
Abstract
We studied the effect of a novel α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA)/kainate antagonist, YM90K [6-(1H-imidazol-1-yl)-7-nitro-2, 3(1H, 4H)-quinoxalinedione monohydrochloride], in a focal cerebral ischemia model using anesthetized cats. Cats were subjected to permanent occlusion of the middle cerebral artery (MCA) for 6 h, then killed and examined histologically. The amount of ischemic damage was assessed in 12 stereotaxic coronal sections. Treatment with
The excessive increase of glutamate in the synaptic cleft following ischemic is considered to play a critical role in the development of neuronal damage. Evidence for this comes from the finding that ischemic cell damage in the hippocampus is protected by surgical transection of ipsilateral glutamatergic afferents to the hippocampus in global cerebral ischemia (Wieloch et al., 1985; Onodera et al., 1986; Jørgensen et al., 1986). Glutamate acts on two distinct subtypes of postsynaptic ionotropic receptors, namely, the N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA)/kainate (or non-NMDA) subtypes (Watkins et al., 1990). Of these two subtypes, the NMDA receptor was first postulated to mediate glutamate neurotoxicity since the ion channel of this subtype is highly permeable to Ca2+ (MacDermott et al., 1986; Uematsu et al., 1989a). In fact, neuroprotective effects of NMDA antagonists have been demonstrated in models of focal cerebral ischemia (Park et al., 1988a, b ; Bullock et al., 1990; Gill et al., 1991; Uematsu et al., 1991), although their neuroprotective effects in global cerebral ischemia have been controversial (Fleischer et al., 1989; Michenfelder et al., 1989; Sterz et al., 1989; Lanier et al., 1990; Buchan et al., 1991b; Nellgård et al., 1991; Nellgård and Wieloch, 1992). However, accumulating evidence suggests that the AMPA/kainate receptor is also involved in neuronal damage following cerebral ischemia. AMPA/kainate antagonists, such as 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline (NBQX) and GYKI 52466 [1-(4-aminophenyl)-4-methyl-7,8-methylene-dioxy-5H-2,3-benzodiazepine hydrochloride], have shown neuroprotective effects in both transient global (Sheardown et al., 1990, 1993; Buchan et al., 1991a; Diemer et al., 1992; Le Peillet et al., 1992; Nellgård and Wieloch 1992; Li and Buchan, 1993) and focal cerebral ischemia models in rodents (Xue et al., 1994; Gill et al., 1992; Smith and Meldrum, 1992). Another AMPA/kainate antagonist, LY-293558, has been proven effective in reducing neuronal damage in a cat focal cerebral ischemia model (Bullock et al,. 1994).
We have recently developed a novel and potent AMPA/kainate antagonist, YM90K [6-(1H-imidazol-1-yl)-7-nitro-2, 3(1H, 4H)-quinoxalinedione monohydrochloride] (Ohmori et al., 1994), and reported that it possesses more potent neuroprotective effect than does NBQX in a gerbil global cerebral ischemia model (Shimizu-Sasamata et al., 1996). In this model, YM90K was effective even when administered 6 h after induction of ischemia. This agent also has a neuroprotective effect against neuronal damage in a rat focal cerebral ischemia model (Shimizu-Sasamata et al., 1993). In the present study, we examined the neuroprotective effect of YM90K against the neuronal damage induced by focal cerebral ischemia in anesthetized cats.
MATERIALS AND METHODS
Male ICo: Eur (Tif) cats (IFFA-CREDO, L'arbresle, France), weighing 2.7–3.5 kg, were used for study. Each cat was anesthetized with ether, tracheotomized, and connected to a ventilator delivering a mixture of nitrous oxide (70%)/oxygen (30%) (vol/vol) containing 1–1.5% halothane in a closed circuit. Femoral arteries and veins were cannulated bilaterally to monitor arterial blood pressure and obtain arterial blood samples for blood gas determination, and to administer drugs and obtain blood samples for determination of plasma drug levels, respectively. Anesthesia was maintained until the end of investigation. After immobilization with gallamine triethiodide (1 mg/kg i.v.), normocapnia (Paco2 close to 32 mmHg) and immobilization were maintained by adjusting the stroke volume of the respirator and by administering gallamine triethiodide (0.5 mg/kg i.v.) every 2 h, respectively, throughout the investigation. Cats were kept warm with a heating pad and lamp; rectal temperatures were monitored. To monitor EEG, screw electrodes were implanted into the skull on the surface of the middle ectosylvian gyri, where blood flow is mainly supplied by the middle cerebral artery (MCA).
The left MCA was occluded via a modified transorbital approach (O'Brien and Waltz, 1973). The skull was fixed in a stereotaxic frame. Using microsurgical techniques, the left orbit was exenterated and the orbital roof and optic foramen were removed using a dental drill to expose the dura overlying the MCA. Under the surgical microscope, the dura was incised and the MCA was exposed. After dissection of the arachnoid from the MCA, the MCA trunk was occluded with a miniature clip. To standardize the severity of ischemia, only those cats in which mean ipsilateral EEG amplitude decreased to 30% of preocclusion level immediately after induction of ischemia without recovery during the first 10 min of ischemia (Fig. 1), were used. Under this condition, our preliminary data indicated that an occlusion time of 6 h provides maximum volume of ischemic damage.
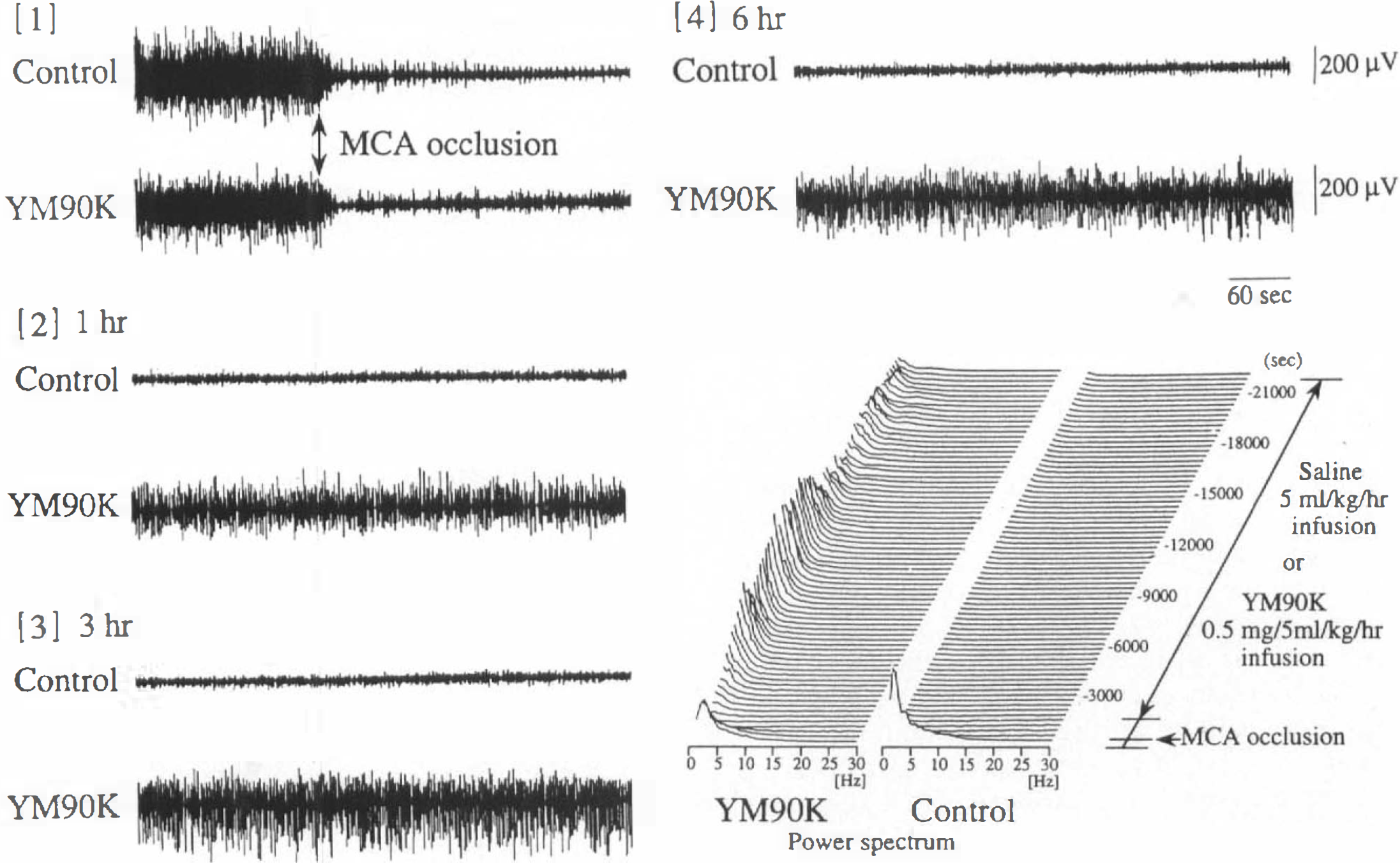
Representative changes in EEG and power spectrum of the left middle ectosylvian cortex saline (5 ml/kg/h)-treated (control) and YM90K (0.5 mg/5 ml/kg/h)-treated cats.
YM90K (0.25 mg/5 ml and 0.5 mg/5 ml) was dissolved in saline alkalized with a few drops of 1 N NaOH solution. This solution was then adjusted to pH 8–9 with 1 N HCl. YM90K was infused i.v., starting 10 min after the MCA occlusion, and continued for 6 h (0.25 mg/5 ml/kg/h or 0.5 mg/5 ml/kg/h). Control animals were infused i.v. with saline (5 ml/kg/h) prepared at pH 8–9. In all
Six hours after occlusion of the MCA, pentobarbital sodium (20 mg/kg) was given i.v. and the cat was killed was without reperfusion by transcardiac perfusion-fixation with saline followed by 10% formalin neutral buffer solution, pH 7.4. After perfusion, the cat was decapitated and the brain removed. The brain was cut into two blocks at the coronal plane through the mammillary body. The anterior part of the brain was cut into four 4-mm thick blocks from the coronal plane. A 5-mm thick block was made by cutting the posterior part of the brain from the coronal plane. Each block was embedded in paraffin and 7–8 μm thick sections were cut at 200-μm intervals. Sections were stained with hematoxylin and eosin, and 12 stereotaxic coronal planes were selected according to the coordinates of Snider and Neimer (1961). Neuronal damage was defined on the basis of the following morphological characteristics: mincrovacuolation, shrinkage of the neuropil, and presence of dark neurons and eosinophilic neurons. Neuronal damage in each coronal section was evaluated by a histologist who was blinded to animal treatment. Areas of ischemic damage in the cerebral hemisphere, cortex, and caudate were calculated by a computer-aided image analyzer system (Luzex III, Nireco Co., Tokyo, Japan). The volume of ischemic damage was calculated from the area of damage of the 12 coronal sections and their stereotaxic coordinates.
In another series of experiments, a thermoprobe (29G, Physitemp Instrument Inc., Clifton, NJ U.S.A.) was inserted into the striatum (A: 18, L: 4, H: +5.5 mm) according to the coordinates of Snider and Niemer (1961) to evaluate the influence of YM90K. on brain temperature under similar experimental conditions.
All data are presented as the means ± SD. For differences among experimental groups, one-way analysis of variance (ANOVA) followed by Dunnett multiple range test or the Kruskal-Wallis H-test followed by Wilcoxon multiple comparison test was used.
RESULTS
Physiological variables, including mean arterial blood pressure (MABP) (mmHg), Paco2 (mmHg), Pao2 (mmHg), pH, and rectal temperature are shown in Table 1. In cats treated with YM90K (0.5 mg/5 ml/kg/h), MABP at 1 h after MCA occlusion was slightly, but significantly, decreased compared with that of saline-treated cats. Additionally, pH values of the YM90K (0.25 mg/5 ml/kg/h)-treated cats, in which no neuroprotective effect was observed, were slightly, but significantly, decreased at 5 and 6 h after occlusion. There were no differences among the three groups for any other variables at any time. To examine whether or not the neuroprotective effect of YM90K was due to its lowering effect on the brain temperature, brain temperature was monitored under the same conditions. Brain temperature was reduced by 0.6°C at 10 min after MCA occlusion; it returned to the preocclusion level by 30 min after occlusion and gradually increased in parallel with rectal temperature in both groups (Fig. 2). There were no differences in brain temperature between cats treated with YM90K (0.5 mg/5 ml/kg/h), in which the volume of ischemic damage was significantly reduced, and those treated with saline (Fig. 2).
Effect of YM90K in physiological variables
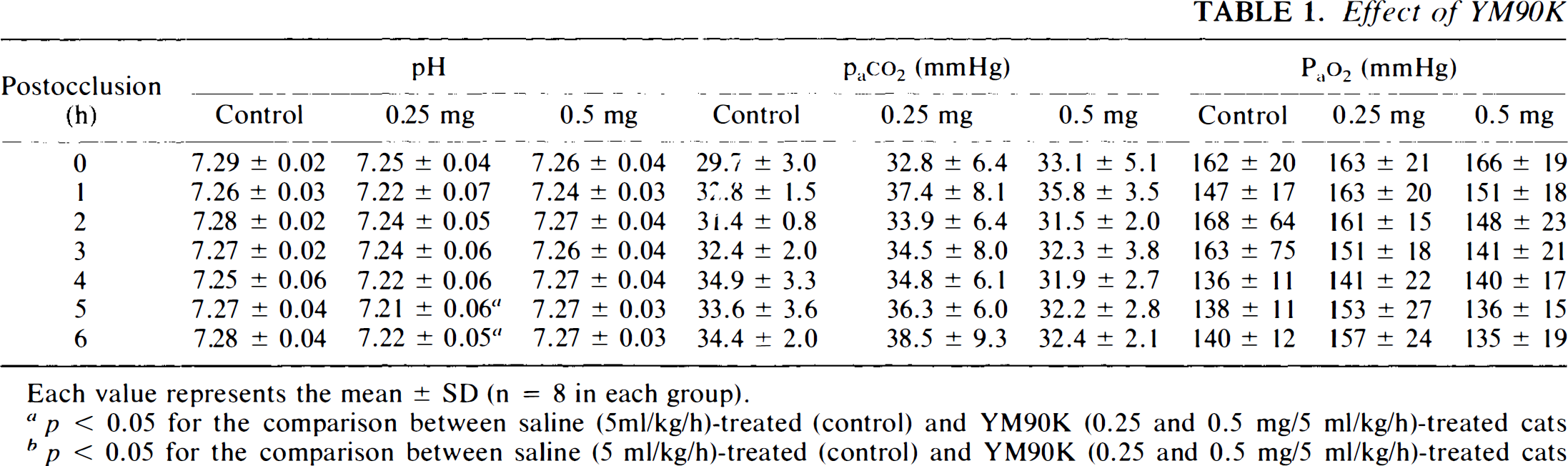
Each value represents the mean ± SD (n = 8 in each group).
p < 0.05 for the comparison between saline (5ml/kg/h)-treated (control) and YM90K (0.25 and 0.5 mg/5 ml/kg/h)-treated cats (one-way ANOVA followed by Dunnett multiple range test).
p < 0.05 for the comparison between saline (5 ml/kg/h)-treated (control) and YM90K (0.25 and 0.5 mg/5 ml/kg/h)-treated cats (Kruskal Wallis H-test followed by Wilcoxon multiple comparison test).
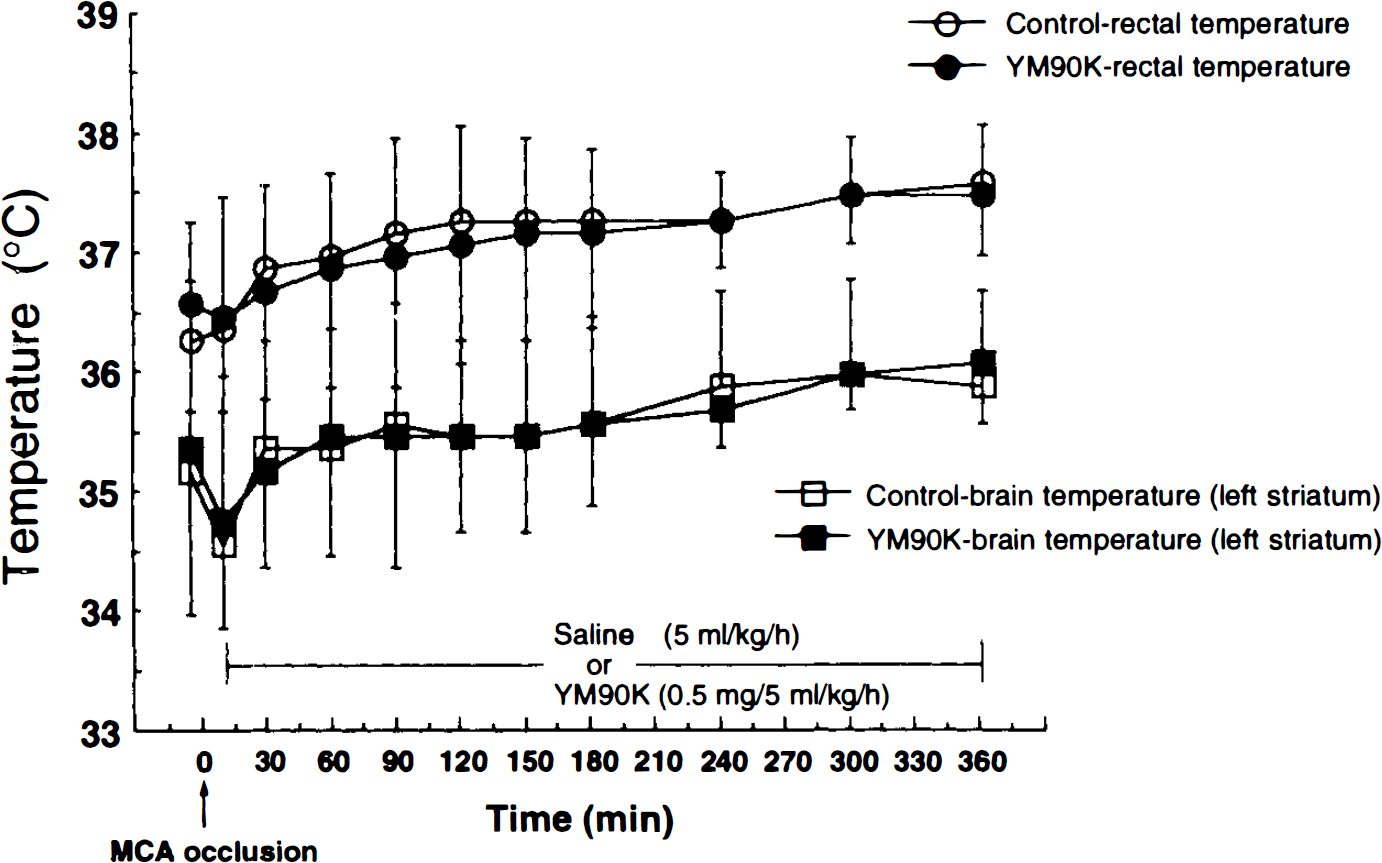
Brain and rectal temperatures in saline (5 ml/kg/h)-treated (control) and YM90K (0.5 mg/5 mg/kl/h)-treated MCA cats. Each value represents the mean ± SD (n = 3 in each group).
Effect of YM90K on neuronal damage
Administration of YM90K starting 10 min into ischemia reduced the volume of ischemic damage in the cerebral hemisphere and cortex (Table 2) in a dose-dependent manner. At a dose of 0.5 mg/5 ml/ kg/h, YM90K reduced the volume of ischemic damage by ∼40% in both brain regions, and these reductions were statistically significant (F = 3.52, p < 0.05, and F = 3.60, p < 0.05, respectively, ANOVA followed by Dunnett multiple range test) compared with saline-treated cats (Table 2). In contrast, YM90K showed no protective effect on ischemic damage in the striatum (Table 2). In the cerebral hemisphere and cortex, YM90K (0.5 mg/5 ml/ kg/h) reduced the area of ischemic damage at all coronal sections, and the reduction of ischemic damage areas in the coronal sections of 1–9-mm stereotaxic distance were statistically significant (Figs. 3A and B ). Areas of ischemic damage in the striatum were not ameliorated by treatment of YM90K (Fig. 3C). In this experiment, only those cats, in which mean ipsilateral EEG amplitude immediately decreased to <30% of the preocclusion level without recovery during the first 10 min of ischemia, were used. When the recovery of EEG amplitude was observed, it took place within at least 3 min after MCA occlusion. Thus, the decrease of EEG continued for 6 h in all control animals in the case of no recovery of EEG during the first 10 min of ischemia. However, in six of eight cats treated with YM90K (0.5 mg/5 ml/kg/h), recovery of EEG was observed.
Effect of YM90K on the volume of ischemic damage at 6 h after MCA occlusion in cats

Each value represents the mean ± SD.
p < 0.05 for the comparison between saline-treated and YM90K-treated cats (one-way ANOVA followed by Dunnett multiple range test).
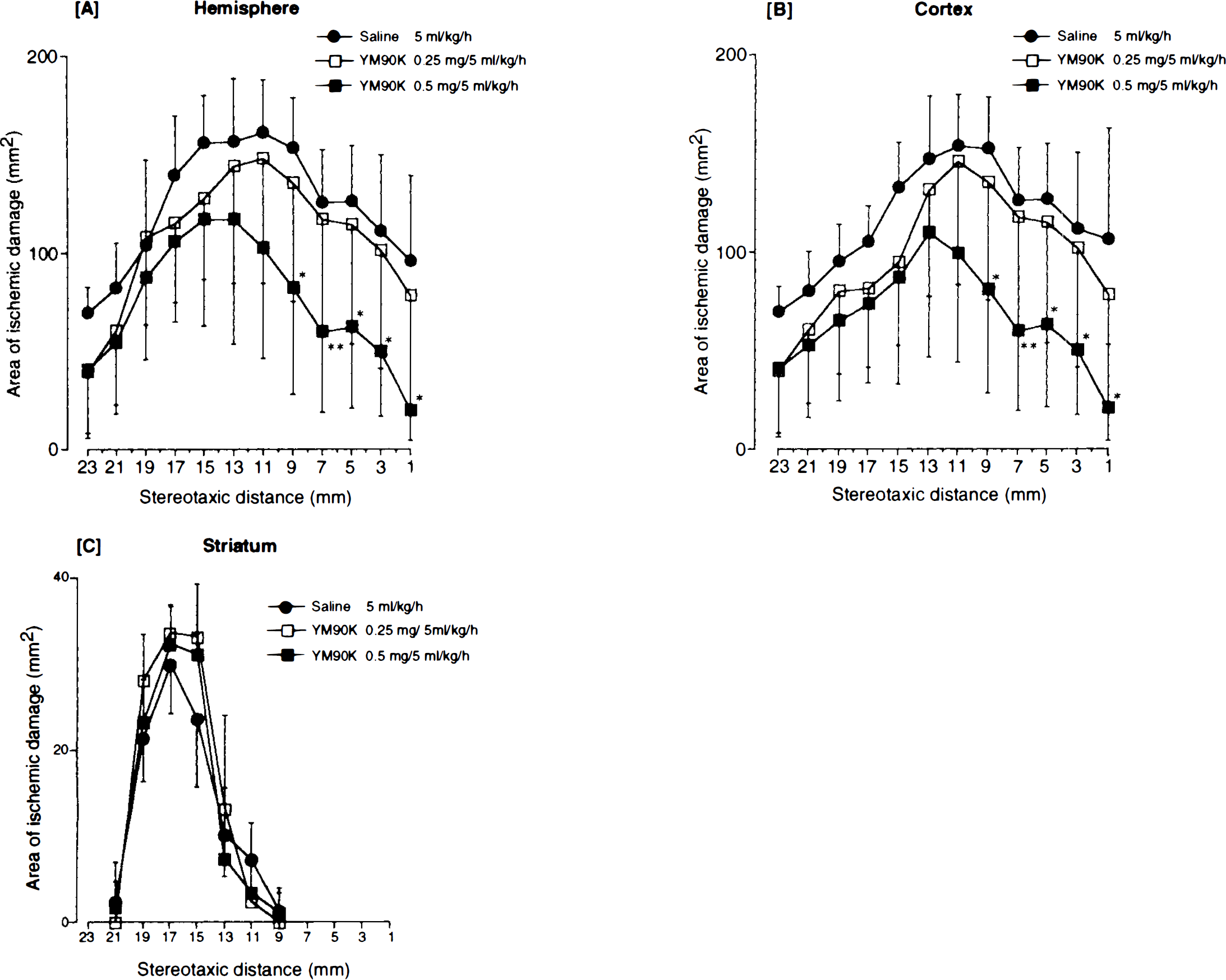
The area of ischemic damage in the cerebral hemisphere
Plasma levels of YM90K (0.25 and 0.5 mg/5 ml/ kg/h) continuously increased, reaching 527 ± 49 ng/ml and 1,242 ± 146 ng/ml, respectively, at the end of experiments (Fig. 4). CSF levels of YM90K were 7.0 ± 0.9 ng/ml (0.25 mg/5 ml/kg/h) and 13.8 ± 3.0 ng/ml (0.5 mg/5 ml/kg/h), indicating that CSF/ plasma ratio was ∼1:100.
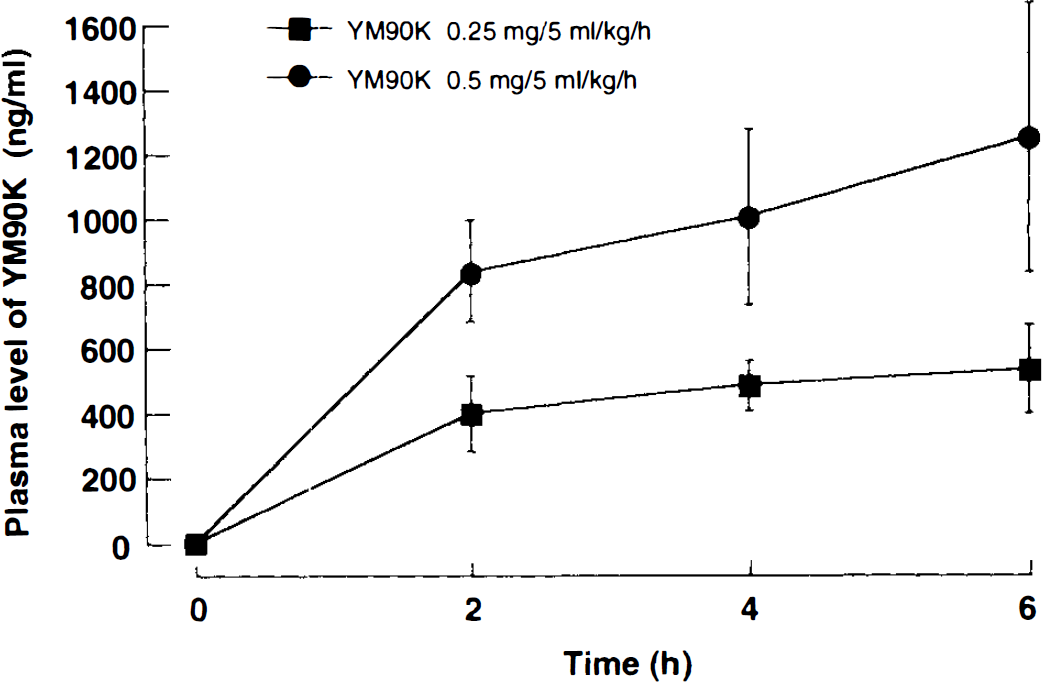
Plasma levels of YM90K during postischemic period (MCA occlusion at time 0). Each value represents the mean ± SD (n = 8 in each group).
In preliminary studies, the effect of YM90K on the behavior of cats was observed. At 0.3–1 mg/5 ml/kg/h, YM90K showed no obvious effect on behavior, but at 3 mg/5 ml/kg/h, it induced sleep (data not shown).
DISCUSSION
The cats used in this study are genetically more homogeneous than conventional mongrel animals. In addition, only those cats, in which mean ipsilateral EEG amplitude immediately after ischemia decreased to 30% of the preocclusion level without recovery during the first 10 min after ischemia, were used. It has been reported that this change in EEG amplitude is a good index for predicting ischemic damage in MCA-occluded cats (Uematsu et al., 1989b). Thus, this protocol provided relatively larger and more uniform infarcts than those of other methods (Ozyurt et al., 1988; Park et al., 1988a; Bullock et al., 1990).
The present study is the first report to indicate efficacy for an AMPA/kainate antagonist when administered after ischemic insult in the cat MCA model. Starting 10 min after MCA occlusion, i.v. infusion of YM90K for 6 h markedly reduced the volume of ischemic damage. Moreover, EEG was also restored in six of eight cats treated with YM90K. This is, presumably, due to a functional recovery of cortical neurons protected by YM90K. These results suggest that the AMPA/kainate receptor plays an important role in the pathogenesis of focal cerebral ischemia in gyrencephalic species. A recent report using LY-293558 in the same model has also suggested this notion (Bullock et al., 1994), although in that study, the compound was administered in bolus 30 min before occlusion, followed by i.v. infusion during postischemic period. AMPA/kainate antagonists, including YM90K, have been suggested to have a wide therapeutic time window (Sheardown et al., 1990, 1993; Li and Buchan et al., 1993; Shimizu-Sasamata et al., 1996); in global cerebral ischemic models in rodents, they exert neuroprotective effects with administration even 6–24 h after insult. In transient focal ischemia in rats, NBQX and GYKI 52466 reduced neuronal damage on delayed treatment (Buchan et al., 1991c; Xue et al., 1994). Moreover, YM90K is effective in the rat permanent MCA model even when administered from 2 h into ischemia (Shimizu-Sasamata et al., unpublished observations). Although we have not examined efficacy in such delayed treatment in the cat MCA model, the present results demonstrating the neuroprotective effect of YM90K by early postischemic treatment suggest its therapeutic potential.
It is well known that the elevation of intracellular Ca2+ concentration, [Ca2+]i, following the excessive synaptic release of glutamate plays a critical role in ischemia-induced cell damage (Siesjö, 1981; Uematsu et al., 1989b). This elevated [Ca2+]i is mediated by many pathways: (a) the NMDA receptor, which is highly permeable to Ca2+, but is blocked in a voltage-dependent manner by Mg2+ (Wong and Kemp, 1991), (b) voltage-sensitive calcium channels (VSCCs) (Miller, 1991), (c) the metabotropic glutamate receptor, which mediates Ca2+ release from intracellular stores (Sugiyama et al., 1987), and (d) the AMPA/kainate receptor lacking the GluR2 subunit, which is also permeable to Ca2+ (Hollmann et al., 1991; Verdoorn et al., 1991). In fact, the NMDA antagonist and the VSCC antagonist reduced ischemic damage in the cat MCA model, and these antagonists also attenuated the elevation of [Ca2+]i (Uematsu et al., 1989c; Uematsu et al., 1991). Blocking of the AMPA/kainate receptor by antagonists, such as NBQX, YM90K, or GYKI 52466, could attenuate the activation of the NMDA receptor and VSCCs following depolarization of the cell membrane. This would reduce Ca2+ entry into the cell via these two pathways. In addition, blocking of the AMPA/kainate receptor would also reduce Ca2+ entry through this receptor lacking GluR2 subunit. The reduction of Ca2+ entry through these pathways by blockade of the AMPA/kainate receptor may be one of the neuroprotective mechanisms of AMPA/kainate antagonists in focal cerebral ischemia.
Monitoring of physiological variables, such as MABP, blood gas, and body temperature, is an essential procedure in the evaluation of neuroprotective agents in cerebral ischemia models, since these parameters may affect neuronal damage (Ginsberg and Busto, 1989). Except for a slight and transient decrease in MABP, YM90K had no effect on any physiological variables monitored. Transient hypotension may be an artifact, since it was only observed at 1 h after the start of infusion, before the plasma level of YM90K had reached a plateau. Since hypothermia during or after ischemia may ameliorate ischemic injury (Busto et al., 1989; Kuroiwa et al., 1990; Welsh et al., 1990; Xue et al., 1992), both brain and rectal temperatures were monitored directly. Neither temperature was influenced by administration of YM90K, indicating that the neuroprotective effect of YM90K is not due to decreased brain temperature.
It has been reported that quinoxalinediones, e.g., NBQX, the most extensively investigated AMPA/kainate antagonist in cerebral ischemia (Sheardown et al., 1990, 1993; Buchan et al., 1991a, Diemer et al., 1992; Nellgård and Wieloch, 1992; Gill et al., 1992), precipitate in the renal tubules due to their low solubility (Xue et al., 1994). This characteristic may limit the clinical usefulness of these compounds. However, the present study showed that YM90K, which also has a quinoxalinedione structure, exerts its neuroprotective effects at a dose as low as 0.5 mg/kg/h, much lower than that at which precipitation in the kidney occurred. This dose also caused no obvious behavioral changes. In fact, although they have similar affinities for the AMPA/kainate receptor (Ohmori et al., 1994), YM90K is more potent than NBQX in cerebral ischemia models (Shimizu-Sasamata et al., 1996). This may be due to the higher permeability of YM90K into the brain than NBQX.
Various studies on the efficacy of glutamate antagonists in focal cerebral ischemia models have shown that the neuroprotective effects of AMPA/kainate antagonists, namely NBQX and LY-293558, are relatively modest as compared with those of NMDA antagonists (Ozyurt et al., 1988; Bullock et al., 1990, 1994; Gill et al., 1991, 1992; Buchan et al., 1992). For example, LY-293558 reduces the volume of ischemic damage in MCA-occluded cats by only ∼20%, while MK-801 reduces it by ∼50% in the same model (Ozyurt et al., 1988; Bullock et al., 1994). In contrast, our present results demonstrated that YM90K reduces the volume of ischemic damage by ∼40%. This suggests that the neuroprotective effects of AMPA/kainate antagonists are not necessarily less potent than those of NMDA antagonists. Since NMDA antagonists have the propensity to induce such adverse effects as psychotomimetic reactions (Koek et al., 1988), cognitive impairment (Morris et al., 1986), and neurotoxicities (Olney et al., 1989, 1991), our present results suggest the advantage of AMPA/kainate antagonists over NMDA antagonists in clinical usage.
In conclusion, we demonstrated that YM90K markedly reduced neuronal damage in focal cerebral ischemia in cats at a dose that is free from side effects. The efficacy of YM90K in gyrencephalic species further suggests its therapeutic potential in the treatment of stroke in humans.
Footnotes
Acknowledgment:
We thank Drs. T. Watanabe and Y. Teraya for their determination of plasma levels of YM90K, and Drs. T. Hanada and T. Shishido for their preparation of brain slices.