
Abstract
Axonal injury and dysfunction in white matter (WM) are caused by many neurologic diseases including ischemia. We characterized ischemic injury and the role of glutamate-mediated excitotoxicity in a purely myelinated WM tract, the mouse optic nerve (MON). For the first time, excitotoxic WM injury was directly correlated with glutamate release. Oxygen and glucose deprivation (OGD) caused duration-dependent loss of axon function in optic nerves from young adult mice. Protection of axon function required blockade of both α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) and kainate receptors, or removal of extracellular Ca2+. Blockade of N-methyl-D-aspartate receptors did not preserve axon function. Curiously, even extended periods of direct exposure to glutamate or kainate or AMPA failed to induce axon dysfunction. Brief periods of OGD, however, caused glutamate receptor agonist exposure to become toxic, suggesting that ionic disruption enabled excitotoxic injury. Glutamate release, directly measured using quantitative high-performance liquid chromatography, occurred late during a 60-mins period of OGD and was due to reversal of the glutamate transporter. Brief periods of OGD (i.e., 15 mins) did not cause glutamate release and produced minimal injury. These results suggested that toxic glutamate accumulation during OGD followed the initial ionic changes mediating early loss of excitability. The onset of glutamate release was an important threshold event for irreversible ischemic injury. Regional differences appear to exist in the specific glutamate receptors that mediate WM ischemic injury. Therapy for ischemic WM injury must be designed accordingly.
Introduction
Axonal injury and dysfunction are responsible for much of the disability associated with spinal cord injury, stroke, multiple sclerosis, and certain other neurologic diseases that target white matter (WM) (Goldberg and Ransom, 2003). The pathophysiology of WM injury is finally attracting attention, and studies to date indicate that the process is unexpectedly complex (Agrawal and Fehlings, 1997; Follett et al, 2000; McDonald et al, 1998; Ransom et al, 1990; Sanchez-Gomez and Matute, 1999; Stys et al, 1990; Tekkök and Goldberg, 2001; Wrathall et al, 1992). The different cellular elements of WM are separately under attack during insults such as ischemia. At the same time, the challenged elements are interactive during energy deprivation in complex ways that are not well understood. Axons can be injured directly by ionic mechanisms leading to toxic accumulation of intracellular Ca2+ (Fern et al, 1995; Ouardouz et al, 2003; Stys et al, 1990; Underhill and Goldberg, 2006; Wolf et al, 2001), oligodendrocytes, and the myelin they manufacture, can be damaged by glutamate (Alberdi et al, 2002; Li et al, 1999; Matute et al, 1997; McDonald et al, 1998; Rosenberg et al, 1999; Sanchez-Gomez and Matute, 1999) and, undoubtedly, astrocytes and microglia will also be shown to be involved.
There is also a growing sense that the mechanisms of WM injury may vary from one WM area of the brain to another (Tekkök and Ransom, 2005; Tekkök and Goldberg, 1999). This question has not been well explored, but has important implications in terms of the ultimate goal of designing therapeutic strategies. For example, there could be significant regional differences in the glutamate receptors that participate in the injury process (Brand-Schieber and Werner, 2003; Gallo and Russell, 1995). Finally, it is clear that the process of WM injury involves a temporal sequence of events. The discovery that both ionic and excitotoxic mechanisms operate during WM ischemia begs the question of the time course of these events. Do they occur simultaneously (i.e., in parallel) or sequentially (i.e., in series)? Although this has been well studied in gray matter subjected to ischemia, it has not been considered in WM. To address these issues, we characterized ischemia-induced WM injury using the mouse optic nerve (MON), a purely myelinated WM tract. Advantages of this preparation of central nervous system (CNS) WM include that it is extremely stable (i.e., > 12 h) in vitro and that ischemic injury can be quantitatively assessed using electrophysiologic recording techniques. Both ionic and excitotoxic injury mechanisms operate in this structure, and the pharmacology of the glutamate receptors was carefully analyzed. In addition, we investigated whether these mechanisms have a similar time course, or whether one mechanism acts to trigger the other. A preliminary account of this work has been published elsewhere (Tekkök and Ransom, 2004).
Materials and methods
Preparation of Optic Nerves and Oxygen Glucose Deprivation
As described previously (Tekkok et al, 2003), MONs were obtained from anaesthetized adult male Swiss Webster mice (age 4 to 8 weeks). Optic nerves were gently freed from their dural sheaths and placed in an interface perfusion chamber (Medical Systems Corp, Greenvale, NY, USA).
Nerves were superfused with artificial cerebrospinal fluid (aCSF) containing (in mmol/L): 124 NaCl, 3.0 KCl, 2.0 CaCl2, 2.0 MgCl2, 1.25 NaH2PO4, 23 NaHCO3, and 10 glucose, bubbled with 95% N2/5% CO2 (O2 was supplied entirely by the gas mixture; see below). All experiments were performed at 37°C. The chamber was continuously aerated by a humidified gas mixture of 95% O2/5% CO2. Suction electrodes back-filled with aCSF were used for stimulation and recording. One electrode was attached to the rostral end of the nerve for stimulation (Isostim 520; WPI, Sarasota, FL, USA). A second suction electrode was attached to the distal end of the nerve to record the compound action potential (CAP). The recording electrode was connected to an Axoclamp2A amplifier, and the signal was amplified 50 times, filtered at 30 kHz, and acquired at 20 kHz. Stimulus pulse (30 μs duration) strength was adjusted to evoke the maximum CAP possible and then increased another 25% (i.e., supramaximal stimulation). MONs were allowed to equilibrate for at least 15 mins in the chamber in normal aCSF (containing 2 mmol/L Ca2+) before experiments. During experiments, the supramaximal CAP was elicited every 30 secs.
Oxygen and glucose deprivation (OGD) was induced by switching to glucose-free aCSF (replaced with equimolar sucrose to maintain osmolarity) and a gas mixture containing 95% N2/5% CO2. To ensure that no oxygen was inadvertently delivered by the superfusate, aCSF was always bubbled with 95% N2/5% CO2. OGD was applied for 15, 30, 45, 60, and 90 mins to determine the effects of OGD on axonal conduction and recovery. After OGD, control aCSF and O2 containing gas were restored and CAPs were recorded for up to 6 h.
A Ca2+-free aCSF was used in some experiments and contained 200 μmol/L EGTA and 4 mmol/L Mg2+ (to maintain constant divalent cation concentration). Control experiments indicated that CAPs remained essentially unchanged in this solution (with normal glucose and oxygen) for at least 90 mins.
To explore the role of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA)/kainate receptors in OGD-induced axon function loss in MONs, the antagonists 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(f)-quinoxaline-2,3-dione (NBQX, 30 μmol/L; Tocris, Ellisville, MO, USA; dissolved in dimethyl sulfoxide (DMSO) as 30 mmol/L stock, care being taken to prevent light exposure) or 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 μmol/L; Sigma, St Louis, MO, USA; dissolved in DMSO as 10 mmol/L stock, care being taken to prevent light exposure) were used. To characterize and specify the subclass of AMPA/kainate receptors involved in OGD-induced injury, selective antagonists were used: the noncompetitive AMPA receptor antagonist GYKI 52466 hydrochloride (30 μmol/L; Sigma, St Louis, MO; dissolved in methanol as 30 mmol/L stock or Tocris, Ellisville, MO, USA; dissolved in 1 eq HCl as 15 mmol/L stock), the Ca2+-permeable AMPA receptor antagonist napthylamine spermine (NAS, 10 μmol/L; gift from Daicel Inc., Fort Lee, NJ, USA; dissolved as 10 mmol/L stock in dd H2O), and the very selective, low-affinity kainate receptor antagonist NS-102 (10 to 20 μmol/L; Sigma, St Louis, MO, USA; dissolved in DMSO as 10 mmol/L stock).
Three types of N-methyl-D-aspartate (NMDA) receptor blockers were used: MK-801 (10 μmol/L; ion channel blocker, dissolved in dd H2O as 10 mmol/L stock), 7-chlorokynurenic acid (7-CKA, 50 μmol/L; glycine binding site blocker, dissolved in DMSO as 50 mmol/L stock), or DL-2-Amino-7-phosphonoheptanoic acid (DL-AP7) (100 μmol/L; NMDA binding site blocker, dissolved in dd H2O as 100 mmol/L stock). All glutamate receptor blockers were applied starting 30 mins before, during, and 30 mins after OGD.
To activate AMPA/kainate receptors, the following agonists were used: 1 mmol/L glutamate (Sigma, St Louis, MO, USA; added to aCSF), or 500 μmol/L kainate (Sigma, St Louis, MO, USA; added to aCSF fresh daily) or 100 μmol/L (S)-AMPA (Tocris, Ellisville, MO, USA; dissolved as 10 mmol/L stock in dd H2O), with or without 30 μmol/L cyclothiazide (CTZ; Tocris, Ellesville, MO, USA; dissolved as 30 mmol/L stock in DMSO) to remove AMPA receptor desensitization.
The role of Na+-dependent glutamate transport as a potential source of glutamate release was tested using the transport blocker DL-threo-beta-benzyloxyaspartic acid (DL-TBOA) (200 μmol/L; Tocris Ellisville, MO, USA; dissolved as 100 mmol/L stock in 100 mmol/L NaOH) or PDC (L-trans-pyrrolidine-2,4-dicarboxylic acid, 1 mmol/L; Tocris, Ellisville, MO, USA; dissolved as 100 mmol/L stock in 1 N NaOH).
Glutamate Measurements
Glutamate release from MON into the superfusate under normoxic and ischemic conditions was measured using high-performance liquid chromatography (HPLC) (Ye et al, 2003). Briefly, amino acids were precolumn derivatized with o-phthalialdehyde (OPA, Sigma, St Louis, MO, USA), separated and measured using standard techniques. Samples of extracellular perfusion fluid were collected continuously such that every vial contained 2 mins of superfusate, and glutamate content was measured in every other vial (i.e., every 4 mins). Collected samples were centrifuged at 16,000 g for 3 min and supernatants were transferred to HPLC analysis. Glutamate measurements, normalized to baseline glutamate release (i.e., the average for 15 mins under control conditions immediately before OGD or TBOA treatment), were made from MONs treated identically to those studied electrophysiologically and results were plotted against time. The rate and release pattern of glutamate was monitored for 30 mins before OGD (15 or 60 mins) and was continued for at least 30 mins after the end of OGD.
Data Analysis
Optic nerve function was monitored quantitatively as the area under the supramaximal CAP. Original data were normalized by setting the mean of initial baseline values (measured over 15 mins) to a value of 1.0. Results from several nerves were pooled, averaged, and plotted against time. All data are presented as mean ± standard errors (± s.e.m.). In time course plots, s.e.m.s are only shown every 3 mins for the sake of clarity. Significance was determined by Student's t-test, one-way or two-way analysis of variance (ANOVA) followed by post-Tukey test (GraphPad Prism 4).
Results
Oxygen and Glucose Deprivation Caused Irreversible Axonal Injury
Prior studies on the mechanisms of WM injury in optic nerve have used anoxia as the insult (Stys et al, 1992). Ischemia (i.e., OGD) is more clinically relevant and was used for this reason. The effects of different durations of OGD on optic nerve excitability and integrity were determined. Under control conditions, axon function (quantified as the area under the CAP) remained stable over many hours in MONs at 37°C (see e.g., Figure 6). After 1 h of stable control CAPs, varying durations of OGD were imposed. OGD caused an initial transient increase in the CAP area (average increase = 14.2% ± 2.9% after 3.9 ± 0.5 mins, n = 6) primarily because of an increase in the second and third peaks and slight prolongation of the CAP duration (data not shown). After this initial increase, the CAP area fell steeply during OGD to undetectable levels in an average of 26.1 ± 2.6 mins (Figure 1A). During the briefest period of OGD, that is, 15 mins, the CAP area was never entirely lost. The extent of irreversible injury detected upon return to control levels of oxygen and glucose varied in strict relationship to the duration of OGD (Figure 1). For example, after 15 or 60 mins of OGD, the CAP area recovered to 88.0% ± 3.5% (n = 6, open squares) or 20.8% ± 3.4% (n = 12, open diamonds) of control, respectively (Figures 1A and 1B). It should be noted that after OGD, the CAP area recovered to a stable level that did not change significantly over the next 6 h. An exception to this was seen with 90 mins of OGD. The CAPs showed a transient recovery within the first 5 to 10 mins and then irreversibly vanished in three of the five MONs. Figure 1A shows the stable recovery seen in two of the MONs after 90 mins OGD.
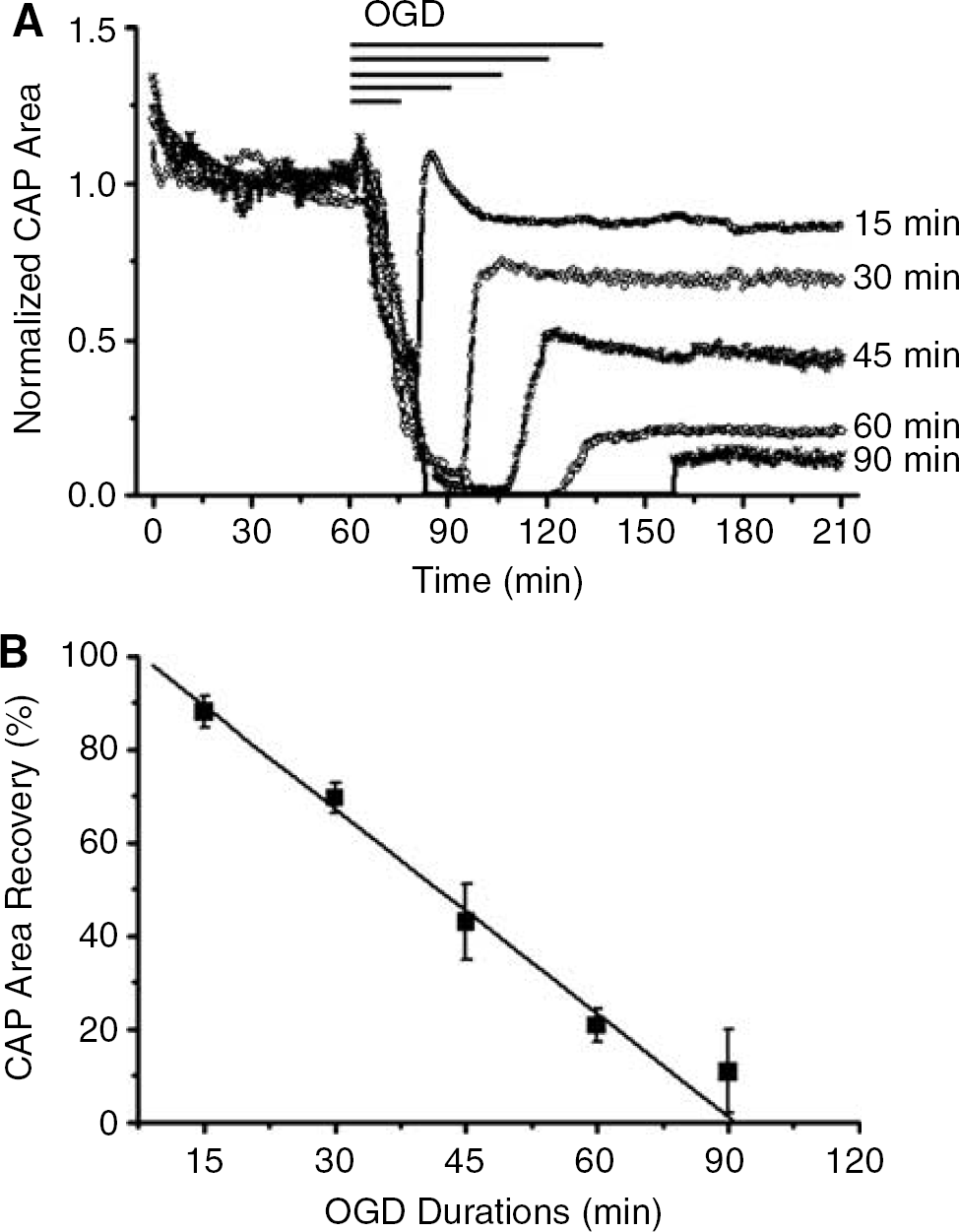
Loss of WM function and degree of recovery depend on the duration of OGD. (
Oxygen and Glucose Deprivation-Induced White Matter Injury was Ca2+-Dependent
In several models of WM injury, including anoxic optic nerve injury (Stys, 2004), irreversible loss of excitability and structural evidence of axon damage strongly depend on the presence of extracellular Ca2+ during the insult period (Alberdi et al, 2002; Stys et al, 1990; Tekkök and Goldberg, 2001; Wolf et al, 2001). Experiments were performed to test if the damaging effects of OGD in MON shared a similar critical dependency on extracellular Ca2+. Recovery of the CAP area was dramatically improved when OGD (60 mins) was applied in Ca2+-free aCSF (containing 200 μmol/L EGTA and 4 mmol/L Mg2+) (Figure 2). The application of Ca2+-free aCSF had no effect on the baseline CAP responses, even for periods up to 90 mins (data not shown). Curiously, the CAP area declined more rapidly during OGD in Ca2+-free conditions; time to CAP failure in OGD was 17.0 ± 3.9 mins (n = 6) in Ca2+-free solution compared with 26.1 ± 2.6 mins (n = 12) in control solution.
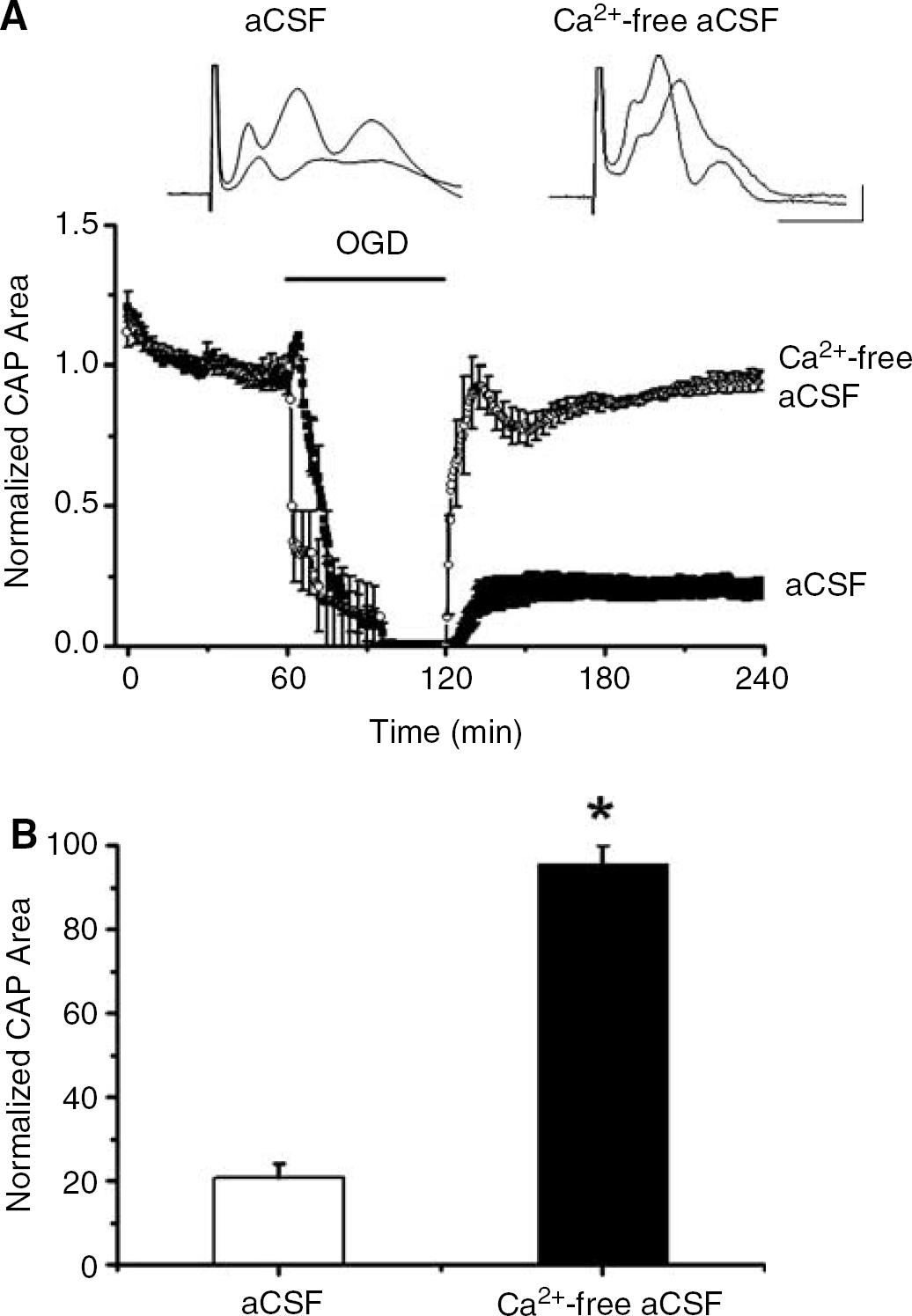
OGD-induced WM injury depends on extracellular Ca2 +. (
α-Amino-3-Hydroxy-5-Methylisoxazole-4-Propionic Acid/Kainate Receptor Antagonists Reduced White Matter Injury caused by Oxygen and Glucose Deprivation
Evidence in other WM tracts indicates that overactivation of AMPA/kainate receptors plays a role in ischemic WM injury (Agrawal and Fehlings, 1997; Li and Stys, 2000; Matute, 1998; McDonald et al, 1998; Tekkök and Goldberg, 2001; Wrathall et al, 1992), but this has not been investigated in MONs. To assess the contribution of AMPA/kainate receptor activation on WM injury, we recorded the CAPs in MONs pretreated with NBQX or CNQX, broad-spectrum AMPA/kainate receptor blockers. Blockade of AMPA/kainate receptors with 30 μmol/L NBQX (Figure 3A) or 10 μmol/L CNQX (Figure 4D) led to partial preservation of the CAP area during OGD (26.6% ± 13.9% with NBQX and 19.3% ± 10.2% with CNQX) followed by improved CAP recovery compared with outcome in normal aCSF (NBQX recovery = 71.9% ± 8.5%, n = 8, P < 0.001; CNQX recovery = 62.1% ± 1.5%, n = 3, P < 0.001).
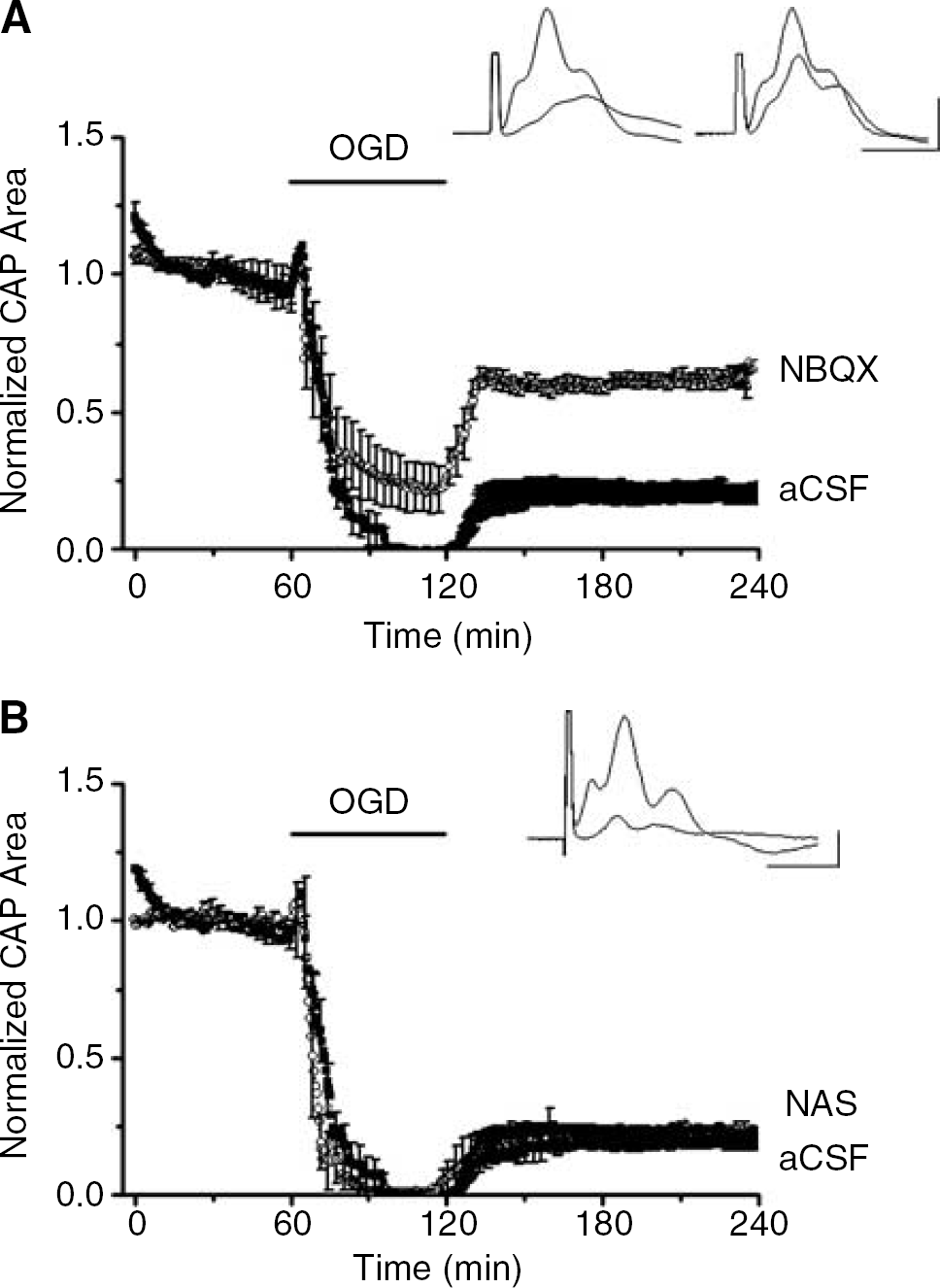
Broad-spectrum AMPA/kainate blockade, but not selective blockade of Ca2+-permeable AMPA receptors, decreased OGD-induced WM injury. (
We next characterized the subclass of glutamate receptors that mediated the OGD-induced injury in MONs. As OGD-induced WM injury depended on extracellular Ca2+, the Ca2+-permeable AMPA receptor was a prime candidate. MONs were pretreated with a stable synthetic polyamine, 1-napthyl acetyl spermine (NAS), to selectively block Ca2+-permeable AMPA receptors (Figure 3B). The behavior of the CAP area during and after OGD, however, was not significantly affected by NAS. Considering that activation of all AMPA receptors may contribute to OGD-induced loss of WM function, another group of MONs were pretreated with 30 μmol/L GYKI 52466, a general AMPA receptor blocker. This general blockade of AMPA receptors also failed to significantly improve recovery of the CAP area (26.5% ± 4.4% of control, n = 10, P = 0.310, Figure 4A).
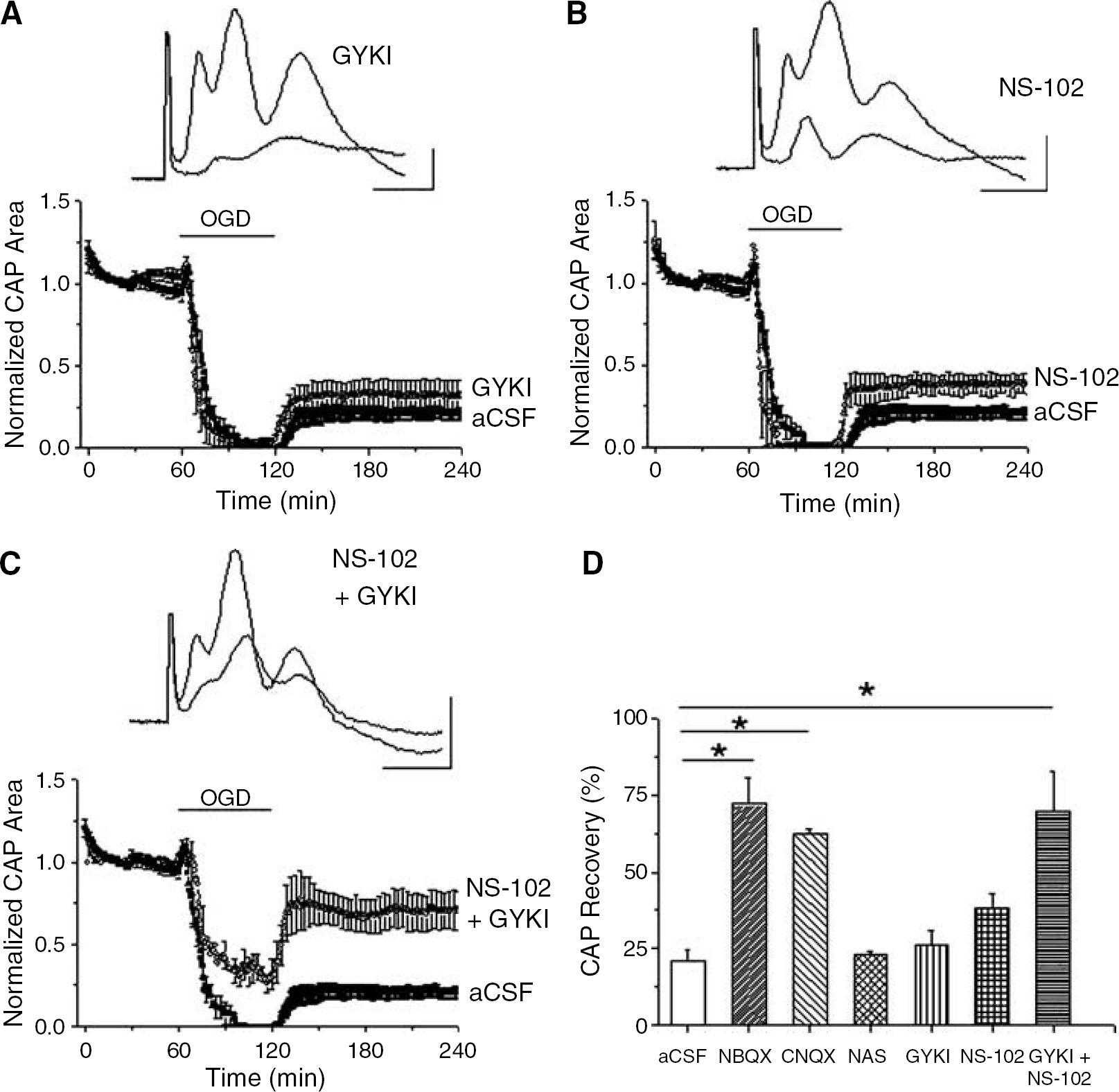
Blockade of both AMPA and kainate receptors was necessary to reduce OGD-induced WM injury. MONs were superfused with (
To explore the involvement of the kainate subtype of glutamate receptors, MONs were exposed to OGD in aCSF containing 10 (n = 4) or 20 μmol/L (n = 2) NS-102, a selective kainate receptor blocker. The CAP area fell more rapidly during OGD in the presence of NS-102 compared with standard OGD (12.0 ± 3.8 mins compared with 26.1 ± 2.6 mins (P = 0.026). There was a small but significantly better recovery of the CAP area to 37.9% ± 6.2% (n = 6, P = 0.045, Figure 4B).
Although blockade of AMPA/kainate receptors provided remarkable protection of axon function, selective blockade of AMPA, or Ca2+-permeable AMPA receptors or kainate receptors failed to replicate the same level of protection from OGD-induced injury. This suggested that subgroups of receptors might interact in producing WM injury. To test this hypothesis, selective antagonists were applied together during OGD. MONs were superfused with a mixture of 30 μmol/L GYKI 52466 and 10 μmol/L NS-102 to block AMPA and kainate receptors (Figure 4C). Indeed, this combination produced comparable protection from OGD-induced injury with that seen with NBQX or CNQX (Figures 4C and 4D). The CAP area fell gradually to only 31.9% ± 8.7% of control at the end of OGD and the CAP area recovered to 69.6% ± 13% of control (n = 6, P < 0.001). These results indicated that activation of either the AMPA or the kainate receptors was sufficient to mediate OGD-induced WM injury in MONs.
The N-Methyl-D-Aspartate Class of Glutamate Receptors did not Protect Against Oxygen and Glucose Deprivation-Induced White Matter Injury
White matter (WM) is generally thought of as devoid of synapses and, in the absence of neuron cell bodies and dendrites, also devoid of NMDA receptors (Gallo and Russell, 1995; Tekkök and Goldberg, 2001). Recently, however, small numbers of NMDA receptors were shown to be located on developing oligodendrocyte processes in rodents and to be activated under pathologic conditions (Micu et al, 2006; Salter and Fern, 2005). We therefore investigated the role of NMDA receptors during OGD-induced WM injury in adult MONs using NMDA receptor antagonists (Figure 5). Three different NMDAR antagonists were used: a pore blocking agent, MK-801 (10 μmol/L (n = 15) or 20 μmol/L (n = 2)), a glycine binding site blocker, 7-CKA (50 μmol/L, n = 5) or an NMDA binding site blocker, DL-APV (100 μmol/L, n = 5). These NMDA receptor blocking agents did not cause any baseline effects on CAP area. Exposing MONs to 60 mins OGD in the presence of 10 μmol/L MK-801 (Figure 5A) caused a similar initial increase in CAP area (15.2% ± 5.8% in 6.5 mins, n = 15) followed by loss of function in 22.4 ± 1.9 mins. The CAP area recovered to 20.5% ± 2.9% of control, similar to recovery after 60 mins OGD under standard conditions (20.8% ± 3.4%, n = 12, P = 0.948). Likewise, neither 7-CKA (17.9% ± 4.5%, n = 5, P = 0.623) nor DL-APV (28.2% ± 8.2%, n = 5, P = 0.357) improved CAP recovery after OGD. These results indicated that NMDA receptors were not significantly involved in OGD-induced injury in adult WM.
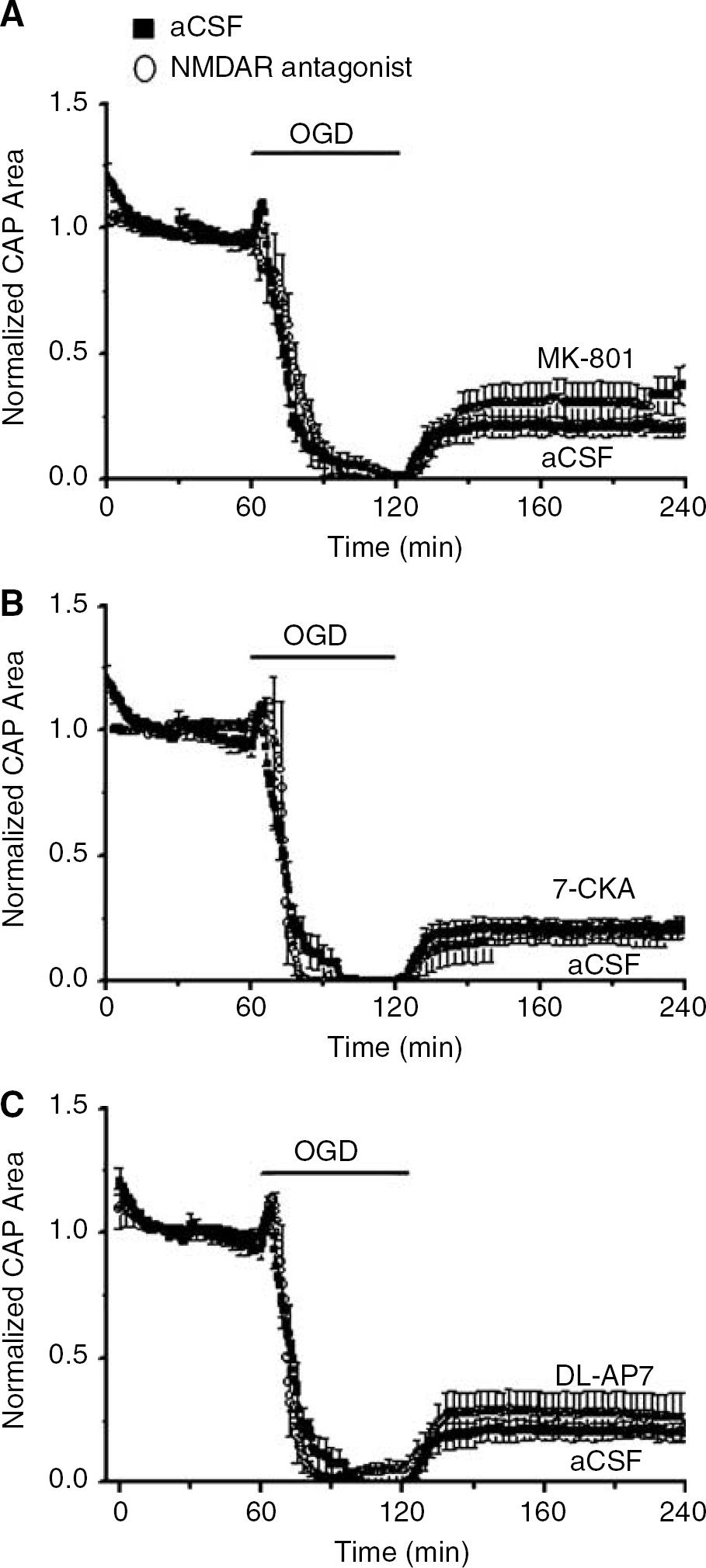
NMDA receptors did not reduce OGD-induced WM injury. Three types of NMDA blocker were used: Blockade of (
Activation of α-Amino-3-Hydroxy-5-Methylisoxazole-4-Propionic Acid/Kainate Receptors was not Sufficient to Cause White Matter Injury
α-Amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) and kainate receptor blockade reduced OGD-induced WM injury in MONs begging the question of whether injury can be induced by directly exposing MONs to glutamate or related agonists. Under control conditions, the CAP area remained stable for at least 7 h in aCSF at 37C (Figure 6, n = 4). Other groups of nerves were exposed to various agonists for 7 h (after 1 h of baseline CAP recording). MONs superfused with 1 mmol/L glutamate (Figure 6) showed a small but significant CAP area increase after 2 h, but not loss of CAP, even when possible AMPA receptor desensitization was blocked with 30 μmol/L CTZ (David et al, 1996). Application of glutamate + CTZ did block the transient increase in the CAP area observed with glutamate application alone (Figure 6). As astrocytes take up glutamate very effectively (Anderson and Swanson, 2000), it was possible that glutamate concentrations within the MON were at low levels. As a safeguard against this possibility, the non-transported agonists, kainate (500 μmol/L) or AMPA (500 μmol/L), were applied (Figure 6). AMPA was without effect, with or without CTZ, and kainate produced a small decrease in the CAP area but this was only seen after 5 h of application. Simply activating AMPA or kainate receptors failed to replicate OGD-induced WM injury in MONs.
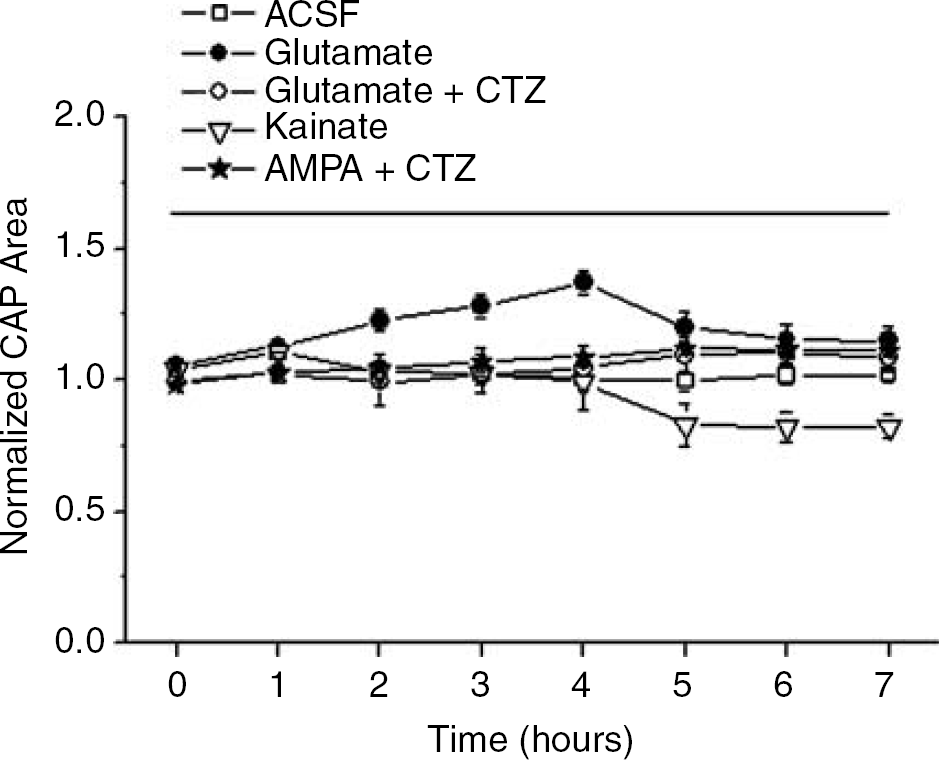
Activation of AMPA or kainate receptors was not sufficient to replicate OGD-induced loss of function and/or injury in WM. CAPs were recorded in MONs in normal aCSF (open squares) or in aCSF containing 1 mmol/L glutamate (open circles), or 1 mmol/L glutamate and 30 μmol/L CTZ (closed circles), or 500 μmol/L kainate (open triangles), or 100 μmol/L AMPA and 30 μmol/L CTZ (stars) for 7 h. Even prolonged applications of glutamate agonists did not depress CAP area.
Application of Glutamate Agonists Damaged White Matter in the Presence of Oxygen and Glucose Deprivation
Although direct application of glutamate or its agonists had little or no effect on WM integrity, activation of their cognate receptors was clearly involved in OGD-mediated WM injury (Figures 3 and 4). How might these apparently discordant observations be reconciled? One possibility could be that OGD leads to metabolic changes that predispose to AMPA/kainate receptor-mediated injury. This idea was tested by determining if a brief period of OGD (i.e., 15 mins) would be more damaging in the presence of glutamate agonists (which by themselves had no toxic effects). Indeed, in the presence of kainate (500 μmol/L), 15 mins OGD caused complete and rapid loss of the CAPs in 8.5 ± 1.7 mins and the CAP recovery was slower and reduced (46.9% ± 6.8%, n = 5) compared with the CAP area recovery observed after 15 mins of OGD alone (88% ± 3.5%, n = 9; P < 0.001, one-way ANOVA, post-Tukey test; Figure 7). The endogenous transmitter that mediated OGD-induced injury was probably glutamate. MONs exposed to 15 mins OGD in 1 mmol/L glutamate exhibited more rapid loss of the CAP (9 ± 3.6 mins) than with OGD alone (26.1 ± 2.6 mins, P < 0.001), but there was no significant difference with regard to the CAP area recovery (Figures 7B and D). As glutamate may cause rapid desensitization of AMPA receptors, a set of experiments was performed with 30 μmol/L CTZ to remove desensitization (Figure 7C). Under these conditions, the CAP area recovery was decreased compared with OGD alone (65.2% ± 5.7% versus 88% ± 3.5%, P < 0.001, one-way ANOVA, post-Tukey test). Experiments with CTZ (30 μmol/L) alone were performed with the presumption that this might enhance the ‘toxicity’ of endogenously released glutamate. In the presence of CTZ, OGD was associated with accelerated loss of the CAPs but no significant reduction in recovery (Figure 7D, 70.7% ± 5.9%). The CTZ experiments indicated that AMPA receptor desensitization played a small but measurable role in limiting the injury that followed exogenous glutamate application during OGD.
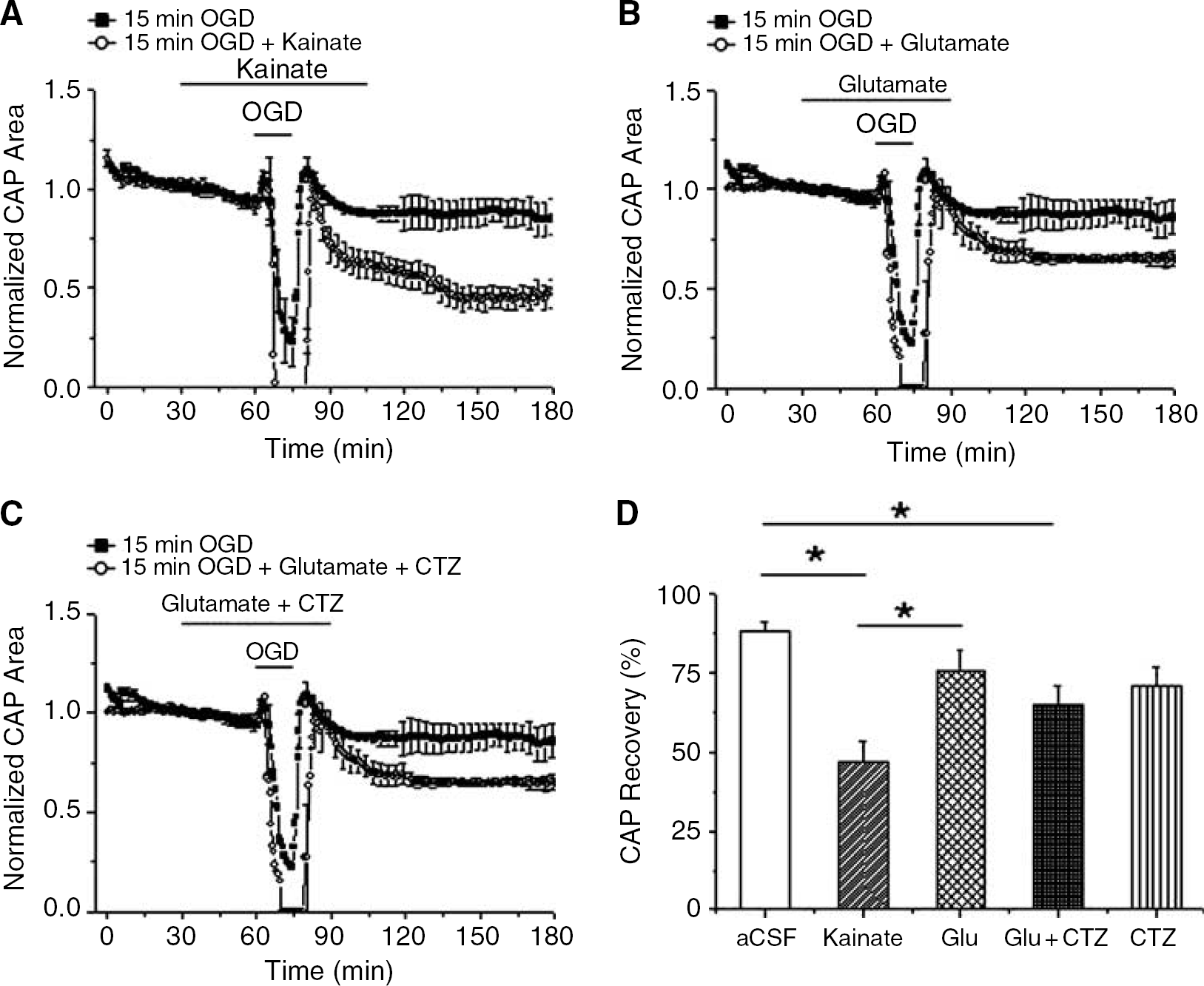
Concomitant activation of AMPA/kainate receptors during brief energy deprivation replicated OGD-induced loss of WM function and induction of injury. MONs were exposed to 15 mins OGD in normal aCSF (closed squares in
Oxygen and Glucose Deprivation in Mouse Optic Nerve Caused Duration-Dependent Glutamate Release Mediated by Reverse Na+-Dependent Glutamate Transport
If glutamate is released during OGD in MON, a plausible mechanism of release is the reversal of Na+-dependent glutamate transport (Anderson and Swanson, 2000). Astrocytes express the greatest density of these transporters and OGD can cause a gradual dissipation of the transmembrane Na+ gradient in astrocytes (Rose and Ransom, 1996) setting up conditions for reverse exchange and glutamate release (Longuemare et al, 1999). In MONs, DL-TBOA (200 μmol/L), a potent competitive Na+-dependent glutamate transporter blocker, significantly improved the CAP area recovery after 60 mins of OGD (Figure 8, 50.5% ± 4.1%, n = 9, P < 0.001). Similar protection was observed with 1 mmol/L PDC, a transportable antagonist that inhibits glutamate release at the cytoplasmic surface (data not shown). These results confirmed the involvement of reverse glutamate transport in OGD-induced WM injury, and also showed that the excitotoxic pathway can be blocked at the point of transmitter release.
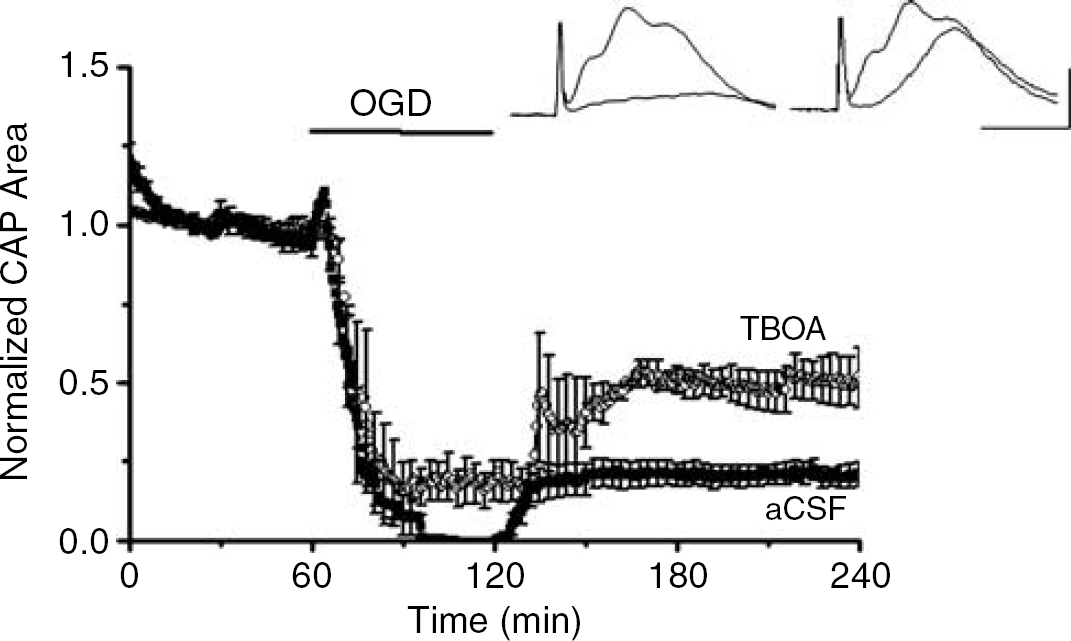
Blockade of Na+-dependent glutamate transporter reduced OGD-induced WM injury. DL-TBOA (200 μmol/L), a potent and competitive blocker of Na+-dependent glutamate transporter, effectively improved the CAP area recovery after 60 mins of OGD (open circles 50.5% ± 4.1%, n = 9, P < 0.001).
To further validate the electrophysiologic findings supporting the hypothesis that glutamate release and excitotoxicity participate in OGD-induced injury in MON, direct measurements of glutamate release from MONs were performed using quantitative HPLC (Figure 9) (Ye et al, 2003). Glutamate measurements were made under conditions similar to all of the electrophysiologic experiments. Using a pair of MONs, baseline release levels at 37°C were determined for 30 mins before exposure to OGD by sampling small volumes of perfusion solution passed over the nerves every 2 mins. For comparison, all release data were normalized to the average control release levels that were in the range of 4 to 6 nmol/L (n = 10; data not shown). It should be noted that the absolute concentrations of glutamate measured in the bath do not represent actual concentrations of glutamate in the nerve; released glutamate is diluted by the bath solution as much as 1,000- to 10,000-fold. During OGD, glutamate release remained very stable at the baseline level for the initial 30 mins. At around 30 mins of OGD, release steadily increased reaching maximal levels that were at least fivefold higher compared with basal levels (i.e., approximately 20 to 30 nmol/L) at the end of 60 mins OGD. On return to control aCSF, glutamate levels declined rapidly and reached baseline levels within 30 mins. These experiments suggested that glutamate release occurred only after approximately 30 mins of continuous OGD. Caution is necessary in interpreting these results, however, because glutamate is only detected in these experiments after it has escaped from the extracellular space of WM, a complex compartment that is separated from the bath by a significant diffusion barrier. Nevertheless, additional circumstantial evidence favored the idea that glutamate release in this WM tract was a relatively late consequence of OGD. We reasoned that if glutamate was released ‘early’ during OGD, its appearance in the bath might be delayed simply because of diffusion. If so, a brief period of OGD (i.e., 15 mins) should still be associated with a late increase in bath glutamate. This hypothesis was tested and disproved. OGD for 15 mins resulted in no detectable increase in bath glutamate over an extended 60-mins period of post-OGD monitoring (Figure 9B, n = 4). Furthermore, experiments with hippocampal slices under identical conditions revealed that glutamate release began approximately 5 mins after the onset of OGD (data not shown), confirming that the delayed nature of glutamate release in WM is a specific response of WM to OGD.
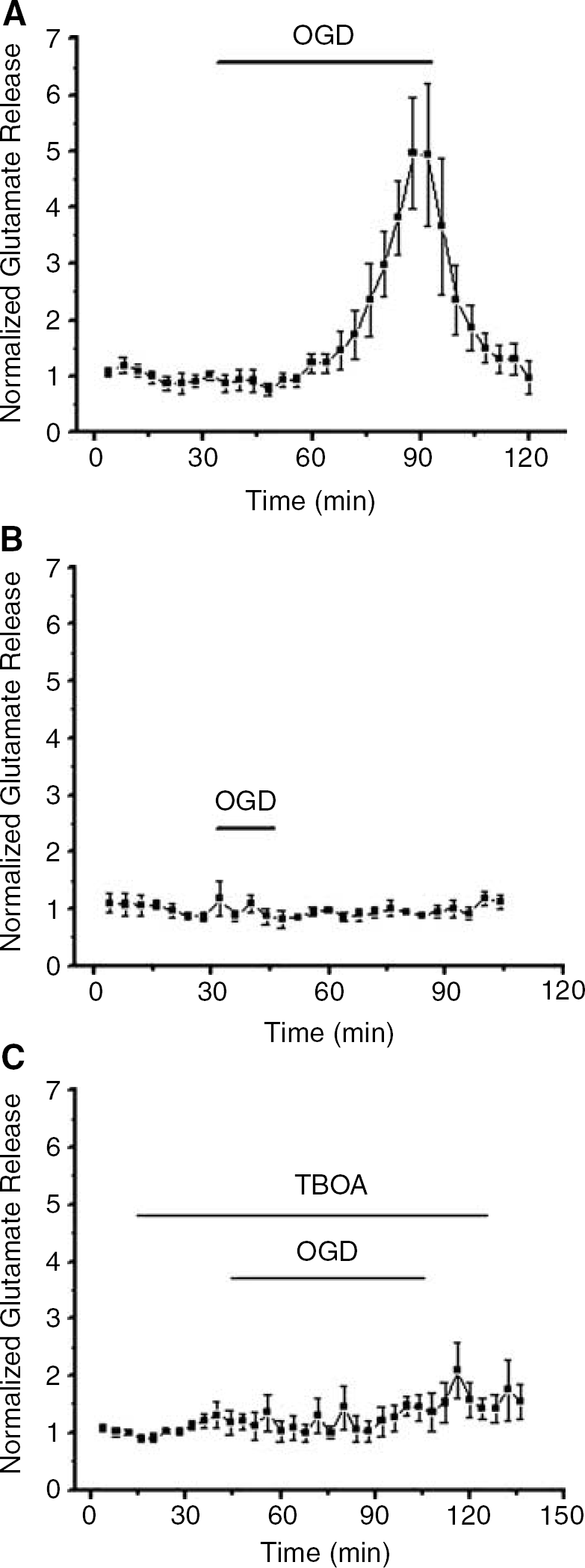
OGD caused slow onset glutamate release in MON via reversal of the Na+-dependent glutamate transporter. (
As TBOA was strongly protective of WM injury (see above), it was hypothesized that this agent would reduce glutamate release. Indeed, pretreating MONs with DL-TBOA (200 μmol/L) prominently reduced glutamate release during 60 mins of OGD (Figure 9C). Moreover, this result indicated that reverse Na+-dependent glutamate transport is the main mechanism of glutamate release during OGD in MON.
Discussion
Our results showed that glutamate receptors participated in ischemic injury in a completely myelinated WM tract, the MON. This was not anticipated from our earlier studies on rat optic nerve that documented the involvement of reverse Na+/Ca2+ exchange and activation of Ca2+ channels in WM injury (Stys et al, 1992; Fern et al, 1995). Moreover, ischemic WM injury in MON was unique compared with other WM regions because either AMPA or kainate receptor activation was sufficient to cause injury (see Table 1; (Agrawal and Fehlings, 1997; Li et al, 1999; Li and Stys, 2000; Rosenberg et al, 1999; Tekkok and Ransom, 2005; Tekkök and Goldberg, 1999, 2001; Wrathall et al, 1996; Wrathall et al, 1992). These data, therefore, establish the important principle that mechanisms of ischemic WM injury are regionally variable. The explanation(s) for regional differences in WM injury are not yet understood at a cellular level. Regional differences in WM oligodendrocytes and/or axons are logical possibilities but differences in astrocytes, cells rich in glutamate, should also be considered.
Glutamate receptors mediating injury in white matter

(−): Kainate receptors are not specifically tested but are unlikely to play a large role because specific AMPA blockade was highly protective.
Data obtained only by using nonspecific non-NMDA receptor: antagonists are available.
AMPA, α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid; NA, not available; NMDA, N-methyl-D-aspartate.
Glutamate release from MON was directly measured and increased at least fivefold late during the 60-mins period of OGD. Importantly, glutamate release was not detected for brief periods of OGD and ischemic injury could not be reproduced by glutamate receptor activation unless accompanied by a brief OGD, suggesting that toxic glutamate accumulation is delayed in comparison with the initial ionic changes mediating the early loss of excitability and injury. Simultaneous blockade of AMPA and kainate receptors, removal of extracellular Ca2+ or blockade of Na+-glutamate transport reduced ischemic WM injury. Blockade of NMDA receptors, however, did not reduce ischemic WM injury. Based on these results, and others, we propose that ischemia disrupts glial and axonal ion gradients leading to glutamate release by reverse transport and overactivation of AMPA and kainate receptors on oligodendrocytes. Axon dysfunction per se may proceed independent of glutamate release (Stys et al, 1992), but it appears to be enhanced by glutamate-mediated oligodendrocyte injury (cf. Tekkök and Goldberg, 2001).
Mouse Optic Nerve as a Model for Studying White Matter Ischemic Injury
Rat optic nerve (RON) has been widely used to study WM injury owing to anoxia (Stys et al, 1990, 1992), aglycemia (Brown et al, 2001) and ischemia (Garthwaite et al, 1999). MON, however, has important advantages over RON as an in vitro model for studying WM injury. The diameter of the adult RON is about twice that of the adult MON (650 ± 60.2 μm versus 281 ± 29.9 μm) and metabolism may be limited by inadequate glucose diffusion (bath (glucose) = 10 mmol/L) into the RON, especially when glucose utilization would be increased as during anoxia (Tekkök et al, 2003). Taking into account this technical point and considering the promise of future transgenic mice to analyze specific injury pathways, we used MONs for these experiments.
Another technical point should be mentioned. It is sometimes assumed that anoxia and ischemia are nearly equivalent insults as they relate to induction of cerebral injury. While this may be true for some areas of the brain, axon function is partially resistant to anoxia in MON (Tekkök et al, 2003) and corpus callosum (Tekkök and Ransom, 2004). Ischemia (i.e., OGD), therefore, is the more appropriate insult for models of WM injury relevant to stroke. Indeed, OGD produced very reliable, duration-dependent irreversible injury of MON (Figures 1A and 1B). As previously emphasized, these studies indicated that gray matter was more sensitive to ischemia than WM. For example, 30 mins of OGD caused a permanent loss of only 30% of the CAP but total loss of synaptic function in a gray matter area such as the hippocampus (Tekkök and Ransom, 2004).
Excitotoxic White Matter Injury Involved α-Amino-3-hydroxy-5-Methylisoxazole-4-Propionic Acid and Kainate but N-Methyl-D Aspartate Receptors
In agreement with previous studies on mechanisms of WM injury, nonspecific glutamate receptor blockade was highly protective (Agrawal and Fehlings, 1997; Li et al, 1999; Li and Stys, 2000; Tekkök and Goldberg, 2001) (Figures 3 and 4). However, our results diverge from previous studies (Li and Stys, 2000; Matute, 1998; Tekkök and Goldberg, 2001) in that OGD caused MON injury by activating either AMPA or kainate receptors; hence, blockade of both receptor subtypes was necessary for axon function preservation. By comparison, kainate receptors do not appear to participate in ischemic injury in the corpus callosum, a WM tract containing both myelinated and unmyelinated fibers (Tekkök and Goldberg, 2001); Table 1). In this preparation, NAS is highly protective against ischemic injury (Tekkök and Goldberg, 1999) while it offers no protection at all in MON (Figure 3B). These differences in glutamate receptor pharmacology of WM injury may reflect regional differences in the receptors themselves (e.g., degree of expression or subunit composition), regional variability in the amount, or onset of glutamate release during ischemia, or the existence of regionally diverse oligodendrocytes. The latter point is particularly noteworthy given that the ratio of myelinating to non-myelinating oligodendrocytes vary between areas where all axons are myelinated (like the MON) compared with areas containing many, perhaps a preponderance of non-myelinated axons (such as the corpus callosum).
Recently, NMDA receptors were shown to be located on developing oligodendrocyte processes in rodents (Micu et al, 2006; Salter and Fern, 2005). In prior studies on mature WM, however, blockade of NMDAR receptors during anoxia (Ransom et al, 1990) or ischemia (Tekkök and Goldberg, 2001; Yam et al, 2000) or glutamate application (Li et al, 1999) did not offer functional or histologic protection against these insults. The only discernable effect of NMDA receptor antagonism (i.e., MK-801) was slowing of the initial loss of the CAP during OGD in corpus callosum slices (Tekkök and Goldberg, 2001). Nevertheless, we carefully searched for involvement of NMDA receptors in OGD-induced MON injury. Although we found the NR1 subunit, the functional subunit of NMDA receptors, on adenomatous polyposis coli (APC) (+) mature oligodendrocytes in the MON (data not shown), none of the three NMDA receptor blockers tested in our study improved recovery or shape of the CAPs after 60 mins of OGD (Figure 5). These results support the tenet that in mature animals NMDA receptor antagonism provides effective protection only in gray matter.
Functional AMPA and kainate receptors are expressed on oligodendrocytes (Gallo et al, 1994) and, it would appear, on the myelin they generate (Li and Stys, 2000). Although glutamate receptor subunit 4 (GluR4) is observed in axons (Li and Stys, 2000), it seems unlikely that functional receptors are expressed on the axolemma; for example, direct application of non-transported glutamate agonists such as AMPA, even when desensitization was blocked, failed to have immediate effects on the CAP area (Figure 6). Therefore, it is likely that activation of AMPA and kainate receptors mediates injury and death of oligodendrocytes during OGD (Li et al, 1999, 2000; Tekkök and Goldberg, 2001; Tekkok and Ransom, unpublished results). Injury from OGD exposure was almost entirely blocked when OGD was administered in the absence of bath Ca2+ (Figure 2). This suggested that glutamate released during OGD activated receptors on oligodendrocytes that are Ca2+ permeable (e.g., Sanchez-Gomez and Matute, 1999). Alternatively, the observed Ca2+ dependence could be secondary to receptor-mediated disruption of the oligodendrocyte transmembrane Na+ gradient leading to reverse Na+/Ca2+ exchange (Stys et al, 1992), or depolarization and activation of L-type Ca2+ channels in oligodendrocytes (Alberdi et al, 2002; Kirischuk et al, 1995).
The consequences of oligodendrocyte injury in myelinated WM is readily understood. The paranodal loops of myelin pull away from nodes during energy disruption, disabling saltatory conduction. If the injury to the oligodendrocyte is sublethal, the paranodal loops may reapply themselves with restoration of function. The effects of oligodendrocyte injury and death on non-myelinated axons are much less intuitive, but indirect evidence suggests that loss of oligodendrocytes increases axon injury (Tekkök and Goldberg, 2001). Indeed, the presence of oligodendrocytes and myelin seems to dictate the mode of axon injury during OGD. In isolated, unmyelinated axons OGD-induced injury is mediated by Na+ and Ca2+ overload independent of excitotoxicity (Underhill and Goldberg, 2006).
Glutamate is Necessary but not Sufficient to Explain Ischemic White Matter Injury
The extent of irreversible MON injury increased linearly in direct proportion to the duration of OGD (i.e., Figure 1). This behavior implied a pathologic process(es) that was smoothly progressive over ∼ 90 mins of OGD required to reach maximum injury. In 1992, we reasoned that this pattern represented progressive Ca2+ accumulation in depolarized and Na+-loaded axons, that is, the advance of a single pathologic process (Stys et al, 1992). However, our data and that of other WM researchers (see review (Stys, 2004)) now strongly favor the participation of at least two related but distinct injury mechanisms: ionic rearrangements owing to energy depletion and loss of glutamate homeostasis with increase in extracellular glutamate leading to toxic activation of AMPA and kainate receptors on oligodendrocytes. Blockade of these receptors during OGD was clearly protective. And just as clearly, activation of these receptors in WM under control conditions did not lead to injury (Figure 6). Injury resulted only when application of glutamate receptor agonists was coupled with OGD. Indeed, normal intact brain tissue is quite resistant to glutamate toxicity as long as the energy supply is not compromised owing, presumably, to efficient glutamate uptake by Na+-dependent glutamate transporters and subsequent maintenance of internal Ca2+ homeostasis. In addition, glutamate activation of metabotropic glutamate receptor (mGluR) may also be neuroprotective masking the injury expected on direct activation of AMPA/kainate receptors (Small et al, 1996).
These findings suggested that OGD produced an essential first stage of dysfunction consisting of ionic disruption in axons and glial cells that primed WM for glutamate toxicity. In the absence of normal amounts of ATP, ion gradients disrupted by exogenous glutamate agonists cannot be restored and, in fact, augment the developing ionic disruption by opening additional ion channels permeable to Na+ and K+. At the same time, glutamate uptake is failing and beginning to operate in reverse, adding glutamate to the extracellular space, rather than removing it. Our observation of reduced injury, and greatly reduced glutamate release, in the presence of TBOA (Figures 8 and 9C), supports that OGD-induced excitotoxicity depended on glutamate release by reverse Na+-dependent glutamate transport. Such transporters are mainly expressed on astrocytes (Anderson and Swanson, 2000), implying that astrocytes are critically involved in toxic glutamate accumulation in this WM preparation. The sequence proposed is supported by the relatively late release of glutamate from MON during OGD (Figure 9). Virtually no glutamate was released during a short period of OGD (i.e., 15 mins) even though ion gradients have collapsed sufficiently in that time frame such that the CAP area was markedly reduced. At this stage, nearly complete recovery is possible. Once the duration of OGD progressed to detectable glutamate release, however, irreversible injury inevitably ensued.
Based on these experimental results, one may deduce that the degree of WM injury from a given period of OGD might depend on the proximity of gray matter. Gray matter releases glutamate more quickly (in approximately 5 mins) and in much greater abundance (more than 20-fold increase, data not shown) than WM. WM in the vicinity of gray matter, therefore, is likely to suffer more rapid and severe injury during OGD because it will ‘see’ higher (perhaps much higher) concentrations of glutamate than would be the case for WM that is distant from gray matter. WM in the forebrain of rodents would be susceptible to this effect because the WM tracts are thin and very close to the flanking gray matter. In humans, however, there is a great expansion of WM in the forebrain (approximately 50% by volume compared with approximately 13% for rodents) and WM can experience ischemia independently of gray matter ischemia. By this reasoning, WM that directly abuts cortex should suffer more injury than deep WM during ischemia. This intriguing hypothesis has not yet been tested.
Footnotes
Acknowledgements
The authors thank Brianna Mbow and Taylor J Abel for expert technical assistance and Deicel Inc, NJ for the gift of NAS.