
Abstract
Nitric oxide (NO) influences infarct size after focal cerebral ischemia and also regulates neurogenesis in the adult brain. These observations suggest that therapeutic approaches to stroke that target NO signaling may provide neuroprotection and also enhance brain repair through cell replacement. However, ischemic injury and neurogenesis are both affected differently depending on which isoform of NO synthase is the source of NO. In addition, ischemia itself stimulates neurogenesis, and ischemia-induced neurogenesis may be regulated differently than neurogenesis in nonischemic brain. To determine how neuronal NO synthase affects ischemia-induced neurogenesis, transient focal cerebral ischemia was produced in wild-type mice and in knockout mice lacking neuronal NO synthase, and BrdU incorporation and doublecortin immunoreactivity were measured in the principal neuroproliferative regions of the adult brain. Knockout of neuronal NO synthase reduced infarct size and increased both basal and ischemia-induced neurogenesis, suggesting that NO from this source is an inhibitory regulator of neurogenesis in the ischemic brain. 7-Nitroindazole, an NO synthase inhibitor that preferentially affects the neuronal isoform, also increased neurogenesis in rats when administered by the intracerebroventricular route. Selective inhibition of neuronal NO synthase may have the potential to both reduce infarct size and enhance neurogenesis in stroke.
Introduction
Ischemia-induced neurogenesis produces cells that may have the capacity to replace dead neurons, because these new cells migrate to sites of brain ischemia (Arvidsson et al, 2002; Jin et al, 2003; Nakatomi et al, 2002), and interfering with their production worsens outcome from experimental stroke (Raber et al, 2004). However, ischemia-induced neurogenesis generates fewer cells than are lost in stroke, suggesting that pharmacological augmentation of neurogenesis may be required to optimize its potential to contribute to brain repair. The use of exogenous factors to enhance neurogenesis for therapeutic purposes seems plausible, because several growth factors can stimulate neurogenesis (Cameron et al, 1998). Some growth factors also have independent, neuroprotective effects that decrease infarct size (Jin et al, 2004; Sun et al, 2003; Wang et al, 2004), which may allow them to reduce the severity of ischemic injury while also promoting brain repair. However, treatment with growth factors is limited by difficulties inherent in delivering them to the brain and by concerns about oncogenic or tumor angiogenic effects.
Several drugs in current clinical use can also stimulate neurogenesis, including antidepressants (Santarelli et al, 2003) and drugs that act through nitric oxide (NO) signaling pathways (Chen et al, 2003; Zhang et al, 2002). Because NO also regulates the severity of cerebral ischemic injury (reviewed in Keynes and Garthwaite, 2004), treatment directed at NO signaling could, in principle, produce favorable effects on both the extent of ischemic brain injury and the effectiveness of brain repair. NO synthase (NOS) gene-knockout studies have shown that NO generated by neuronal (nNOS) and inducible (iNOS) isoforms exacerbates (Huang et al, 1994; Nagayama et al, 1999), whereas endothelial NOS (eNOS) attenuates (Huang et al, 1996) ischemic neuronal injury (reviewed in Samdani et al, 1997). Accordingly, specific inhibitors of nNOS or iNOS may have therapeutic potential in this setting.
The localization of nNOS to sites of neuronal proliferation and migration in the hippocampal dentate gyrus (DG) (Islam et al, 2003), forebrain subventricular zone (SVZ) and rostral migratory stream (RMS) (Moreno-Lopez et al, 2000; Shariful Islam et al, 1998), and olfactory bulb (OB) (Chen et al, 2004) suggests a role for NO in neurogenesis. However, this role appears to be complex, depending on which NOS isoforms are involved, which neuroproliferative regions are examined, and what phase of neurogenesis—proliferation or neuronal differentiation—is considered. nNOS-knockout mice showed an increase in the number of newborn cells, labeled with BrdU, in DG, SVZ, RMS, and OB (Packer et al, 2003), consistent with an inhibitory effect of nNOS on basal neurogenesis. In iNOS-knockout compared with wild-type mice, the number of BrdU-labeled cells in DG was reduced ipsilateral, to an ischemic lesion, pointing to iNOS as a positive mediator of ischemia-induced (but not basal) neurogenesis (Zhu et al, 2003). Endothelial NOS-knockout mice have defects in the production of hematopoietic and endothelial progenitors (Aicher et al, 2003), as well as decreased proliferation of neuronal precursors in DG (Reif et al, 2004).
Pharmacological studies have also addressed the role of NOS in neurogenesis, although NO donors and NOS inhibitors are less isoform-selective than genetic models. The NO donor, (Z)-1-[N-(2-aminoethyl)-N-(2-ammonioethyl) aminio]diazen-1-ium-1,2-diolate (DETA/NONOate), increased BrdU labeling in the rat DG and SVZ in vivo, as well as the number of BrdU-labeled cells that expressed neuronal marker proteins, and also enhanced ischemia-induced neurogenesis (Zhang et al, 2001). In contrast, NO donors decreased proliferation of murine SVZ cells in vitro (Cheng et al, 2003; Matarredona et al, 2004), accompanied in some cases by enhanced neuronal differentiation. The effects of NOS inhibitors on neurogenesis have also been studied. Intraventricular infusion of Nω-nitro-L-arginine methyl ester (L-NAME) in rats increased BrdU labeling in DG, SVZ, RMS, and OB, without changing the percentage of newly generated cells that expressed neuronal markers (Packer et al, 2003). In another study, L-NAME and 7-nitroindazole (7-NI), administered systemically, increased the number of cells that incorporated BrdU in SVZ, RMS, and OB, but not DG, and decreased the percentage of new cells with phenotypic neuronal features (Moreno-Lopez et al, 2004). Finally, aminoguanidine, which preferentially inhibits iNOS, blocked ischemia-induced, but not basal, BrdU labeling in DG (Zhu et al, 2003).
These findings indicate that the outcome of strategies designed to attenuate ischemic brain damage, while stimulating brain repair through neurogenesis, is not easily predictable. Inhibiting eNOS is detrimental in ischemic injury (Huang et al, 1996) and its effect on neurogenesis is unknown. Inhibiting either nNOS (Huang et al, 1994) or iNOS (Nagayama et al, 1999) should afford neuroprotection, but inhibiting iNOS may also adversely affect outcome by blocking ischemia-induced neurogenesis (Zhu et al, 2003). Inhibiting nNOS might be expected to increase basal neurogenesis (Packer et al, 2003), but how this would affect ischemia-induced neurogenesis is unknown, and pharmacological data suggest it might be harmful (Zhang et al, 2001).
To begin to address these issues, with the aim of identifying a combined neuroprotective and neurogenesis-promoting approach to cerebral ischemia that targets NO signaling, we investigated the effect of nNOS gene deletion on ischemia-induced neurogenesis in a mouse middle cerebral artery occlusion (MCAO) model of stroke.
Methods
Neuronal Nitric Oxide Synthase-Knockout Mice
Experiments were approved by local committee review and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Neuronal NOS-deficient (nNOS −/− ) mice (strain B6;129S4-Nos1 tm1Plh /J, stock number 002633), backcrossed to genetic homogeneity, were obtained from The Jackson Laboratory (Bar Harbor, ME, USA) and bred in-house. Male mice were used exclusively and littermates served as controls. Mice were genotyped by polymerase chain reaction (PCR) of tail-tip genomic DNA using the allele-specific PCR primers: (1) 5′- CTT GGG TGG AGA GGC TAT TC-3′ (forward primer lying upstream of the insertion site), (2) 5′- AGG TGA GAT GAC AGG AGA TC-3′ (reverse primer lying downstream of the insertion site), (3) 5′-TCA GAT CTG ATC CGA GGA GG-3′ (lying at the end of the LTR of the insertion sequence), and (4) 5′-TTC CAG AGC GCT GTC ATA GC-3′. A 280-bp DNA fragment was amplified by primers (1) and (2) from the knockout allele and a 117-bp fragment was amplified by primers (3) and (4) from the wild-type allele. The PCR protocol included a 10 mins preincubation at 94°C, followed 35 cycles of 30 secs each at 94°C, 30 secs at 58°C, and 1 mins at 72°C.
Focal Cerebral Ischemia
Transient focal cerebral ischemia was induced by intraluminal MCAO using a nylon monofilament suture, as described (Parmentier-Batteur et al, 2002). Neuronal NOS-knockout mice or wild-type littermates weighing 25 to 30 g were anesthetized with 1.5% to 2% isoflurane in 30% oxygen and 70% nitrous oxide with a vaporizer (VetEquip.Inc., Pleasanton, CA, USA). The rectal temperature was controlled at 37°C±0.5°C with a thermostat-controlled heating blanket. The skin overlying the anterior neck was incised in the midline, and the left external carotid artery was exposed, ligated with a 6–0 silk suture and dissected distally, and its branches were electrocoagulated. The left internal carotid artery was then isolated and separated from the vagus nerve. A 5–0 surgical monofilament nylon suture (Devis and Geck, Manati, Puerto Rico) blunted at the tip was introduced into the left internal carotid artery through the external carotid stump and advanced 9 to 10 mm past the common carotid bifurcation. The middle cerebral artery (MCA) was occluded for 45 mins and then reperfused by removing the suture. Arterial blood gases and mean arterial blood pressure were monitored until 30 mins after the onset of reperfusion. After recovering from anesthesia, mice were maintained in an air-conditioned room at 20°C.
Administration of the Neuronal Nitric Oxide Synthase Inhibitor, 7-Nitroindazole
Adult male Sprague–Dawley rats weighing 280 to 320 g were anesthetized with 2% isoflurane in 70% N2O/30% O2 and implanted with an Alzet 1003D osmotic minipump (Alza Corporation, Mountain View, CA, USA). The cannula was placed in the left lateral ventricle, 3.7 mm deep to the pial surface, −0.6 mm anteroposterior relative to the bregma, and 1.3 mm lateral to the midline. Rats were treated for 3 days with 1 μL/h of either artificial cerebrospinal fluid (aCSF) containing 128 mmol/L NaCl, 2.5 mmol/L KCl, 0.95 mmol/L CaCl2 and 1.99 mmol/L MgCl2 (n=4) or the nNOS inhibitor, 7-NI (25 mmol/L in aCSF; R&D Calbiochem, San Diego, CA, USA) (n=5). Rats were killed 3 or 7 days later.
BrdU Labeling In Vivo
BrdU (50 mg/kg in saline; Sigma, St Louis, MO, USA) was administered twice daily by the intraperitoneal route, for 1 day (2 total doses) in mice or for 3 days (6 total doses) in rats, after ischemia.
BrdU Immunohistochemistry in Brain Sections
Brains (3 to 5 per condition) were removed after perfusion with saline and 4% paraformaldehyde in phosphate-buffered saline (PBS). Adjacent 50 μm sections, corresponding to coronal coordinates interaural 8.7 to 10.2 mm, bregma −0.30 to bregma −1.2 mm (SVZ), and interaural 4.48 to 5.86 mm, bregma −4.52 to bregma −3.14 (DG), were cut with a cryostat and stored at −80°C. Sections were incubated in 2 mol/L HCl at 37°C for 30 mins, and rinsed in 0.1 mol/L boric acid (pH 8.5) at room temperature for 10 mins. Sections were then incubated in blocking solution (2% goat serum/0.3% Triton X-100/0.1% bovine serum albumin (BSA) in PBS) for 1 h at room temperature, and with mouse monoclonal anti-BrdU (1:200, Roche Applied Science, Indianapolis, IN, USA) at 4°C overnight. Sections were washed with PBS, incubated with biotinylated goat-anti-mouse secondary antibody (1:200; Vector Laboratories, Burlingame, CA, USA) for 1 h at room temperature, washed, and placed in avidin–peroxidase conjugate solution (Vector Laboratories) for 30 mins. The horseradish–peroxidase reaction was detected with 0.05% diaminobenzidine and 0.03% H2O2. Processing was stopped with H2O, sections were dehydrated through graded alcohols and cleared in xylene, and coverslips were applied using permanent mounting medium (Vector Laboratories).
Colocalization of BrdU with doublecortin (DCX) was assessed by double-label immunocytochemistry using affinity-purified goat polyclonal anti-DCX (1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and staining was visualized with an FITC-labeled and rhodamine-conjugated secondary antibody (Jackson Immuno-Research Laboratories, Inc., West Grove, PA, USA; 1:200). Controls included preabsorbing primary antibody and omitting secondary antibody. For confocal microscopy, a Nikon PCM-2000 laser-scanning confocal microscope and Simple PCI imaging software (Compix Inc., Cranberry Township, PA, USA) were used.
BrdU-Immunopositive Cell Counting
BrdU-positive cells in subgranular zone (SGZ) and SVZ were counted blindly in six diaminobenzidine-stained, 50 μm coronal sections per animal, spaced 200 μm apart. Cells were counted under high-power (× 200) on a Nikon E300 microscope with a Magnifire digital camera, and the image was displayed on a computer monitor. Results were expressed as the average number of BrdU-positive cells per section.
Image Analysis
To quantify cell death and cell migration after MCAO in nNOS wild-type and knockout mice, images of Klenow staining from the cortical ischemic penumbra region and images of DCX staining in SVZ and CPu were acquired using Nikon Eclipse-800 Microsoft and Nikon digital camera DXM 1200 and the number of Klenow and DCX-positive cells in each image (× 200 magnification) was determined by image analysis using Simple PCI software (Compix Inc.).
Statistical Analysis
Values were expressed as the mean±s.d. from at least three experiments. The statistical significance of differences between means was evaluated by Student's t-test for single, and by ANOVA followed by post hoc t-tests for multiple, comparisons, with P<0.05 considered significant.
Results
Preischemic arterial blood gas measurements and rectal temperatures were similar in wild-type and nNOS-knockout mice, and were unchanged after 30 mins of MCAO (Table 1). However, infarct size was reduced in nNOS-knockout compared with wild-type mice, consistent with previous findings (Huang et al, 1994), because of a decrease in the medial extent of the cerebral cortical damage along the MCA/anterior cerebral artery (ACA) and MCA/posterior cerebral artery border zones (Figure 1A). The extent of reduction in infarct size that we observed in nNOS-knockout was smaller (∼20%), however, than that (∼40%) observed previously (Huang et al, 1994), perhaps because we employed transient rather than permanent MCAO. At the microscopic level, the decrease in infarct size was manifested by a reduction in cellular injury revealed by cresyl violet staining, caspase-3 cleavage, and Klenow staining (Figure 1B). The number of cells with DNA strand breaks detectable with the Klenow reagent was 32% lower adjacent to the MCA/ACA border zone of nNOS-knockout than in wild-type mice (Figure 1C).
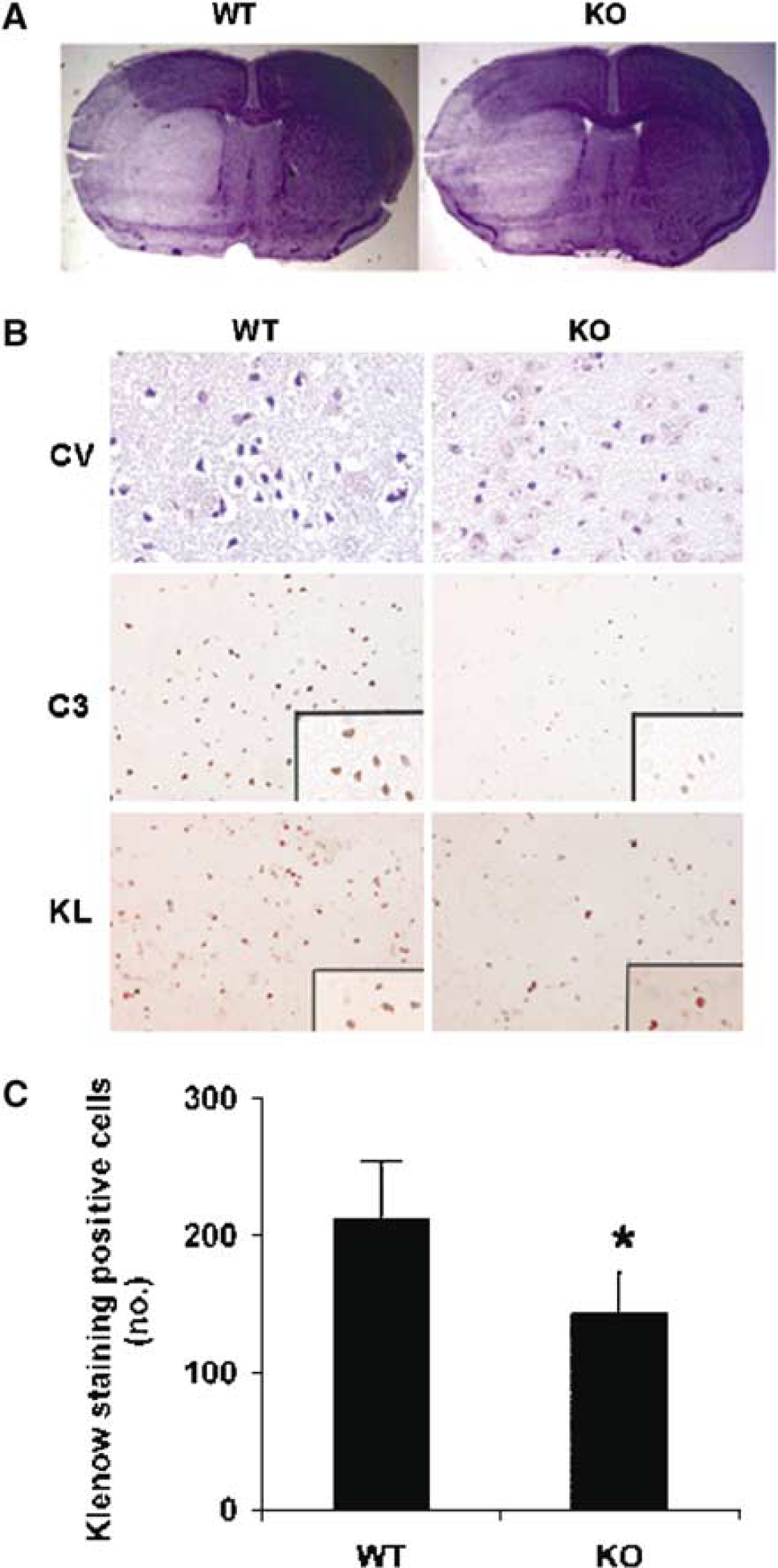
Effect of middle cerebral artery occlusion (MCAO) in wild-type (WT) and neuronal nitric oxide synthase (nNOS)-knockout (KO) mice. The middle cerebral artery (MCA) was occluded with a suture for 45 mins, followed by reperfusion for 3 days. (
Physiological measures in mice before and 30 mins after middle cerebral artery (MCAO)
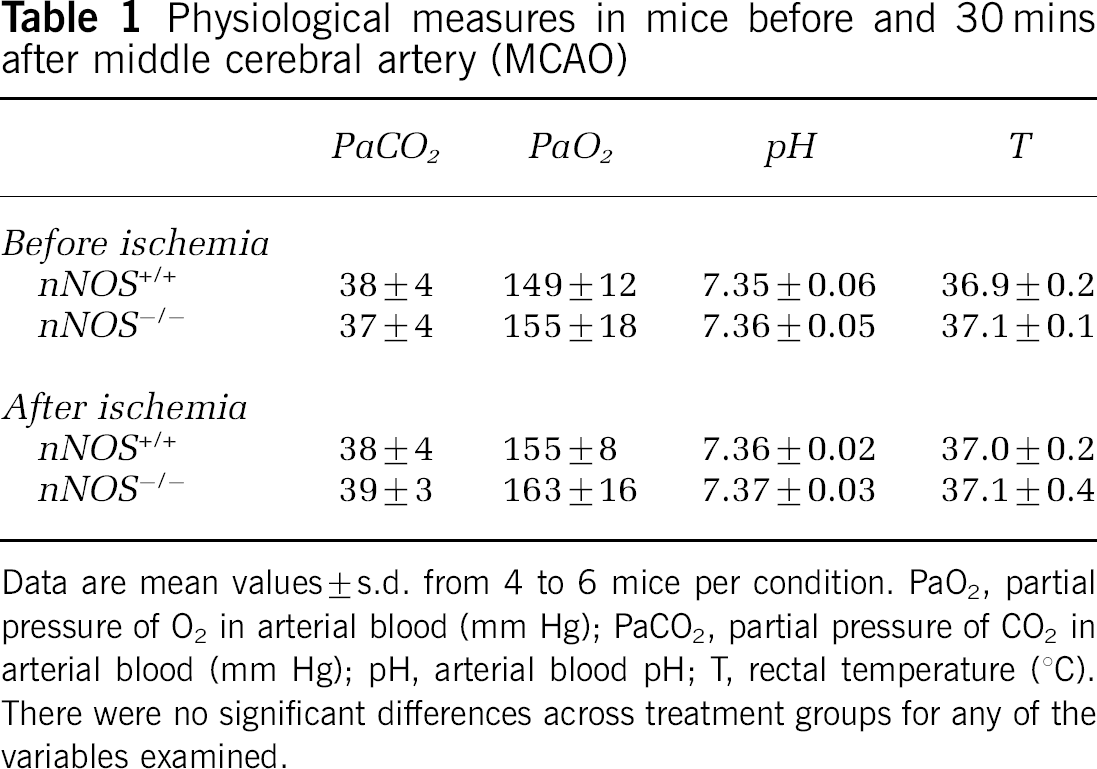
Data are mean values±s.d. from 4 to 6 mice per condition. PaO2, partial pressure of O2 in arterial blood (mm Hg); PaCO2, partial pressure of CO2 in arterial blood (mm Hg); pH, arterial blood pH; T, rectal temperature (°C). There were no significant differences across treatment groups for any of the variables examined.
Immunohistochemistry revealed an increase in the number of BrdU-labeled cells in both DG subgranular zone (SGZ) and SVZ of nNOS-knockout mice (Figures 2A–2E). Approximately 60% to 70% of BrdU-immunopositive cells expressed neuronal phenotype marker proteins such as DCX (Figures 2F–2H). In nonischemic (sham-operated) controls, the number of cells that incorporated BrdU was ∼30% higher in DG-SGZ and ∼45% higher in SVZ of nNOS-knockout mice than wild-type mice (Figures 3A, 3B), although the difference in SGZ did not reach statistical significance. After ischemia, the effect of nNOS deletion was more pronounced. In DG-SGZ, ischemia increased BrdU labeling by 78% at 3 days and by 66% at 2 weeks in nNOS-knockout mice, but by only 37% and 38%, respectively, in wild-type mice. Expressed differently, nNOS deletion increased BrdU labeling by 61% at 3 days and by 66% at 2 weeks in ischemic mice, but by only 23% and 38%, respectively, in nonischemic controls. Findings were similar in SVZ, where ischemia increased BrdU incorporation by 67% (3 days) or 76% (2 weeks) in nNOS-knockout mice, and by 28% (3 days) or 45% (2 weeks) in wild-type mice, and where nNOS deletion increased BrdU labeling by 79% (3 days) or 78% (2 weeks) in ischemic mice, compared with 42% and 46% in nonischemic controls. In summary, nNOS appeared to inhibit basal neurogenesis, at least in SVZ, because BrdU labeling was increased in nonischemic nNOS-knockout compared with nonischemic wild-type mice, and inhibited ischemia-induced more than basal neurogenesis in both DG-SGZ and SVZ.
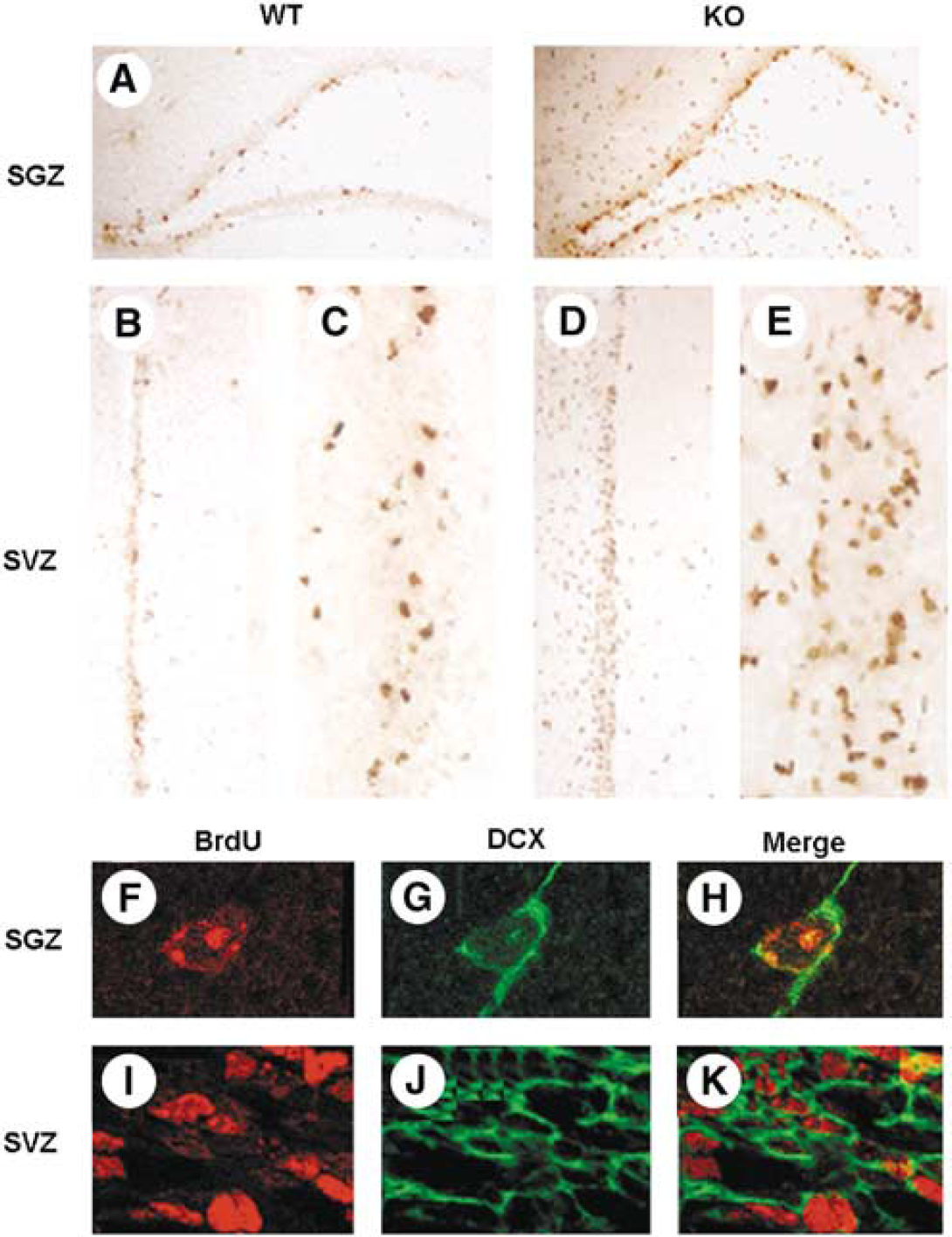
BrdU labeling of cells in neuroproliferative brain regions of ischemic adult wild-type (WT) and neuronal nitric oxide synthase (nNOS)-knockout (KO) mice. Ischemia was produced as described in the legend to Figure 1, mice were administered BrdU (50 mg/kg intraperitoneally twice daily for 3 days) and BrdU-labeled cells in subgranular zone (SGZ) and subventricular zone (SVZ) were detected by immunohistochemistry. (
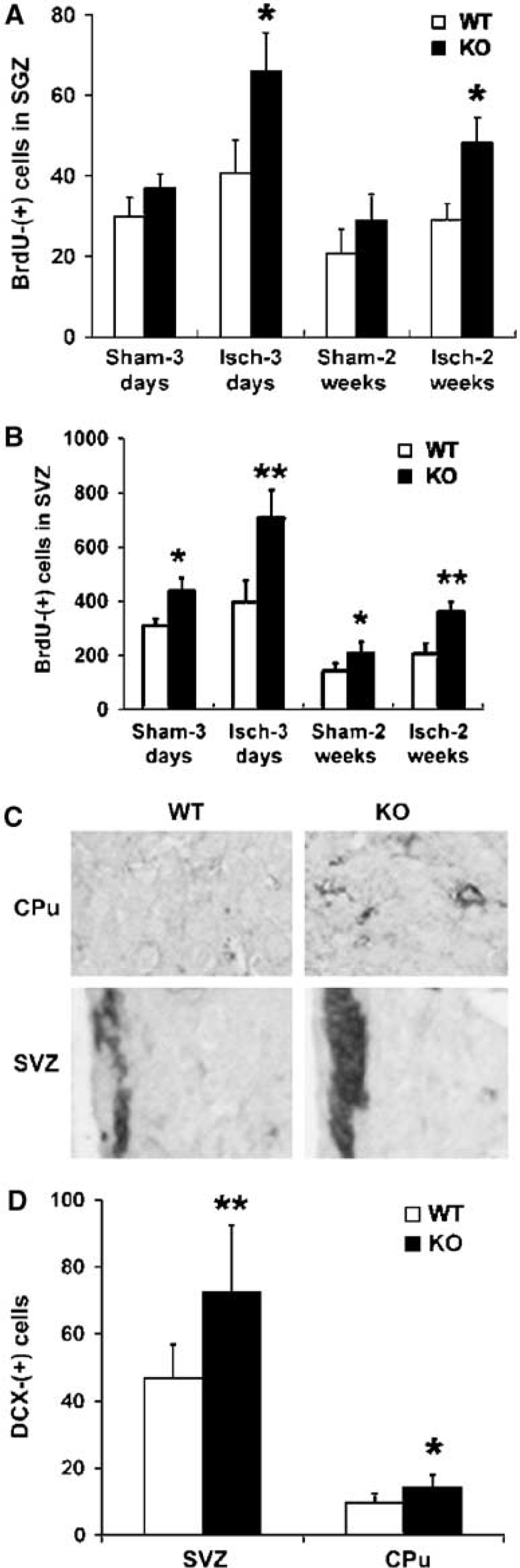
Ischemia-induced neurogenesis is increased in neuronal nitric oxide synthase (nNOS)-knockout mice. Ischemia was produced and BrdU was administered as described in the legends to Figures 1 and 2, and BrdU-labeled cells in SGZ and SVZ from wild-type (WT, unfilled bars) and nNOS-knockout (KO, filled bars) mice were detected by immunohistochemistry 3 days or 2 weeks after-ischemia. (
After focal cerebral ischemia, newborn neurons that arise in the SVZ migrate into ischemic regions of the caudate-putamen and cerebral cortex (Arvidsson et al, 2002; Jin et al, 2003; Parent et al, 2002). To determine the effect of nNOS on ischemia-induced migration of these new neurons, we examined immunohistochemical expression of the immature neuronal marker DCX in SVZ and caudate-putamen (CPu) from wild-type and nNOS-knockout mice after MCAO. Doublecortin expression, like BrdU labeling, was increased after ischemia in SVZ of knockout compared with wild-type mice (consistent with a neuronal lineage of cells associated with ischemia-induced BrdU incorporation), and was increased to a similar extent in caudate-putamen of knockout mice (Figures 3C, 3D). Therefore, whereas nNOS appeared to modify basal and ischemia-induced neurogenesis, as evidenced by the fact that both were increased in nNOS-knockout compared with wild-type mice, nNOS did not seem to affect ischemia-induced migration of newborn neurons. Thus, the number of DCX-immunopositive neurons found in CPu, expressed as a percentage of DCX-immunopositive neurons in the SVZ from which they originate (Jin et al, 2003), was almost identical.
As noted previously, although some studies have employed NOS inhibitors to ascertain the effect of NO on neurogenesis (Cheng et al, 2003; Matarredona et al, 2004; Moreno-Lopez et al, 2004; Packer et al, 2003; Zhang et al, 2001; Zhu et al, 2003), these inhibitors are less NOS isoform-specific than gene knockouts. This is especially a consideration when, as in the case of NOS, different isoforms can produce opposite effects on an end-point like neurogenesis. Nevertheless, pharmacological inhibitors can provide confirmatory evidence that the effects of gene knockout result directly from absence of the targeted gene product, and not from adaptations thereto. For this reason, we also examined the effect of 7-NI, a relatively specific nNOS inhibitor (Southan and Szabo, 1996). BrdU immunoreactivity was increased in SGZ and SVZ of rats administered 7-NI for 3 days by the intraventricular route and killed 3 or 7 days later (Figure 4A). Like BrdU-labeled cells in nNOS-knockout mice, BrdU-labeled cells in 7-NI-treated rats were associated primarily with cells that expressed markers of neuronal lineage, including DCX (Figure 4B). Quantitative analysis of BrdU-labeled cells showed that 7-NI increased labeling in rat SGZ by 40% at 3 days and by 71% at 7 days after BrdU administration, and increased labeling in SVZ by 15% (which was not statistically significant) and 48% at the same time points (Figures 4C, 4D). The time-dependent reduction in the number of BrdU-positive cells seen in controls in this experiment and the experiments illustrated in Figure 3 are likely because of physiological death of BrdU-labeled cells or dilution of the label through cell division. Although the effects of nNOS gene knockout in mice and pharmacological inhibition of NOS in rats are not directly comparable, the results shown in Figures 3A, 3B and 4C, 4D may suggest that SVZ neurogenesis was more prominently affected in the former and SGZ neurogenesis in the latter. However, the relative roles of different treatments and different species in this apparent disparity are unclear.
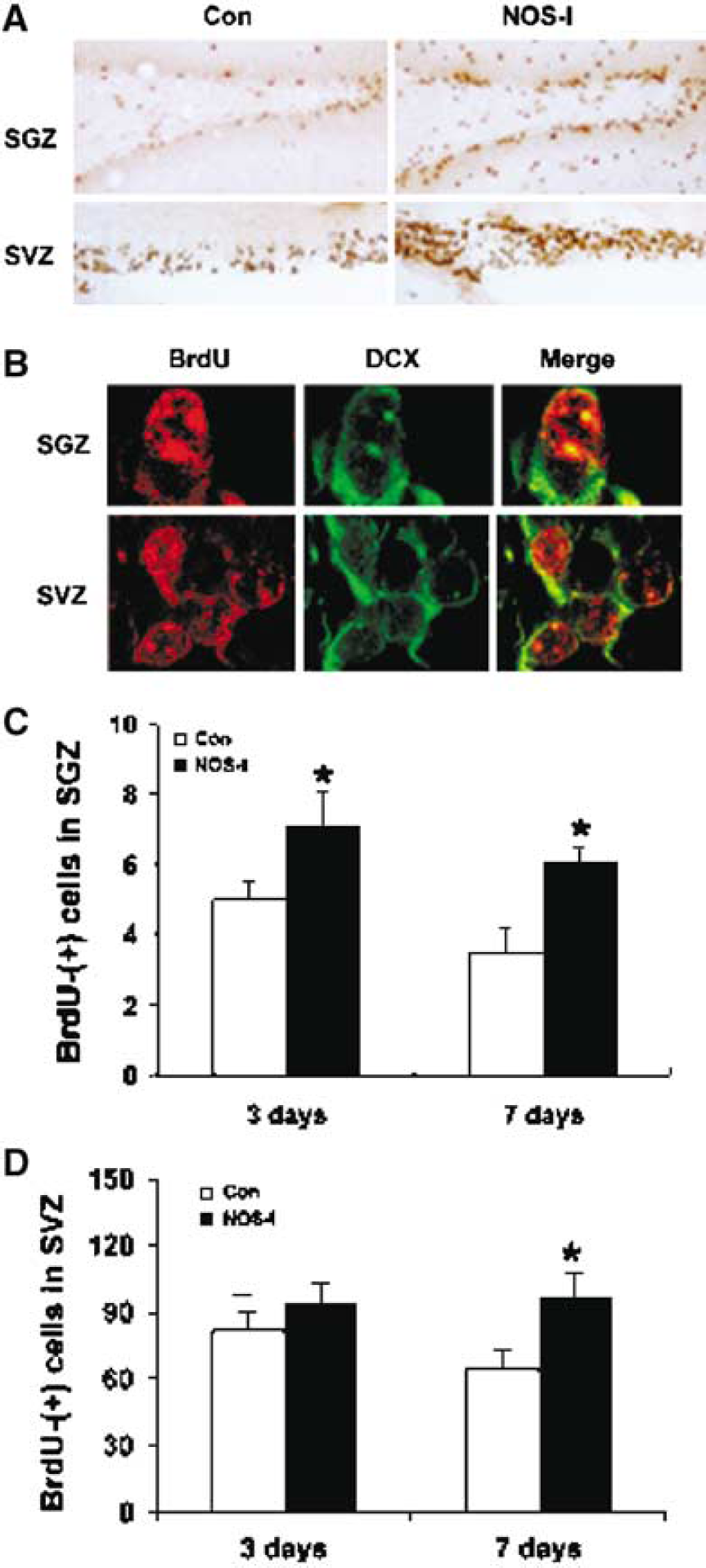
Effect of the neuronal nitric oxide synthase (nNOS)-preferring NOS inhibitor, 7-nitroindazole (7-NI), on neurogenesis in subgranular zone (SGZ) and subventricular zone (SVZ) of rat brain. Vehicle (Con) or 7-NI (NOS-I; 25 mmol/L i.c.v. at 1 μL/h) and BrdU (50 mg/kg intraperitoneally twice daily) were administered for 3 days, and BrdU was localized 3 or 7 days later by immunohistochemistry. (
Discussion
The main finding of this study is that ischemia-induced neurogenesis is increased in the brains of nNOS-knockout mice, implicating NO produced by nNOS as an inhibitory regulator of ischemic neurogenesis. Strokes are reduced in size in nNOS mice (Huang et al, 1994), as we confirmed, which might have attenuated ischemia-induced neurogenesis by decreasing its stimulus. However, deletion of nNOS increases neurogenesis under nonischemic conditions (Packer et al, 2003), making it difficult to predict the net effect of nNOS on neurogenesis in the ischemic brain. This is an important issue because pharmacological interventions that modify neurogenesis could have therapeutic value in diseases associated with neuronal loss, such as stroke, and NO signaling pathways are potential targets for such an approach. Although we investigated ischemia-induced neurogenesis and others have examined basal neurogenesis, our results are most consistent with previous studies favoring an inhibitory effect of nitric oxide (Packer et al, 2003). NO donors have been reported to increase neurogenesis as well (Zhang et al, 2001), and the reason for this discrepancy is unclear. One possibility is that NO donors simulate the effect of a mixture of NOS isoforms, including iNOS, which stimulates ischemia-induced neurogenesis (Zhu et al, 2003) and eNOS, the effect of which is unknown. In contrast to NOS, NO donors may generate NO at nonphysiologic sites. Alternatively, lifelong deletion of nNOS in knockout mice may produce adaptations that make it difficult to compare them with animals subjected to acute pharmacological manipulations. Finally, basal and ischemia-induced neurogenesis could be regulated differently.
It is a commonplace that BrdU labeling, which we used in this study, does not suffice to establish that a cell is of recent provenance, and is also uninformative as to phenotype. Therefore, we used DCX, a marker protein specific for immature neurons, to confirm the neuronal character of BrdU-labeled cells. However, even the colocalization of BrdU with neuronal markers cannot exclude the possibility that the cells in question will undergo programmed cell death at their site of origin, which is thought to be the fate of most new neurons generated in the uninjured adult brain. To address this issue, we measured BrdU labeling in SGZ and SVZ for up to 2 weeks after-ischemia, and also counted DCX-expressing cells in the ischemic striatum, where newborn neurons from SVZ migrate after ischemia (Jin et al, 2003; Parent et al, 2002). The increased BrdU labeling observed in nNOS-knockout mice persisted for at least 2 weeks, and was accompanied by the appearance of new (DCX-immunopositive) neurons in the striatum, where they might be capable of replacing lost cells. One question that the present study cannot answer relates to the functional fate of newborn neurons in the ischemic brain of nNOS-knockout mice. As in most studies of adult neurogenesis, it is unclear how many newly generated cells become mature, functional neurons that can integrate into the brain's circuitry, or otherwise contribute to recovery. However, the recent demonstration (Raber et al, 2004) that radiation-induced ablation of neuronal precursor cells in the adult rodent brain worsens outcome from cerebral ischemia argues that the progeny of these cells may be functional.
In addition to NO produced by nNOS, other inhibitory regulators of neurogenesis have been identified. These include corticosteroids (Gould et al, 1992), excitatory amino acids (Cameron et al, 1995) (but also see Deisseroth et al, 2004), and inflammatory cytokines (Vallieres et al, 2002). Inhibitory regulators may help prevent gratuitous neurogenesis under physiological conditions, but for neurogenesis to increase after injury, their effects must be overcome. This could be accomplished by reduced production of inhibitors alone, increased production of positive regulators of neurogenesis, or both.
The isoform-dependence of NOS effects on neurogenesis is reminiscent of the similar dependence observed for NOS effects on infarct size (Huang et al, 1994, 1996), and indicates that the cell type or site of origin, or both, determine what the effect will be. In the case of ischemia, it is possible to imagine that eNOS affects primarily blood vessels, and that the resulting vasodilatation increases blood flow and thereby reduces the severity of ischemia. It seems less obvious how nNOS- and iNOS-derived NO may exert opposite effects on ischemia-induced neurogenesis, although nNOS could act directly on neurons, whereas the effect of iNOS might be mediated through the release of neurogenesis-promoting factors from non-neuronal (e.g., glial) cells that are targets for NO.
Our results underscore the complexity of the NO signaling system as a potential target for therapeutic neurogenesis, and suggest that selective inhibition of nNOS might be a promising strategy, at least in the setting of stroke. Other approaches that have been proposed include the use of sildenafil, a phosphodiesterase type 5 inhibitor that, like NO, increases brain cyclic GMP levels (Zhang et al, 2002) and statins (Chen et al, 2003), which stimulate eNOS.