
Abstract
We examined by immunohistochemistry the expression of ionotropic glutamate receptor subunits (GluRs) in glial cells of the rat dorsal hippocampus 3 to 28 days after transient forebrain ischemia. In general, the expression of GluRs at all time points studied underwent a drastic reduction that was primarily restricted to the CA1 region. In addition to the disappearance of GluRs as a result of neuronal cell death, we observed their expression in reactive glial cells, The time course of expression and the subunits involved were different for astrocytes and microglia. Reactive astrocytes exhibited kainate, GluR5–7, and N-methyl-D-aspartate (NMDA), NR2A/B, receptor subunits, both of which were maximally expressed approximately 4 weeks after ischemia. In contrast, reactive microglia expressed GluR4 and NRI subunits, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA), and NMDA receptor subtypes, respectively, with maximal expression observed between 3 and 7 days after ischemia. These results demonstrate that specific types of GluRs are expressed in reactive glial cells after ischemia and that, overall, their expression levels peak around or after the periods of maximal astrogliosis and microgliosis. Thus, modulation of GluR expression may be one of the molecular components accompanying the gliotic process.
Transient forebrain ischemia results in delayed neuronal death in the CA1 region of the hippocampus 48 to 72 h after injury, but neurons in CA3 are relatively spared (Kirino, 1982; Pulsinelli et al., 1982; Kirino et al., 1984). Cell death is accompanied by morphologically well-defined reactive astrocytosis (Petito et al., 1990) and a microglial reaction (Morioka et al., 1991). Two days after ischemia, astrocytes in the region of necrosis enlarge and increase their expression of glial fibrillary acidic protein (GFAP), a response that persists for longer than a month (Petito et al., 1990). The microglial reaction also involves important morphological changes. It can be detected as early as 20 min after ischemia and is maximal 4 to 6 days after ischemia (Morioka et al., 1991). In addition, after an ischemic insult, astrocytes and microglia upregulate many proteins (for reviews, see Eddleston and Mucke, 1993; Kreutzberg, 1996).
Ischemia-induced neuronal damage involves a disturbance in calcium homeostasis, an excessive release of glutamate, and an increase in oxygen free-radical formation (Choi and Rothman, 1990; Meldrum and Garthwaite, 1990; Schurr and Rigor, 1992; Lipton and Rosenberg, 1994). Glutamate is the most widely distributed excitatory neurotransmitter in the brain and also has been implicated in degenerative processes associated with neurological diseases. Signal transmission at glutamatergic synapses is mediated by ionotropic and metabotropic receptors that have now been molecularly characterized (Hollmann and Heinemann, 1994). According to molecular, pharmacological, and electrophysiological criteria, ionotropic glutamate receptors are classified as α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA), kainate, and N-methyl-
Glutamate receptors also have been found in glial cells in vitro, in brain slices from immature animals, and in the adult brain (for recent reviews, see Blankenfeld et al., 1995; Gallo and Russell, 1995; Steinhäuser and Gallo, 1996). Activation of these receptors on glial cells produces a large variety of responses, including membrane depolarization (Bevan, 1990) and release of neurotransmitters (Martin, 1992) and growth factors (Eddleston and Mucke, 1993), suggesting that they may have an active role in brain signaling and repair. Glial cells of the CNS express ionotropic GluRs of the AMPA/kainate and NMDA types (for recent reviews, see Blankenfeld et al., 1995; Gallo and Russell, 1995; Steinhäuser and Gallo, 1996) however, the pattern of expression of receptor subtypes shows regional variations and does not always correlate with observations obtained in culture. In the immature/juvenile hippocampus, the responses of glial cells to glutamate application are mediated by AMPA/kainate receptors (Jabs et al., 1994; Steinhäuser et al., 1994; Porter and McCarthy, 1995; Seifert and Steinhäuser, 1995) and also by NMDA and metabotropic receptors (Porter and McCarthy, 1995). AMPA/kainate receptors with some Ca2+ permeability are present in a subpopulation of undifferentiated glial cells, presumably GFAP-negative immature astrocytes, of the mouse hippocampal slice (Jabs et al., 1994; Steinhäuser et al., 1994; Seifert and Steinhäuser, 1995) and in most astrocytes in the stratum radiatum of the rat hippocampal slice (Porter and McCarthy, 1995). In addition, about half of the astrocytes in the latter preparation respond to perfusion of NMDA and the pharmacology of the responses (Porter and McCarthy, 1995) resembles that observed in neurons (Collingridge and Watkins, 1994).
These findings raise the possibility that changes in the extracellular glutamate concentration occurring during ischemia (Benveniste et al., 1984, 1989) and the subsequent neuronal cell death occurring in this pathological condition may have a profound effect on the expression of glutamate receptor subunits (GluRs) in glial cells in the resulting damaged area. In the present study, we analyzed the expression of ionotropic GluRs in the CA1 region after transient forebrain ischemia. Several receptor subunits are expressed in glial cells in the damaged area, and their expression peaks around or after the periods of maximal astrogliosis or microgliosis.
MATERIALS AND METHODS
Surgery
Thirty male Wistar rats (250–300 g body weight) were used. Transient forebrain ischemia was induced by the four-vessel occlusion method (Pulsinelli and Brierley, 1979). The rats were anesthetized with pentobarbital (25 mg/kg, i.p. injection), and the vertebral arteries were electrocauterized in the alar foramina of the first cervical vertebra. Immediately after this procedure, an incision was made to isolate the common carotid arteries, and polyethylene cuffs were placed loosely around each artery without interrupting the blood flow. The incision then was closed with surgical clips, and the animals were allowed to recover from the anesthesia overnight. The next day, the rats were lightly reanesthetized with ether, and the surgical clip was removed. When the level of anesthesia was such that the animals did not show spontaneous movement but reacted to pain stimuli, the common carotid arteries were clamped for 30 min. Rats that did not fully loose their righting reflexes or developed seizures following carotid artery occlusion were excluded from the study.
Tissue preparation, immunohistochemistry, and lectin staining
The rats were deeply anesthetized with chloral hydrate and perfused transcardially with fixative at 3, 7, 28, or 90 days after ischemia (n = 5–9 in each group). Tissue was also obtained from control nonoperated rats (n = 4). Fixation solution consisted of 4% paraformaldehyde and 0.1% glutaraldehyde in 0.1 M sodium phosphate buffer, pH 7.4. After fixation, the brains were postfixed overnight at 4°C in 4% paraformaldehyde in the same buffer. The brains were then sectioned at 30 μm on a vibratome and collected in 10 mM sodium phosphate-buffered saline (PBS, pH 7.4). A preliminary evaluation of tissue damage was carried out by counterstaining with toluidine blue several sections from each brain.
We used the following rabbit antibodies to GluR1 (1.5 μg/ml), GluR2/3 (1 μg/ml), GluR4 (1 μg/ml), NR1 (1 μg/ml), and NR2 A/B (4 μg/ml), all from Chemicon (Temecula, CA, U.S.A.). In addition, we used mouse monoclonal antibodies to the GluRs GluR5/6/7 (0.5 μg/ml; PharMingen) and to GFAP (0.5 μg/ml; Boehringer Manheim, Germany). As a negative control, several sections in all experiments were incubated with normal nonimmune rabbit immunoglobulin Gs (IgGs) (4 μg/ml) or mouse IgGs (0.5 μg/ml).
All incubations were carried out with free-floating sections in 24-well culture dishes with gentle shaking. Sections were first treated with 0.1 % H2O2 in PBS at room temperature for 25 min to quench endogenous peroxidase activity and washed three times for 10 min in PBS. Then the sections were preincubated for 1 h in a solution containing 0.5% normal serum of the species in which the secondary antibody was raised, and 0.1% Triton X-100 (the latter omitted in the case of antibodies to NR1 and NRA2/B. Later, the sections were exposed overnight at 4°C to the corresponding primary antibody diluted in the preincubation solution. Antibodies were detected using biotinylated secondary antibodies and the Vectastain ABC-Elite kit (Vector) with 3′,3-diaminobenzidine tetrahydrochloride as a peroxidase substrate. Finally, the sections were mounted onto gelatinized slides, air-dried, dehydrated, and coverslipped.
Lectin staining was carried out by a procedure based on a previously described method (Streit, 1990). Vibratome sections (30 μm thick) were washed in 10 and 20% dimethylsulfoxide (10 min each) diluted in 0.1 M sodium phosphate buffer (pH 6.8) containing CaCl2, MgCl2, MnCl2 (all at 0.1 mM), and 0.1% Triton X-100 (microglia buffer) and then equilibrated for 15 min in the same solution. Subsequently, the sections were treated with 0.6% H2O2 in microglia buffer, rinsed thoroughly in this buffer, and incubated overnight at 4°C with a horseradish peroxidase (HRP) conjugate of Griffonia simplicifolia B4-isolectin (GSA I-B4-HRP, Sigma) diluted at 20 μg/ml in microglia buffer. Lectin binding was developed using 3′,3-diaminobenzidine tetrahydrochloride as a substrate.
RESULTS
The histopathology of the hippocampal CA1 region of the brains used in the immunohistochemical experiments reported here showed that the vast majority of neurons in the pyramidal cell layer were damaged after transient forebrain ischemia (Figs. 1–6). The morphology of astrocytes and microglial cells, stained with a monoclonal GFAP antibody (Fig. 7A–C) and GSA I-B4-HRP lectin (Fig. 8A–C), respectively, was systematically analyzed and found to correspond well with that of reactive astrocytes and microglia.
Histological appearance of the hippocampus from a control rat (
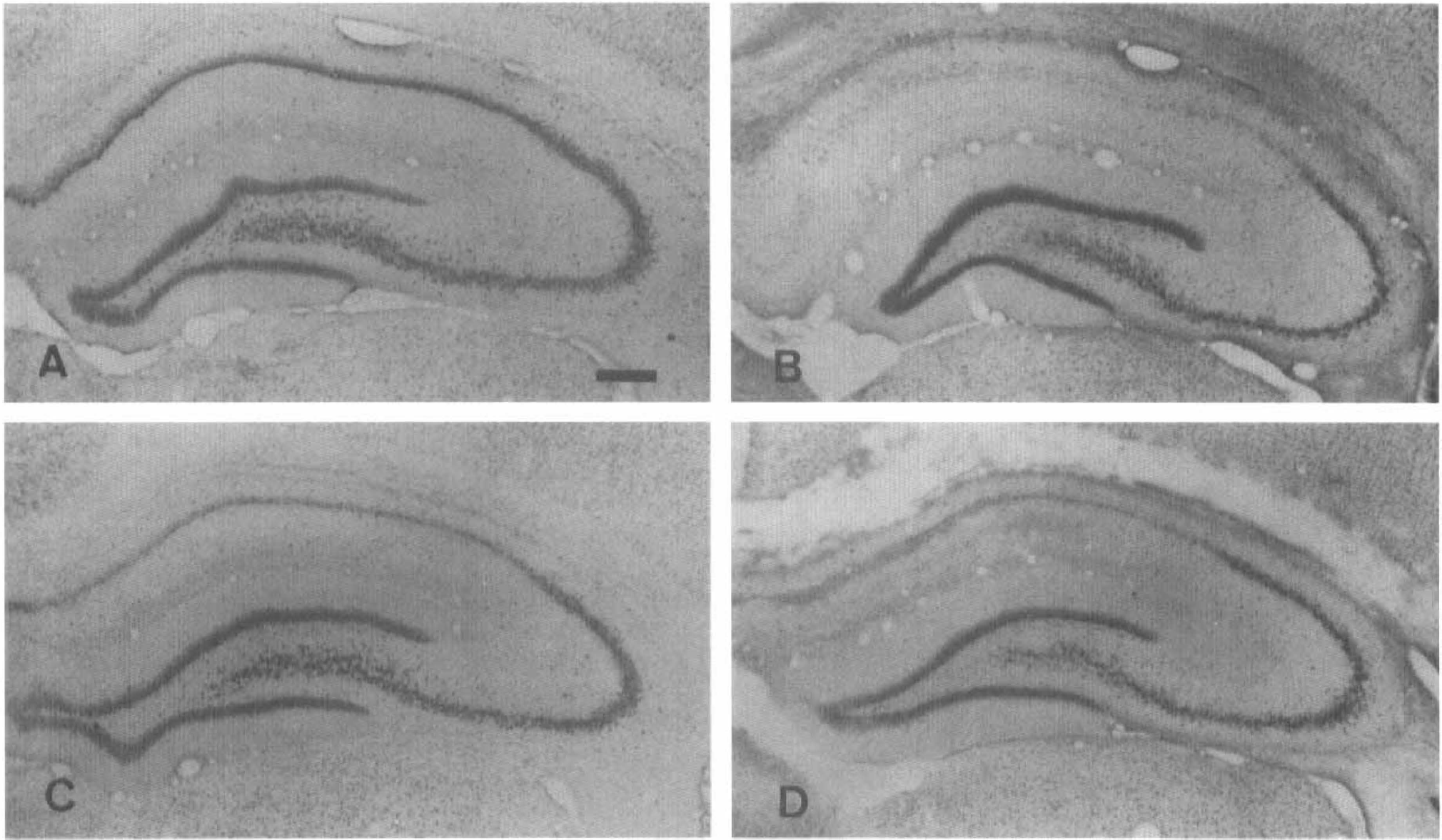
Immunolocalization of the glutamate receptor subunit 1 (GluR1) in control hippocampus (
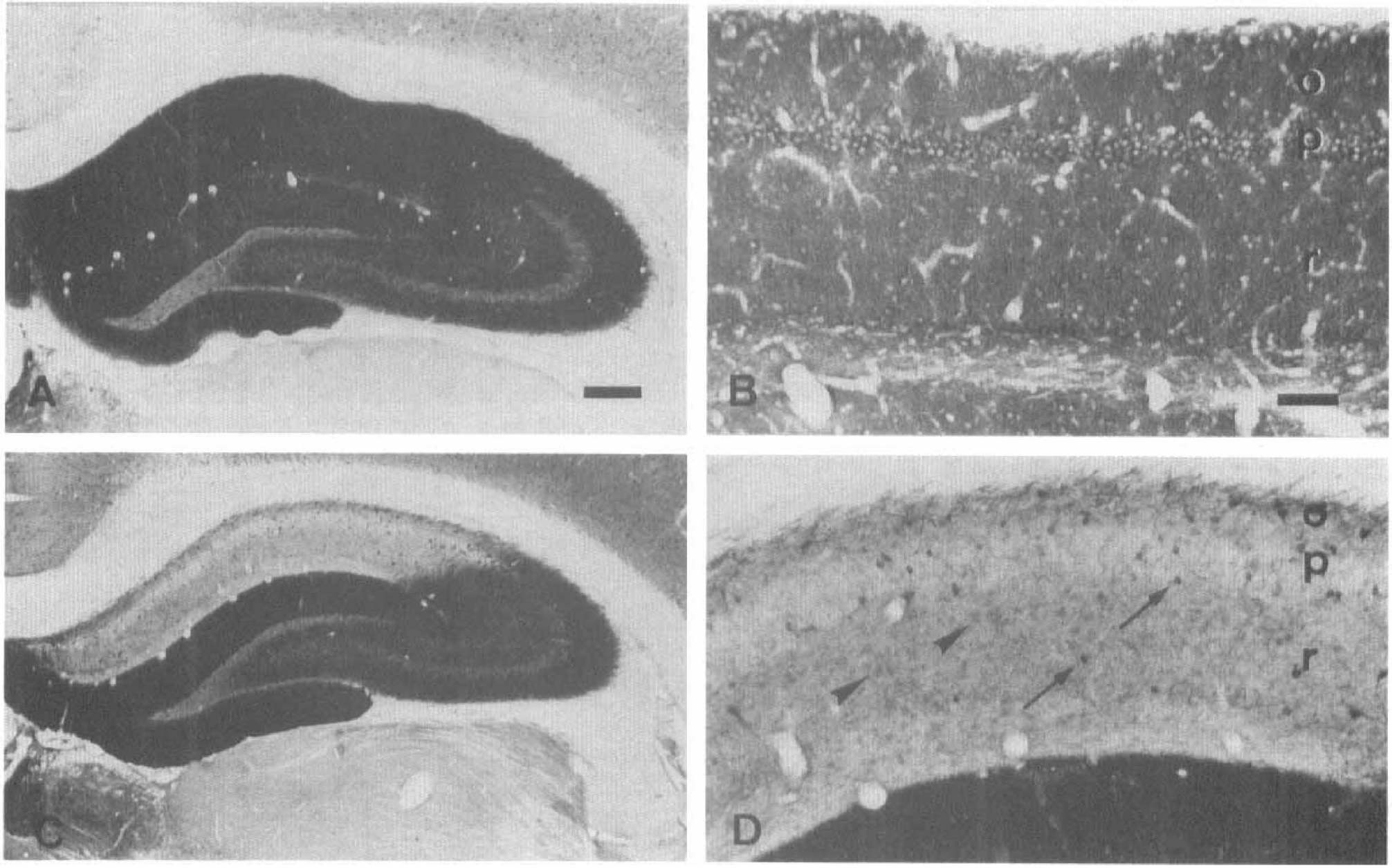
Distribution of the glutamate receptor subunit 4 (GluR4) in control hippocampus (
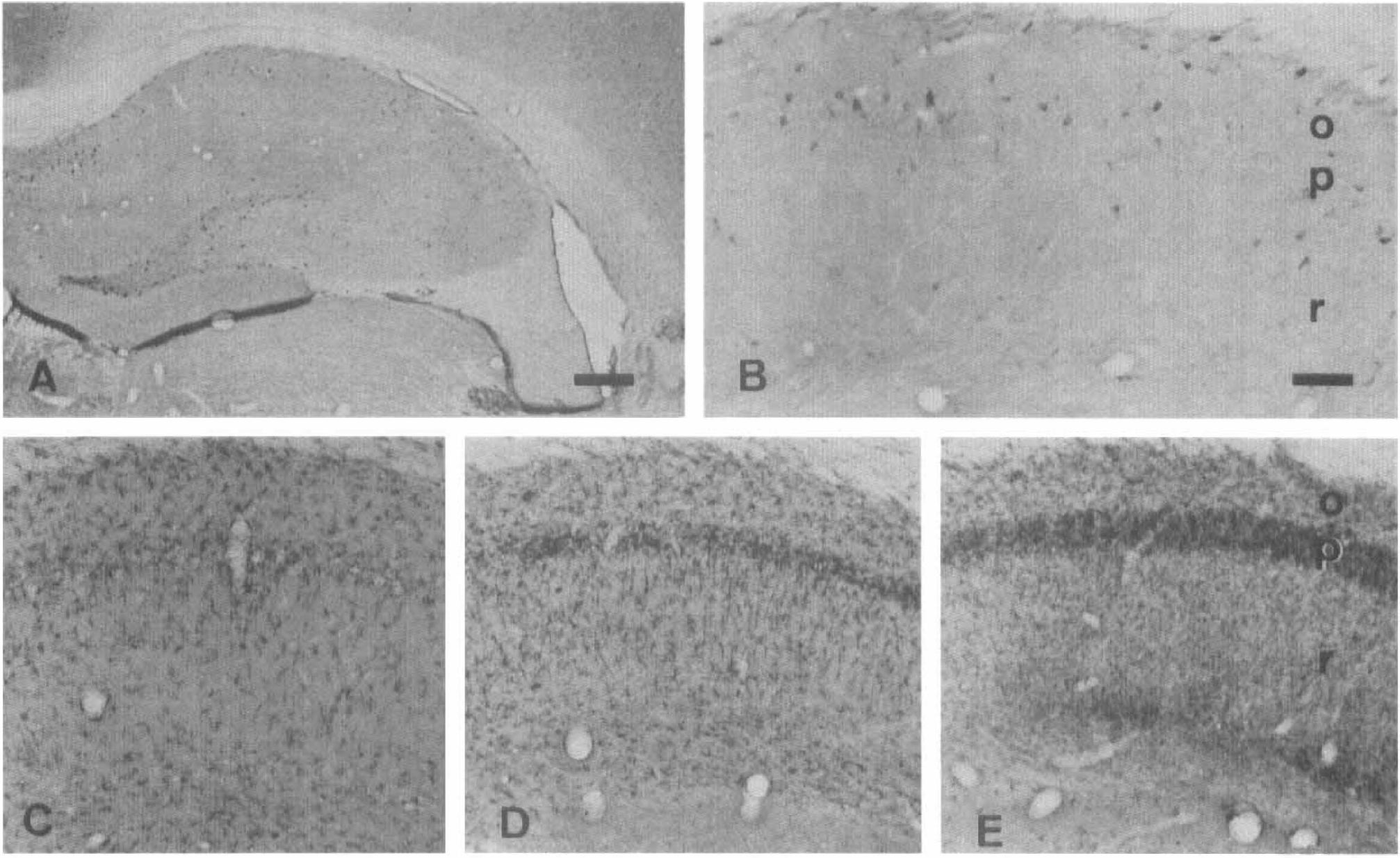
Kainate receptor subunits GluR5–7 in control hippocampus (
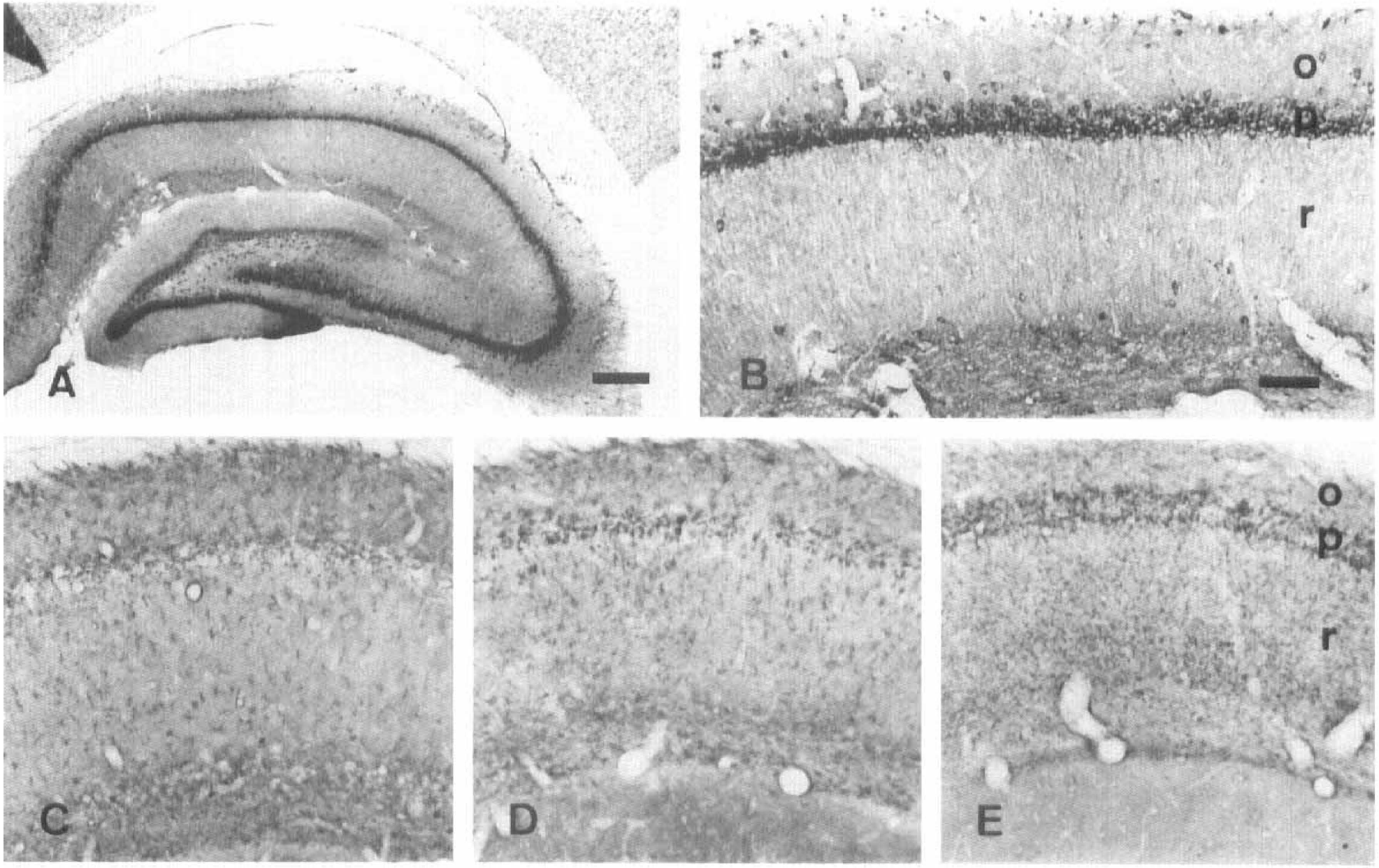
The NR1 NMOA receptor subunit in the hippocampus (
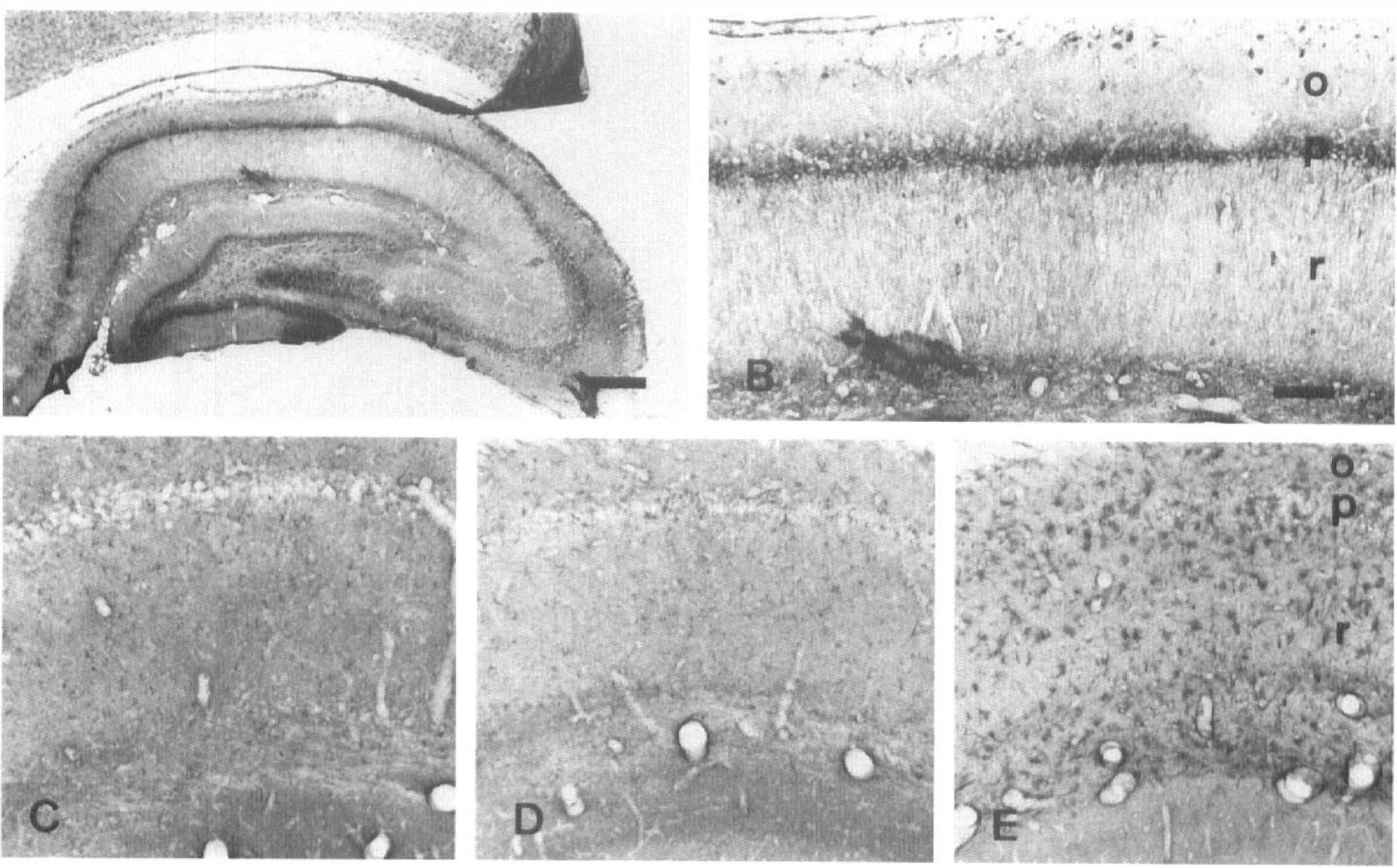
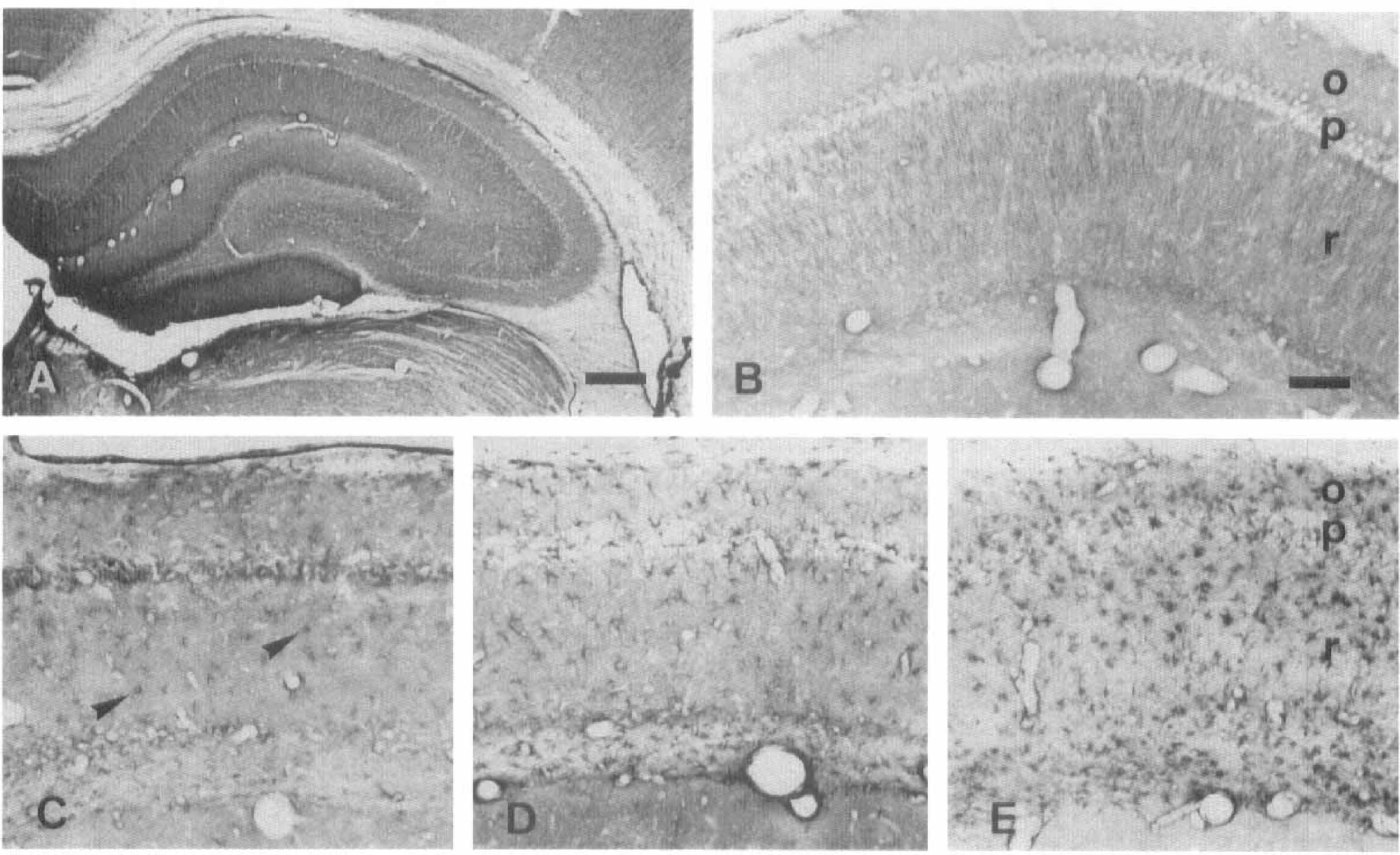
NR2A/B NMDA receptor subunits in the hippocampus (
Our results show that in control hippocampus, the distribution of the various subunits studied (Figures 2–6, two top photographs) coincided with that reported earlier for receptors to AMPA (Petralia and Wenthold, 1992), kainate (Wisden and Seeburg, 1993), and NMDA (Petralia et al., 1994a, b ). Therefore, the results of the present study in control hippocampus will be described briefly and used as a reference for comparison with those observed after transient ischemia.
Antibodies to the GluR1 subunit gave rise to intense immunolabel throughout the neuropil and in pyramidal and nonpyramidal neurons in control tissue (Fig. 2A and B). In contrast, we observed that after ischemia labeling with this antibody was dramatically reduced in the CA1 region, probably as a result of neuronal cell loss caused by the insult, but not in CA3 (Fig. 2C). This reactivity reduction was already maximal at 3 days and largely maintained at 7 and 28 days after ischemia. The most heavily stained elements at all time points examined had the appearance of nonpyramidal neurons and were located in the stratum oriens, pyramidal, and radiatum (Fig. 2D). Interestingly, at 28 days postlesion, but not earlier, we also found lightly reactive cells with a flattened and polygonal morphology (see Fig. 2D) that probably corresponded to hypertrophic astrocytes (see Fig. 7C). The distribution of staining with antibodies to the GluR2/3 subunits was similar to that observed with antibodies to the GluR1 subunit (not shown).
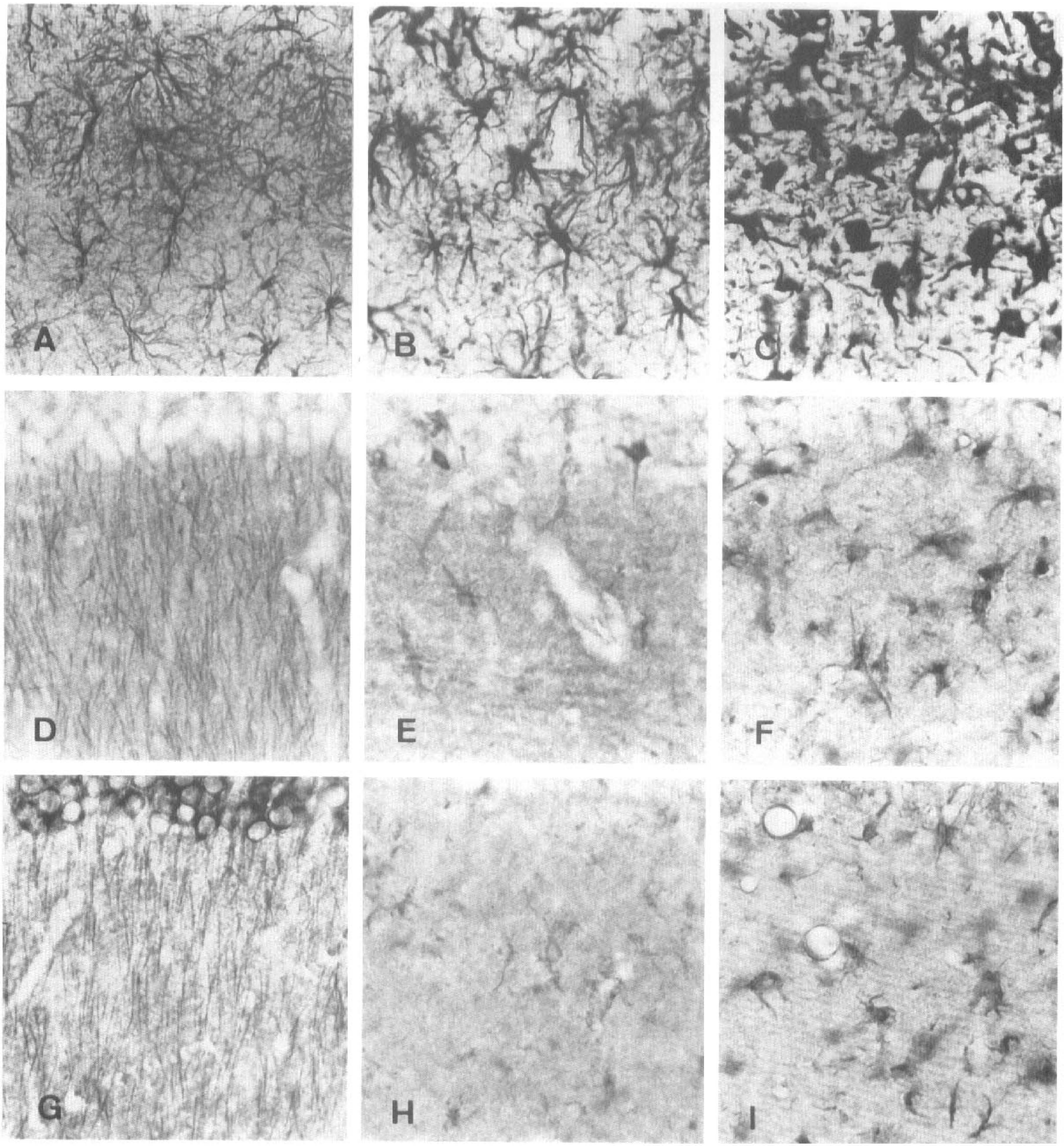
Immunolabeling in the stratum radiatum with antibodies to glial fibrillary acidic protein (GFAP) (
Expression of the GluR4 subunit in the control CA1 region was much lower (Figs. 3A, B, and 8D) than that of the GluR1 and GluR2/3 subunits and appeared to be restricted to neuronal elements. Ischemic damage, however, caused the disappearance of most neuronal staining at all postischemia stages examined, whereas microglial processes were immunoreactive (Figs. 3C–E and 8D–F). The shape and number of these immunolabeled processes varied with the time after injury. At 3 and 7 days postischemia, both bushy and rodlike stained microglial cells were detected (Fig. 8E, F). In the stratum radiatum, the amount of immunolabel observed using GluR4 antibodies peaked at approximately day 7 postischemia and decreased progressively thereafter (Fig. 3C–E). In contrast, labeling with this antibody developed more slowly in microglial cells of the pyramidal cell layer and was still elevated 28 days postischemia (Fig. 3C–E).
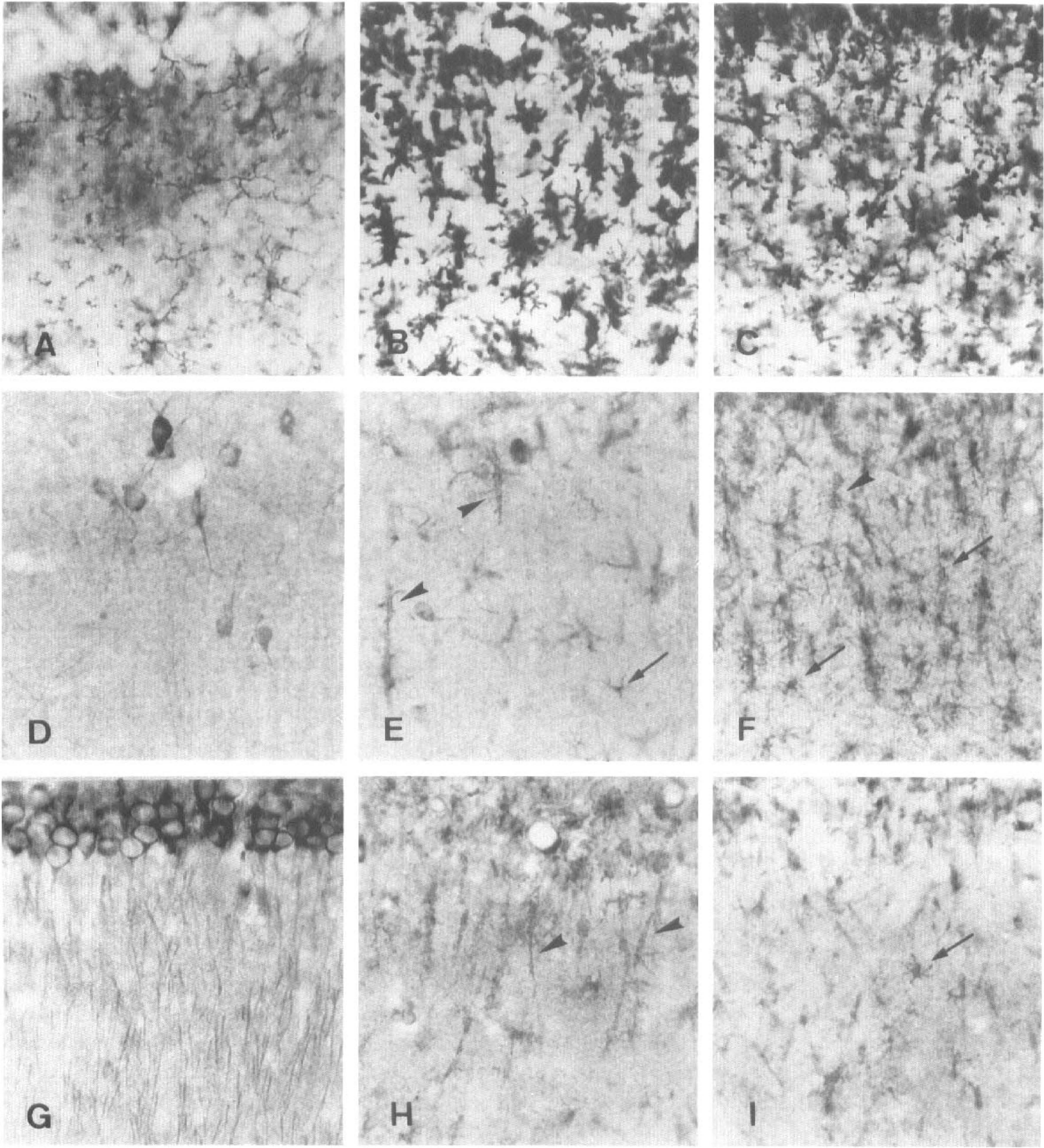
Microglial cells in the stratum radiatum detected with GS I-B4 horseradish peroxidase (HRP) lectin (
Kainate-selective glutamate receptors are made up of subunits GluR5-7 and KA1-2, although the latter two peptides do not form functional receptors by themselves (Hollmann and Heinemann, 1994). Therefore, we focused on examining the distribution of GluR5-7 subunits. To this end, we used an antibody to the GluR5 subunit that crossreacts with the other two members in this subfamily of receptors (Huntley et al., 1993). In controls, this antibody stained cells in the CA1 region and, in particular, stained apical dendrites of pyramidal neurons (Figs. 4A, B, and 7D). This labeling disappeared after transient ischemia, whereas astrocytes in the CA1 region displayed immunostaining as early as 3 days postischemia (Figs. 4C–E; 7E and F). The intensity of the labeling gradually increased in the soma and processes of astrocytes (Fig. 7E and F). It was maximal at approximately 4 weeks (Fig. 7F) and greatly reduced by 3 months postischemia (not shown).
Functional NMDA receptors are heteromeric complexes formed by at least one NR1 subunit together with NR2 subunits (Monyer et al., 1992). In the same study, these authors found that the most abundant NR2 subunits in the hippocampus are NR2A and NR2B. Accordingly, we used antibodies to the NR1 and NR2A/B receptor subunits to investigate their distribution in the hippocampus after transient ischemia. In the normal CA1 field, NR1 (Figs. 5A, B, and 8G) and NR2A/B (Figs. 6A, B, and 7G) were richly expressed in both pyramidal cell bodies and apical dendrites and also were found in neurons in the stratum oriens and radiatum. After forebrain ischemia, the NR1 subunit was observed in cells with a typical microglial morphology (Fig. 8H–I), whereas the NR2A/B subunits were expressed in cells with the characteristic morphological features of astrocytes (Fig. 7H–I); however, the time course of the expression was different. The amount of NR1 immunolabel increased relatively rapidly and reached a maximum between days 3 and 7 postischemia and was largely maintained thereafter (Figs. 5C–E and 8H–I). In contrast, the NR2A/B subunits were maximally expressed several weeks postischemia (Figs. 6C–E and 7H–I).
A summary of the results obtained is presented in Table 1. Distinct GluRs were found to be expressed by different glial cell types following transient forebrain ischemia. In astrocytes, expression of GluRs developed slowly over several weeks and was restricted to the kainate subtype GluR5–7 and the NMDA subtype NR2A/B. In contrast, in microglial cells, the receptor subunits appeared during the first week postlesion and involved the AMPA subunit GluR4 and the NMDA subunit NR1.
Glutamate receptor subunit (GluR) expression in the CA1 region after transient forebrain ischemia

–/+, absence or presence, respectively, of immunolabel using the corresponding antibodies.
DISCUSSION
The present study shows that after transient forebrain ischemia, delayed neuronal death in the CA1 hippocampal region is accompanied by the expression of some ionotropic GluR subunits in glial cells in the damaged area. The expression of subunits is cell-type specific, and the levels of expression are high at the postischemia period in which GFAP immunoreactivity is maximal in reactive astrocytes (Petíto et al., 1990) and in which the microglial reaction is most intense (Morioka et al., 1991).
Ischemic damage induced a severe loss of CA1 pyramidal neurons, comparable to that described earlier (Kirino, 1982; Pulsinelli et al., 1982; Kirino et al., 1984). In contrast, GluR1 immunohistochemistry revealed that nonpyramidal cells were spared in the CA1 region. The appearance and number of GluR1 positive neurons resembled those of GABAergic neurons (Matute and Streit, 1986), suggesting that most GABAergic interneurons in the hippocampal CA1 field of the rat may escape injury caused by transient ischemia, as previously shown in the gerbil (Nitsch et al., 1989; Tortosa and Ferrer, 1993). Indeed, GABAergic neurons have been shown to express high levels of the GluR1 subunit in other areas of the cerebral cortex (Gutiérrez-Igarza et al., 1996). The mechanism by which GABAergic interneurons with GluR1 subunits are protected from dying may be related to the presence in these cells of the Ca2+-binding protein parvalbumin (Tortosa and Ferrer, 1993), which may buffer the excessive Ca2+ influx associated with the sustained activation of receptors containing the GluR1 subunit (Hollmann and Heinemann, 1994).
Although it is well established that hippocampal astrocytes express AMPA/kainate and NMDA receptors in the immature/juvenile brain (Jabs et al., 1994; Steinhäuser et al., 1994; Porter and McCarthy, 1995; Seifert and Steinhäuser, 1995), little is known about the GluRs expressed by glial cells of the adult hippocampus. It has been shown, however, that in other areas of the adult brain, glial cells express a variety of ionotropic GluRs (Matute and Miledi, 1993, 1995; Matute et al., 1994a,b; García-Barcina and Matute, 1995), raising the possibility that this is also the case for the adult hippocampus. The intense immunohistochemical labeling with most antibodies to specific GluR subunits observed throughout the entire extent of all the hippocampal layers in this and other studies (e.g., Petralia and Wenthold, 1992; Petralia et al., 1994a,b) does not allow a clear visualization of stained glial cells, although it suggests that these receptors may also be present in such cells. Interestingly, ischemic damage in the CA1 region was accompanied by the expression of specific GluRs in glial cells. Thus, we observed that GluR5-7 and NR2A/B receptor subunits were present in a few astrocytes during the first week after reperfusion. In contrast, the number of positive astrocytes and the levels of expression of these subunits were significantly increased at 28 days postischemia, a time when GFAP expression is still maximal (Petito et al., 1990). The time course of receptor expression suggests that GluR subunits may be associated with hyperplasic astrocytes. This idea is supported by recent data showing a similar time course in the expression of the β-amyloid precursor protein in the CA1 region after transient ischemia, which is associated with hyperplasic but not with hypertrophic astrocytes (Palacios et al., 1995).
Consistent with the results of the present study, previous evidence indicates that many proteins are upregulated in reactive astrocytes (Eddleston and Mucke, 1993). The mechanisms by which the expression of many of these molecules is upregulated, however, remain largely unknown. Several signaling pathways may account for the resulting expression of GluRs in astrocytes after ischemia. The signaling cascades may be triggered by a transient increase in extracellular glutamate (Benveniste et al., 1984, 1989), the cellular debris generated by neuronal damage, or substances produced by activated microglia (Giulian, 1995). Activation of metabotropic and ionotropic receptors in astrocytes by glutamate can induce profound metabolic and morphological changes in astrocyte physiology (Teichberg, 1991). The time course of the expression of GluR levels is relatively slow and is preceded by the microglial reaction. It is therefore conceivable that GluRs levels are modulated by factors secreted by microglia.
Our results indicate that reactive microglial cells expressed the GluR4 and NR1 subunits and that the levels of expression peak between 3 and 7 days after reperfusion, a time when the microglial reaction is strong following transient forebrain ischemia (Morioka et al., 1991). Although microglial cells express receptors to a variety of neurotransmitters (Kettenmann et al., 1993), the expression of AMPA and NMDA receptors has not been reported in microglial cells (see Steinhäuser and Gallo, 1996). This, together with the fact that the receptors observed in our study are located in cells with morphological features of activated microglia (Morioka et al., 1991), suggest that GluR4 and NR1 subunits are expressed in a subset of proliferating microglia. The physiological consequences of the presence of GluRs in activated microglia might involve the processing and regulation of signals orchestrated by microglia (for review, see Kreutzberg, 1996), which lead to the removal of cellular debris, tissue repair, and neural regeneration.
A critical and still unresolved issue regarding brain repair after injury is whether the induced changes in glial cells are beneficial or detrimental. Because little is known about the functions mediated by GluRs in glial cells, it is difficult at present to assess the contribution of these receptors in reactive glia to the repair process. Interestingly, purines, a family of well-established neurotransmitters and neuromodulators that activate specific receptors in glial cells, can trigger the production of cytokines and trophic factors in astrocytes and in microglia and thus minimize neuronal loss subsequent to brain ischemia and trauma (Neary et al., 1996). It is possible that such a cascade of events is also mediated by GluRs in reactive glia after ischemia. This possibility is supported by the fact that, in addition to the expression of GluRs, ischemia leads to astrocyte-mediated increases in the levels of cytokines and growth factors, such as basic fibroblast growth factor, brain-derived neurotrophic factor, insulin-like growth factor 1, and nerve growth factor (reviewed by Eddleston and Mucke, 1993). The trophic activity provided by these growth factors may be sufficient to protect neurons in the penumbra zone of the ischemic area, and to reverse partially the induced damage in the case of a brief blood supply deficit (e.g., Meyer-Franke et al., 1995; however, see also Koh et al., 1995).
In conclusion, we demonstrated the expression of ionotropic GluRs in glial cells in damaged areas of the hippocampus following ischemia. The expression of these receptors in reactive glial cells may serve to initiate signaling cascades that promote tissue repair.
Footnotes
Acknowledgment:
We thank F. Pérez-Cerdá and J. García-Barcina for reading the manuscript and D. J. Fogarty and J. Kahle for correcting the English version. We also thank one of the anonymous reviewers for valuable suggestions. This study was supported by grants from the Gobierno Vasco (PI94/106) and the Ministerio de Educación y Ciencia (DGCYT, PB94/1377). M.G. held a fellowship from the Gobierno Vasco.