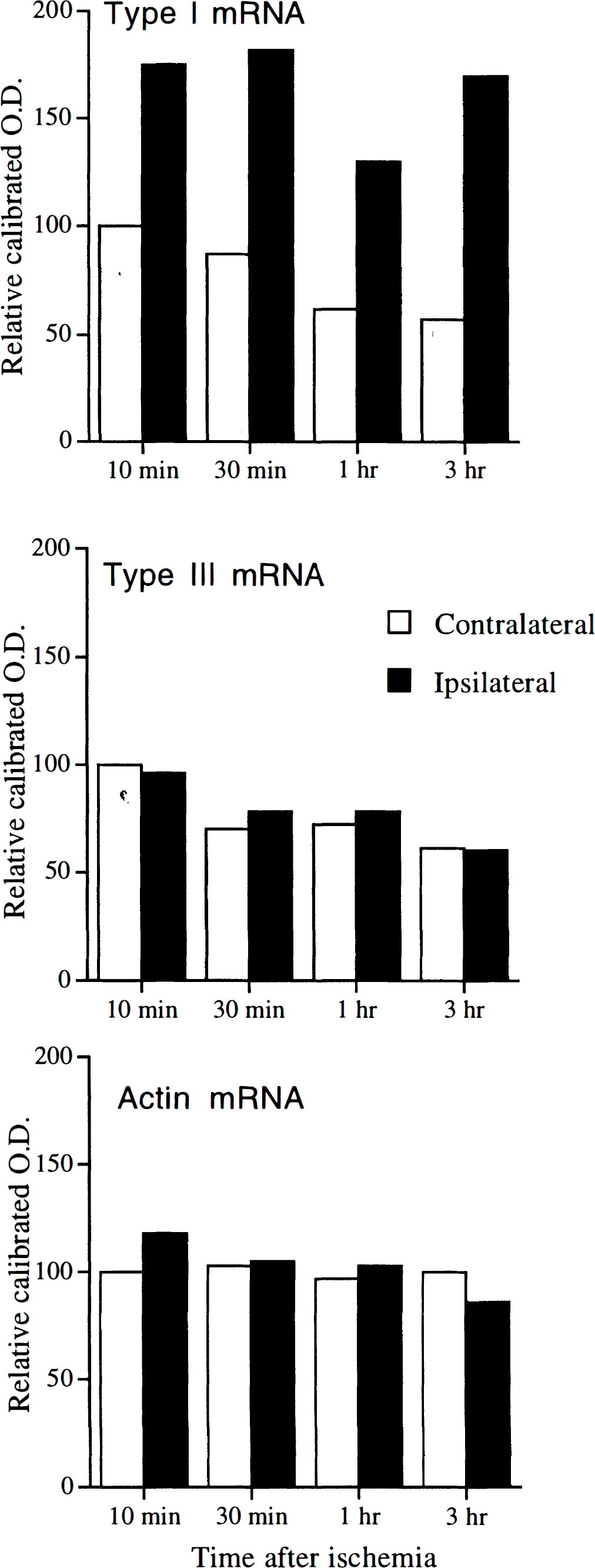
Quantitation of type I nitric oxide synthase (NOS), type II NOS, and actin mRNA after focal ischemia in mice by reverse transcription polymerase chain reaction (PCR). mRNA was measured at 10 min, 30 min, 1 h, and 3 h after middle cerebral artery occlusion. The gels in Fig. 8 were analyzed using NIH Image 1.60 after scanning. The intensity of the bands corresponding to the major PCR products was determined and plotted. Ipsilateral, ischemic forebrain hemisphere; Contralateral, contralateral forebrain hemisphere; OD, optical density.
Type III NOS mRNA showed a similar decline, and there was no difference in type III NOS mRNA levels between ischemic and control sides (Figs. 8B and 9). Type II NOS mRNA could not be detected under these reaction conditions, although the primers were successfully used to detect type II NOS mRNA from the spleen of LPS-treated mice (data not shown). Actin mRNA was used as a control. The levels of actin mRNA showed a slight decline over 3 h, although there was no difference between the ischemic and control sides (Figs. 8C and 9).
DISCUSSION
Focal cerebral ischemia and intrastriatal NMDA microinjection caused a robust increase in [3H]L-NNA binding in mouse brain limited to the anatomical regions at risk. Enhanced binding was observed inSV-129, C57black/6, and type III NOS mutant mice and was displaced by 7-NI, a selective type I NOS inhibitor. Only a small increase was detected in the type I NOS-deficient mouse, and this may have been due to either type III NOS up-regulation or products of the type I NOS gene processed by alternative splicing (Brenman et al., 1996). One such splice variant lacks two exons, constitutes ∼5% of type I NOS transcripts (Ogura et al., 1993), but lacks the ability to synthesize NO from L-arginine (Ogura et al., 1994). Furthermore, alternatively spliced type I NOS has been identified in both mutant and wild-type mice and is ∼80% as active as full-length type I NOS (Brenman et al., 1996). However, it is not known whether this isoform binds [3H]L-NNA.
A decrease in [3H]L-NNA binding below baseline was observed in amygdala and cortex after 3 and 7 days (Fig. 2). Ischemic infarction is well developed in mice 3 and 7 days following such a severe ischemic insult (3-h ischemia) (Yang et al., 1994). We cannot rule out the possibility that the development of ischemic infarction affected the kinetics of [3H]L-NNA binding in this study, although the reduced binding at these later time points probably does represent less NOS protein within dead or dying tissue.
Despite increased mRNA and [3H]L-NNA binding reflecting increased formation of new protein, NOS catalytic activity decreased after NMDA injection and, as reported previously, decreases shortly after ischemia (Yoshida et al., 1995). NO levels, however, increase shortly after ischemia. Malinski et al. (1993) detected enhanced NO production using a porphyrincoated microelectrode and electrochemical detection method. Increased brain nitrite, cyclic GMP levels (Kader et al., 1993), plus the formation of paramagnetic adducts of NO after injection of diethyldithiocarbamate (Mullins et al., 1996) are consistent with enhanced NO production during ischemia. It is difficult to easily resolve this discrepancy, but it might reflect differences between those methods used to measure NO activity in vivo and in vitro or, for example, the presence of NOS tissue inhibitors released during ischemia from sequestered compartments. Regardless, the observed differences in binding between groups (Figs. 3, 7–9; Table 3) were not likely to follow from differences in physiological parameters (e.g., MABP, rCBF) measured before ischemia and during the reperfusion period.
Several published studies suggest that type I NOS expression may be up-regulated after noxious insults in vivo and in vitro. In rats, type I NOS mRNA increases as early as 15 min after permanent focal ischemia (Zhang et al., 1994). In the same study, type I NOS immunoreactivity and NADPH-diaphorase-positive neurons increased in the ischemic regions 1–24 h following ischemia in the rat. Other studies supported these findings (Endoh et al., 1994; Peng et al., 1996). In addition, Kato et al. (1994) found an increase in NADPH-diaphorase activity in CA1 pyramidal neurons following global ischemia. Furthermore, O'Hearn et al. (1995) reported that type I NOS was induced in cerebellar Purkinje cells following excitotoxic insults after ibogaine. Our data suggest that NOS binding sites increased during ischemia and that these increases persisted during reperfusion for several hours before returning toward baseline.
It appears unlikely that the increase in binding after NMDA injection was due to a physical interaction between excitotoxin (Rao and Butterworth, 1996) and radiolabeled ligand or an artifact of ligand binding in ischemic brain. More likely, it was the result of an increase in type I NOS protein, as evidenced by the quantitative RT-PCR data (Figs. 8 and 9) and the increase in Bmax during reperfusion. Moreover, the increase in [3H]L-NNA binding showed a gene dosage effect, being most robust during NMDA excitotoxicity in wild-type > heterozygote > type I NOS mutant brain (Fig. 7).
Type II NOS catalytic activity was below detectable limits following intrastriatal NMDA injection as previously reported early after MCA occlusion in rats (Iadecola et al., 1995; Yoshida et al., 1995). For unexplained reasons [3H]L-NNA does not bind type II NOS despite blocking enzyme activity both in vivo and in vitro. [3H]L-NNA binding in spleen did not change after LPS, and aminoguanidine, a type II NOS inhibitor, weakly displaced the [3H]L-NNA binding after ischemia, indicating that under the conditions of the present study, aminoguanidine was not specific for type II NOS and may not be appropriate to use a marker of type II NOS binding.
In conclusion, our results demonstrate the [3H]L-NNA binding, increases during focal cerebral ischemia or NMDA excitotoxicity, reflecting a possible up-regulation of type I NOS gene expression. Up-regulation of NOS may play an important role in the pathogenesis of ischemic/excitotoxic disorders such as stroke and neurodegenerative diseases.
Footnotes
Acknowledgment:
This research was supported by the Massachusetts General Hospital Interdepartmental Stroke Program Project (NS10828), grants from the NINDS NS2683 (M.A.M.) and NS33335 (P.L.H.), and an unrestricted research award from Bristol-Meyers Squibb (M.A.M.). P.L.H. is an Established Investigator of the American Heart Association.