
Abstract
Tumor necrosis factor alpha (TNF-α) is expressed in the ischemic brain; however, its precise role is not fully understood. We studied the effect of the dimeric form of the type I soluble TNF receptor linked to polyethylene glycol (TNFbp) on focal cerebral ischemia in mice using a permanent middle cerebral arterial occlusion (MCAO) model. TNFbp was applied topically, intravenously, or intraperitoneally. TNFbp binds and inhibits TNF-α. The volume of cortical ischemic lesions was measured by means of 2,3,5-triphenyltetrazolium chloride 24 h after MCAO. TNFbp produced a significant reduction in the cortical infarct volume of vehicle-treated animals (p < 0.001). The reduction in the volume of brain damage was 26% in animals that received 3 mg/kg of TNFbp topically. Further analysis of TNF-α inhibition following acute brain ischemia is indicated.
Tumor necrosis factor alpha (TNF-α) is one of the proinflammatory cytokines and is expressed in ischemic brain (Liu et al., 1994). There is evidence that another proinflammatory cytokine, interleukin-1, contributes to ischemic brain damage (Relton and Rothwell, 1992; Garcia et al., 1995). The role of TNF-α in acute ischemic stroke is not settled at this point. TNF-α in acute stroke not only may be detrimental, it also may be involved in recovery from tissue damage (Barger et al., 1995). However, excessive production of proinflammatory cytokines may be harmful to the brain and can cause secondary tissue damage after focal cerebral ischemia. There are several lines of evidence that TNF-α may play a crucial role in tissue injury during disease processes after release from either activated lymphocytes and macrophages of peripheral origin or astrocytes and microglial cells of the brain parenchyma (Martin et al., 1995; Probert et al., 1995). We have tested the effect of the dimeric form of the type I soluble TNF receptor linked to polyethylene glycol (TNFbp) (kindly provided by Amgen Inc., Boulder, Colorado) on the volume of ischemic brain damage induced by middle cerebral artery occlusion (MCAO) in mice. Species and strain differences in infarct topography after MCAO at a given site depend on variations in vascular anatomy. The anatomical variation of the circle of Willis affects the outcome of major vessel occlusions in animals (Barone et al., 1993). We have found that distal MCAO in BALB/C mice can cause a reproducible cortical infarct. The ability of TNFbp to reduce the expected infarct size was studied in this model.
MATERIALS AND METHODS
Mice
Mature male BALB/C mice, weighing 22–26 g, were used in the study. Animals were obtained from Charles River Labs (Wilmington, MA, U.S.A.) and were given free access to food and water before surgery. Animals were housed and cared for in accordance with the Guide for the Care and Use of Laboratory Animals (DHEW [DHHS] publication no. [NIH] 85-23, rev. 1985, Office of Science and Health Reports, DRR/NIH, Bethesda, MD 20205, U.S.A.). Procedures using animals were approved by the NIH Animal Care and Use Committee.
Permanent middle cerebral artery occlusion in mice
Animals were anesthetized with 3% halothane for induction and 1.5% halothane for maintenance in a 30% O2 and 70% N2O gas mixture via a face mask. The rectal temperature was measured and maintained at 37 ± 0.2°C with a heating blanket. The exposure of the middle cerebral artery was based on the description of Welsh et al. (1987). Briefly, the left temporoparietal region of the head was shaved, and a 5-mm incision was made between the orbit and ear. Under an operating microscope, an incision was made dividing the temporal muscle, and the left lateral aspect of the skull was exposed by reflecting the temporal muscle surrounding soft tissue. A small burr hole (2 mm) was made with a high-speed microdrill through the outer surface of the semitranslucent skull just over the visibly identified middle cerebral artery at the level of the inferior cerebral vein. Saline was applied to the area throughout the procedure to prevent heat injury. The inner layer of the skull was removed with fine forceps, the dura and arachnoid were opened, and left MCAO was performed by electrocoagulation (by means of a small-vessel cauterizer) without damaging the brain surface. If the brain surface was visibly damaged or if the middle cerebral artery bled owing to incomplete artery occlusion/coagulation, the animal was killed and not used. The duration of anesthesia did not exceed 30 min in any of the animals.
Immediately after MCAO, 0.3 or 3 mg/kg body weight of TNFbp (1 μl or 10 μl, respectively) was applied topically. With respect to topical application of TNFbp, the solutions were dripped through the dural and arachnoidal opening and allowed to disseminate for 3 min before turning the animal from its lateral decubitus position. There were seven and eight animals, respectively, in the two groups. Ten animals received the same amount of vehicle (phosphate-buffered saline, pH 7.5) by the same route and constituted the control group. The muscle and soft tissue were replaced, and the skin was closed using 5-0 nylon suture. To test the effect of systemic administration of TNFbp, six animals received intraperitoneal injection of 3 mg/kg body weight of TNFbp, and seven animals received intravenous injection of 3 mg/kg body weight of TNFbp immediately after MCAO.
Measurement of the size of cortical infarcts using 2,3,5-triphenyltetrazolium chloride (TTC)
Twenty-four hours after MCAO, the animals were injected with a lethal dose of thiopental (100 mg per kilogram of body mass, i.p.), and brains were removed, cut into 1-mm-thick coronal sections, and immersed in 2% TTC-saline solution at 37°C for 30 min. The infarcted areas of the brain slice were measured by means of a computer-assisted image analyzing system (NIH image 1.57). The infarct volume was calculated by taking the sum of the infarct area of different brain slices times the thickness of the slices. The contribution of the edema volume to the infarct volume was corrected by subtracting the ipsilateral uninvolved hemisphere volume from the total contralateral hemisphere volume and dividing by the total contralateral hemisphere volume (Swanson et al., 1990; Lin et al., 1993). Infarct volumes in each group were analyzed by analysis of variance. Statistical significance was set at p < 0.05.
RESULTS
The volume of ischemic brain damage averaged 30.4 ± 3.6 mm3 in control vehicle-treated animals (n = 10), 27.6 ± 3.3 mm3 in animals treated with 0.3 mg/kg body weight of TNFbp (n = 7), 22.5 ±3.7 mm3 in those given 3 mg/kg of TNFbp (n = 8), 25.9 ± 4.5 mm3 in the group injected with 3 mg/kg of TNFbp i.p. (n = 6), and 24.4 ± 2.8 mm3 in animals given 3 mg/kg of TNFbp i.v. (n = 7) (mean ± SD) (Fig. 1). TNFbp produced a significant reduction in infarct volumes that was dose-related. The reduction of the volume of brain damage was 26% in animals that received 3 mg/kg of TNFbp, 10% in those given 0.3 mg/kg of TNFbp, 15% in those injected with 3 mg/kg of TNFbp i.p., and 20% in animals administered 3 mg/kg of TNFbp i.v. The representative photographs of brain slices from each of five animals from the control group and the group given topically administered TNFbp 3 mg/kg are shown in Fig. 2. In TNFbp-treated animals, border zones between anterior and middle cerebral arteries and between posterior and middle cerebral arteries appeared to be the zones of recovery compared with control animals.
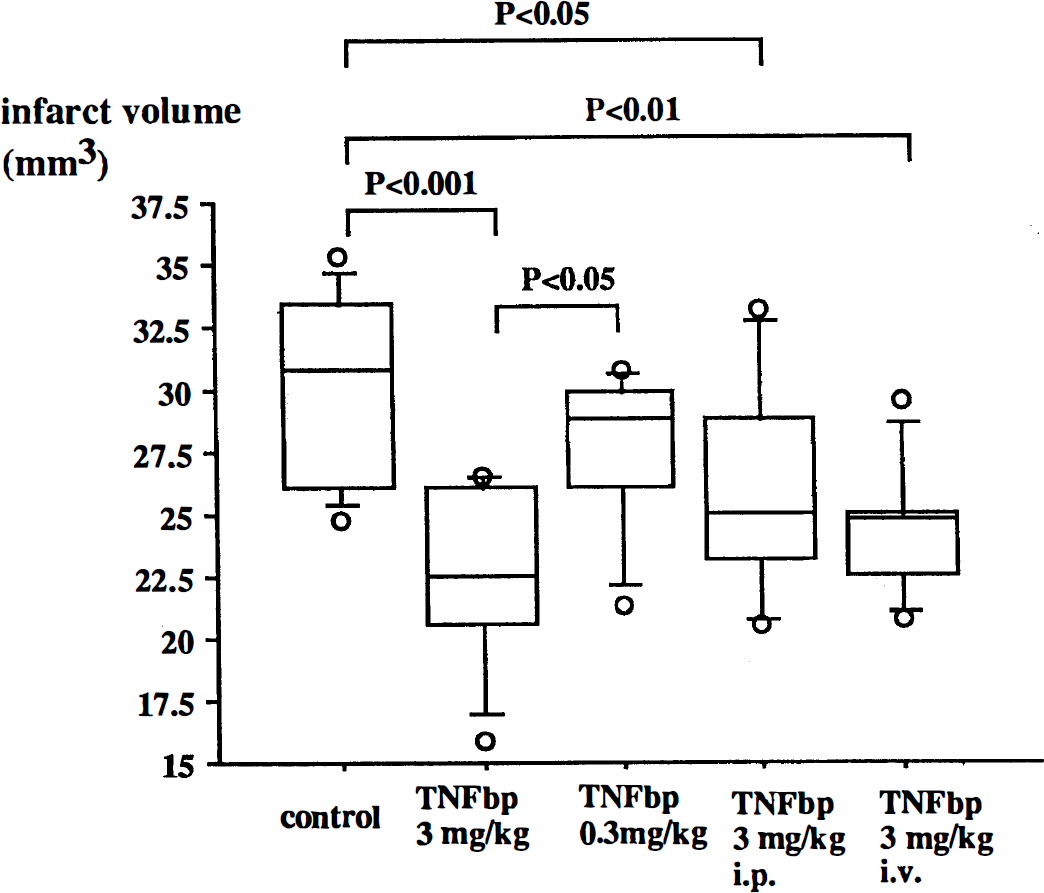
The size of infarcts in each experimental group. Average infarct volumes ± SD with dispersion in each group are shown. The open circles in the graph indicate the lowest and highest values in each group. TNFbp indicates the soluble human dimeric form of the type I receptor for TNF conjugated to polyethylene glycol. Significance was determined by analysis of variance.
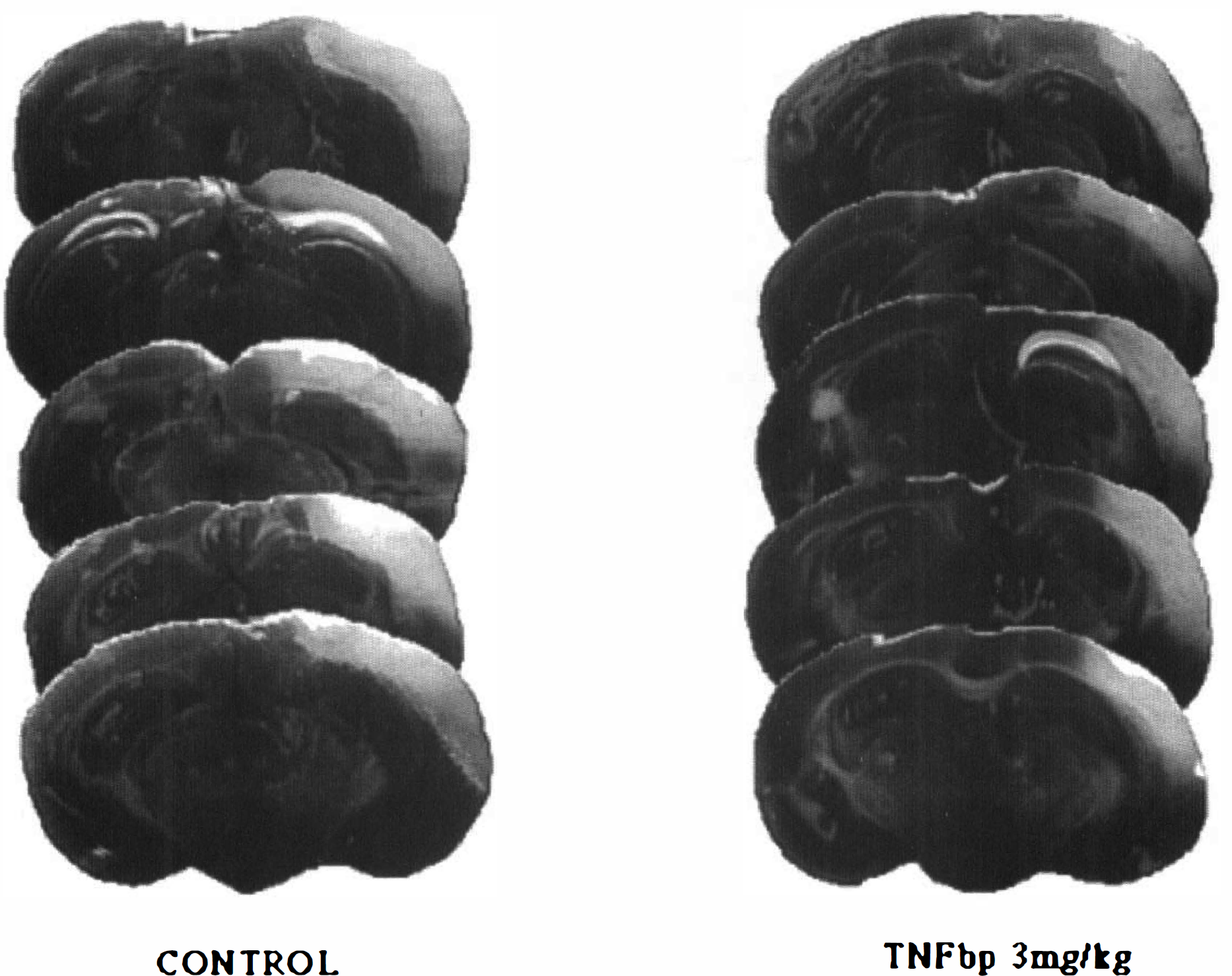
Coronal brain sections from each of five animals in the control group and the group given 3 mg/kg of TNFbp topically, at the level of the dorsal hippocampus, 5.0 mm from the frontal pole, stained by TTC 24 h after MCAO.
DISCUSSION
Evidence from several laboratories indicates that cytokines participate in the process of ischemic brain injury (Rothwell and Relton, 1993; Feuerstein et al., 1994; Garcia et al., 1995; Saito et al., 1996). It has been reported that TNF-α is expressed in ischemic neurons after focal ischemia produced by permanent MCAO (Liu et al., 1994). TNF-α stimulates acute-phase protein production, enhances the permeability of the blood–brain barrier, and induces expression of endothelial adhesion molecules and other inflammatory mediators (Feuerstein et al., 1994). These actions of TNF-α facilitate migration of activated leukocytes into the ischemic brain and might result in secondary damage. However, TNF-α itself is not toxic to neurons, but is instead protective in an in vitro culture system (Barger et al., 1995). The role of TNF-α in ischemia remains to be resolved. Our results support a harmful effect of TNF-α in acute ischemic stroke and indicate that it aggravates outcome. TNFbp used in this study has a high molecular weight (55 kd) and is thought to penetrate the blood–brain barrier poorly. It is reported that the soluble p75 receptor for TNF-α does not cross the blood–brain barrier (Banks et al., 1995). This is the reason that we used the topical and intrathecal application of this drug. Systemic administration of TNFbp has neuroprotective effects; however, they appear to be less potent compared with intrathecal application. Pharmacokinetic studies of TNFbp administered intravenously or subcutaneously to rats indicate that the plasma half-life for this protein is ∼18–36 h (Martin et al., 1995). This long-lasting effect is sufficient to ameliorate the ischemic injury process that usually proceeds for 3–4 h after MCAO in rats (Memezawa et al., 1992). The degree of neuroprotection, about a 25% reduction from the control value, was smaller than that reported previously using the noncompetitive N-methyl-