
Abstract
A short ischemic episode preceding sustained ischemia is known to increase tolerance against ischemic cell death. We report early-onset long-lasting neuroprotection against in vitro hypoxia by preceding selective chemical inhibition of oxidative phosphorylation: “chemical preconditioning.” The amplitude of CA1 population spikes (psap) in hippocampal slices prepared from control animals (control slices) was 31 ± 27% (mean ± SD) upon 45-min recovery from 15-min in vitro hypoxia. In slices prepared from animals treated in vivo with 20 mg/kg 3-nitropropionate (3-np) 1–24 h prior to slice preparation (preconditioned slices), psap improved to 90 ± 15% (p < 0.01). Posthypoxic oxygen free radicals were reduced to 65 ± 10% (mean ± SD) of control in preconditioned slices (p < 0.05). Posthypoxic neuronal density improved from 52 ± 15% (mean ± SD) in control slices to 97 ± 23% in preconditioned slices (p < 0.001). Glibenclamide, an antagonist at KATP-channels, partly reversed increased hypoxic tolerance. We conclude that chemical preconditioning induces early-onset long-lasting tolerance against in vitro hypoxia. Ultimately, this strategy may be applicable as a neuroprotective strategy in humans.
Inhibition of oxidative energy metabolism has been suggested to increase glutamate receptor-mediated neurotoxicity (Novelli et al., 1988; Simpson and Isacson, 1993), which is believed to be of major importance in ischemic cell death (Rothman and Olney, 1986). However, increased neuronal susceptibility to cell death after metabolic impairment may be contrasted by physiologically relevant adaptations. Depending on the time pattern of metabolic perturbations, a short ischemic or hypoxic episode may increase or decrease tolerance toward subsequent severe ischemia in heart and brain (Murry et al., 1986; Schurr et al., 1986; Tomida et al., 1987; Kato et al., 1989, 1991; Kitagawa et al., 1990; Lutz, 1992): ischemic preconditioning. The time pattern seems to be rather critical in heart as well as in brain. Ischemia during the time span used to induce early-onset tolerance in heart muscle causes an aggravation of morphologic lesions in brain (Kato et al., 1991; Li and Kloner, 1994). On the contrary, the time window to induce tolerance against subsequent ischemia by a short preceding ischemic episode is limited to ∼1 h in heart, while it may last a few days in brain (Kato et al., 1991; Li and Kloner, 1994). Several mechanisms have been proposed to mediate increased tolerance against prolonged hypoxia or ischemia. For neuronal tissue, long-lasting tolerance has been related to a change in activity of ATPases, e.g., a reduced activity of Na+/K+-ATPase and increased activity of Mg2+-ATPase (Benzi et al., 1993). Likewise, changes in the activity of respiratory enzymes (Dagani et al., 1984) and expression of heat shock proteins (Kirino et al., 1991; Liu et al., 1993) have been suggested. Early-onset ischemic preconditioning in heart muscle has been related to activation of ATP-regulated potassium channels (Gross and Auchampach, 1992). Involvement of KATP-channels also has been shown in preconditioning of brain after short ischemic episodes (Heurteaux et al., 1995) and chemical hypoxia (Riepe et al., 1996a).
3-Nitropropionate (3-np) inhibits succinic dehydrogenase (SDH) activity by covalent binding (Coles et al., 1979). SDH activity and ATP levels decrease upon treatment with 3-np in mouse cortical explants (Ludolph et al., 1992) and hippocampal slices (Riepe et al., 1996a). Application of 3-np in hippocampal slices has been shown to activate KATP-channels in CA1 pyramidal cells (Riepe et al., 1992). Upon prolonged application, decrease of activity of Na+/K+-ATPase is observed (Riepe et al., 1995). While a strong age dependency of morphological lesions was observed (Brouillet et al., 1993), single dosage of up to 20 mg/kg in animals up to 400 g did not induce histological abnormalities in striatum. Application of 3-np therefore seemed a suitable strategy to chemically induce preconditioning with early onset and long duration.
Ischemic preconditioning already has been applied as a preventive cytoprotective strategy in the in situ human heart in coronary angioplasty (Tomai et al., 1994). Protection was reduced by glibenclamide (Tomai et al., 1994), an antagonist at the KATP-channel (Schmid-Antomarchi et al., 1987). However, intermittent occlusion of cerebral arteries in humans does not seem practicable. In contrast, development of a practical and safe pharmacological strategy to induce preconditioning might lead to a novel neuroprotective treatment in diseases with increased risk for cerebral ischemia.
The goal of this study was to investigate whether increased tolerance against in vitro hypoxia can be induced by chemical inhibition of a selective step of oxidative phosphorylation.
MATERIALS AND METHODS
Preparation of slices
Hippocampal slices were prepared from control male Wistar rats (200–250 g) as described previously (Riepe et al., 1992). Prior to further treatment, slices were incubated for 2 h at 35°C in a static bath with Ringer solution containing NaCl 126 mM, KCl 5 mM, KH2PO4 1.3 mM, MgSO4 1.3 mM, CaCl2 2.4 mM, NaHCO3 26 mM, dextrose 10 mM, bubbled with 95% O2 and 5% CO2. Similar slices were prepared from animals pretreated in vivo with a single intraperitoneal injection of 20 mg/kg wt 3-np as described previously (Riepe et al., 1996a). No obvious impairment of gross clinical parameters was observed upon injection (in particular, no gasping or impairment of movements).
Electrophysiology
Electrophysiological recordings were made in a superfusion chamber. Extracellular recordings were obtained in hippocampal CA1 region with synaptic stimulation of Schaffer collaterals at 0.1 Hz (see Fig. 1). The raw figures showing the population spike amplitude (psap; calculated from minimum to maximum) after each stimulation are shown in Fig. 3B. For statistical analysis, the psap at indicated times was calculated as the average of six subsequent stimulations to account for variations of subsequent stimuli. Extracellular electrodes were filled with Ringer solution and had a resistance of 3–8 MΩ.
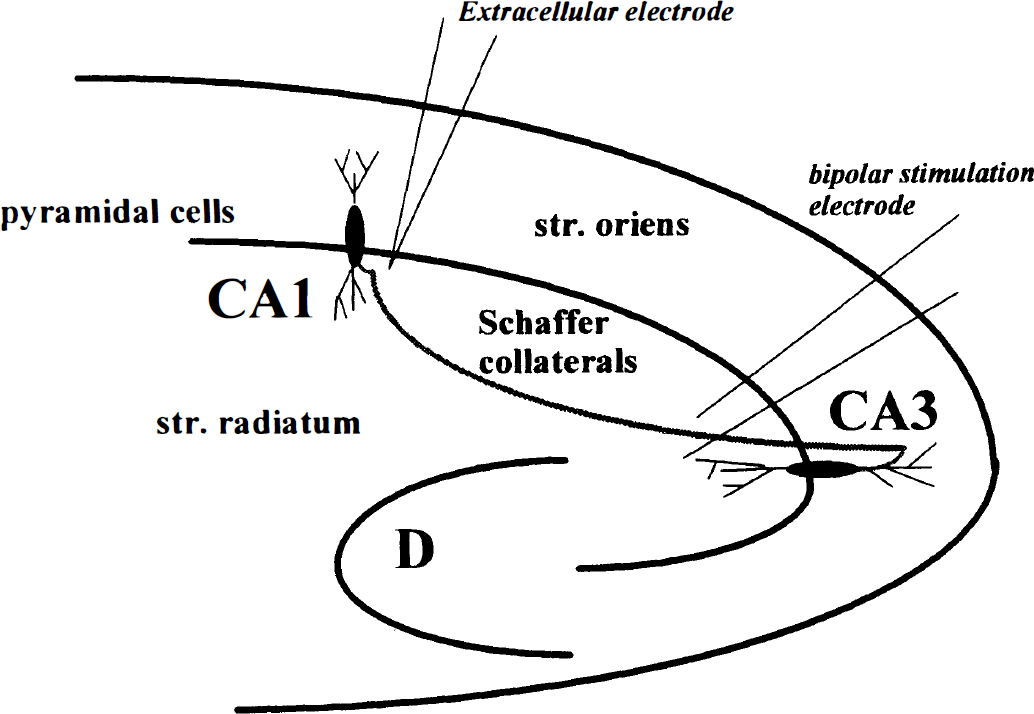
Experimental arrangement for registration of population spike amplitude. The stimulating electrode was placed in the Schaffer collaterals and the recording electrode in hippocampal region CA1.
Histology
After preincubation, slices were transferred for 15 min to a static bath with the Ringer solution bubbled with 95% N2 and 5% CO2. Afterward, slices were incubated for another 6 h in oxygenated Ringer solution. Slices were then fixed overnight in phosphate-buffered saline containing 4% paraformaldehyde plus 0.1% glutaraldehyde and were paraffin embedded. Six-micron sagittal sections were prepared and stained by 0.25% toluidine blue. The number of intact neurons was assessed in a standardized area (3 × 104 μm2) of the CA1 hippocampal region, and necrotic neurons were excluded. Evaluation of tissue damage was evaluated blindly. At least eight tissue sections for each condition were evaluated in two different experiments. Results were expressed as means ± SD and statistically assessed by analysis of variance.
Oxygen free radicals
Parasagittal slices were prepared as described. Successively, they were transferred to a chamber perfused with 37°C Ringer solution containing 10–4 M of the chemoluminescence (CL) enhancer lucigenin. Lucigenin (N,N′-dimethyldiacridinium; Sigma Chemicals) emits blue–green light upon reaction with superoxide. The slice chamber was housed in a double dark box, and the photons emitted from the slice were counted using a Hamamatsu photon counting system (Dirnagl et al., 1995). After loading of lucigenin (∼30–60 min) and recording of a stable CL baseline, slices were made hypoxic for 15 min by switching to a perfusion medium with a Pao2 < 15 mM Hg but otherwise unchanged composition. CL was recorded continuously during hypoxia and 2 h of reoxygenation.
Fluorometric NADH measurement
A pulsed nitrogen laser (λexc = 337 nm, 30 μJ) was used to induce fluorescence. High spatial resolution for excitation and detection was obtained by coupling the laser light into an optical quartz fiber (diameter 200 μm), which leads the excitation light onto the probe and the fluorescence light back to the detector. A dichroic mirror separates the scattered excitation light and the fluorescence light. A narrow band-pass filter, centered at 460 nm (maximum of NADH fluorescence), is used for the spectral resolution. A photomultiplier is used as detector, and an electronic gate provides the time resolution of the signal. Data acquisition is controlled by a standard PC. All results are displayed on-line via the monitor. The optical fiber was mounted to a microprocessor-controlled positioning system. This enabled repeated positioning at selected spots in the CA1 and CA3 pyramidal cell layer with high regional selectivity. Further details of method have been provided by Riepe et al. (1996b).
Statistics
Statistical testing was performed by analysis of variance and Tukey's protected t test. Statistical significance was accepted at p < 0.05.
RESULTS
Histology
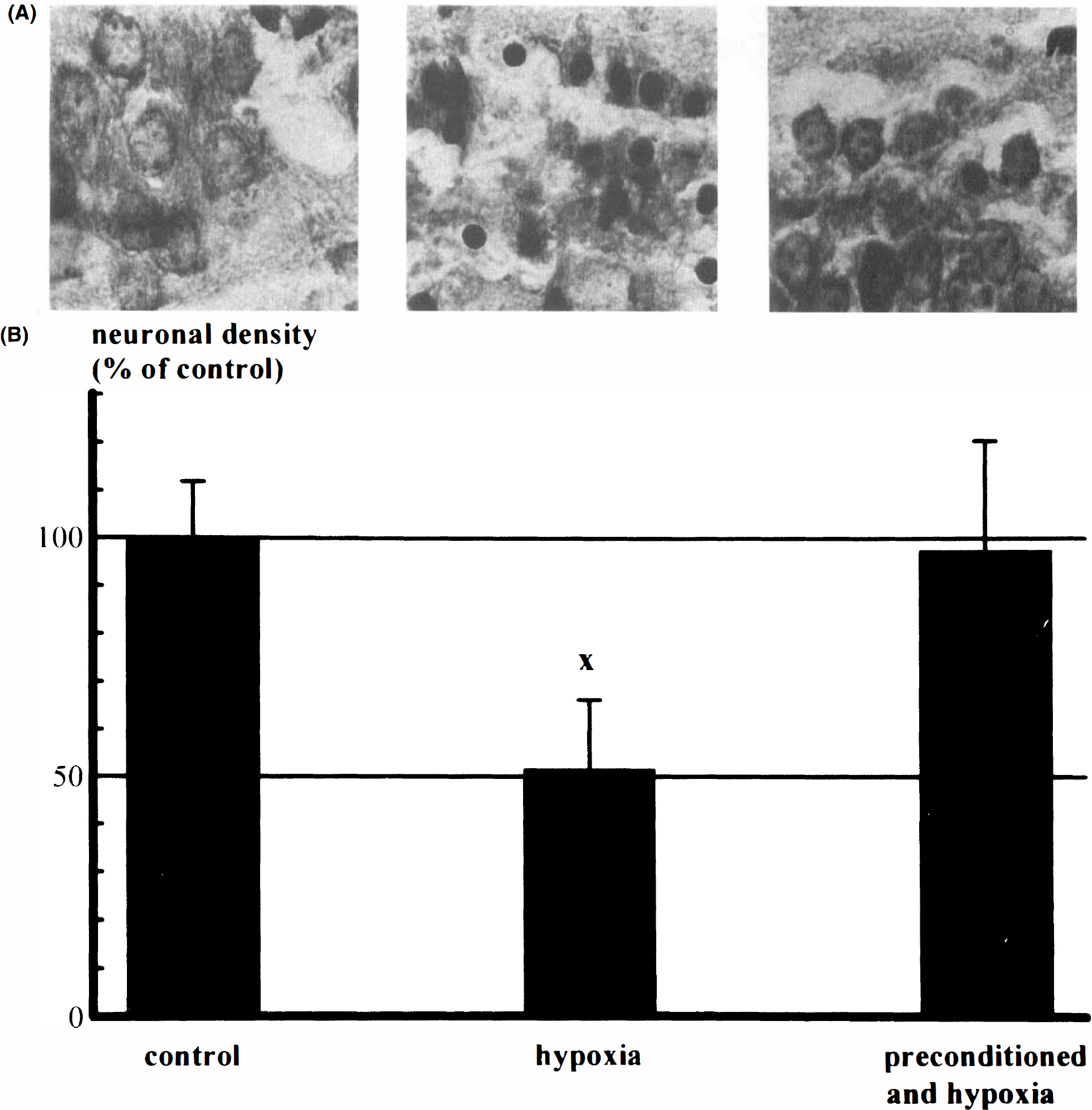
Hippocampal slices were prepared from controls (control slices) and animals pretreated in vivo with 20 mg/kg 3-np at 1–24 h prior to slice preparation (preconditioned slices). The time to onset of in vitro hypoxia was at least 2 h, at most 6 h after preparation of slices. With this treatment, morphology upon 15 min of hypoxia in vitro is preserved in region CA1 (Fig. 2A). Compared with untreated control slices (n = 10), neuronal density after 15-min hypoxia (n = 10) decreased to 52 ± 15% (mean ± SD; p < 0.001 vs. untreated control slices). In preconditioned slices (n = 10), neuronal density after 15-min hypoxia was maintained at 97 ± 23% (p < 0.001 vs. hypoxia alone, no significant difference from controls) (Fig. 2B).
Population spikes upon in vitro anoxia
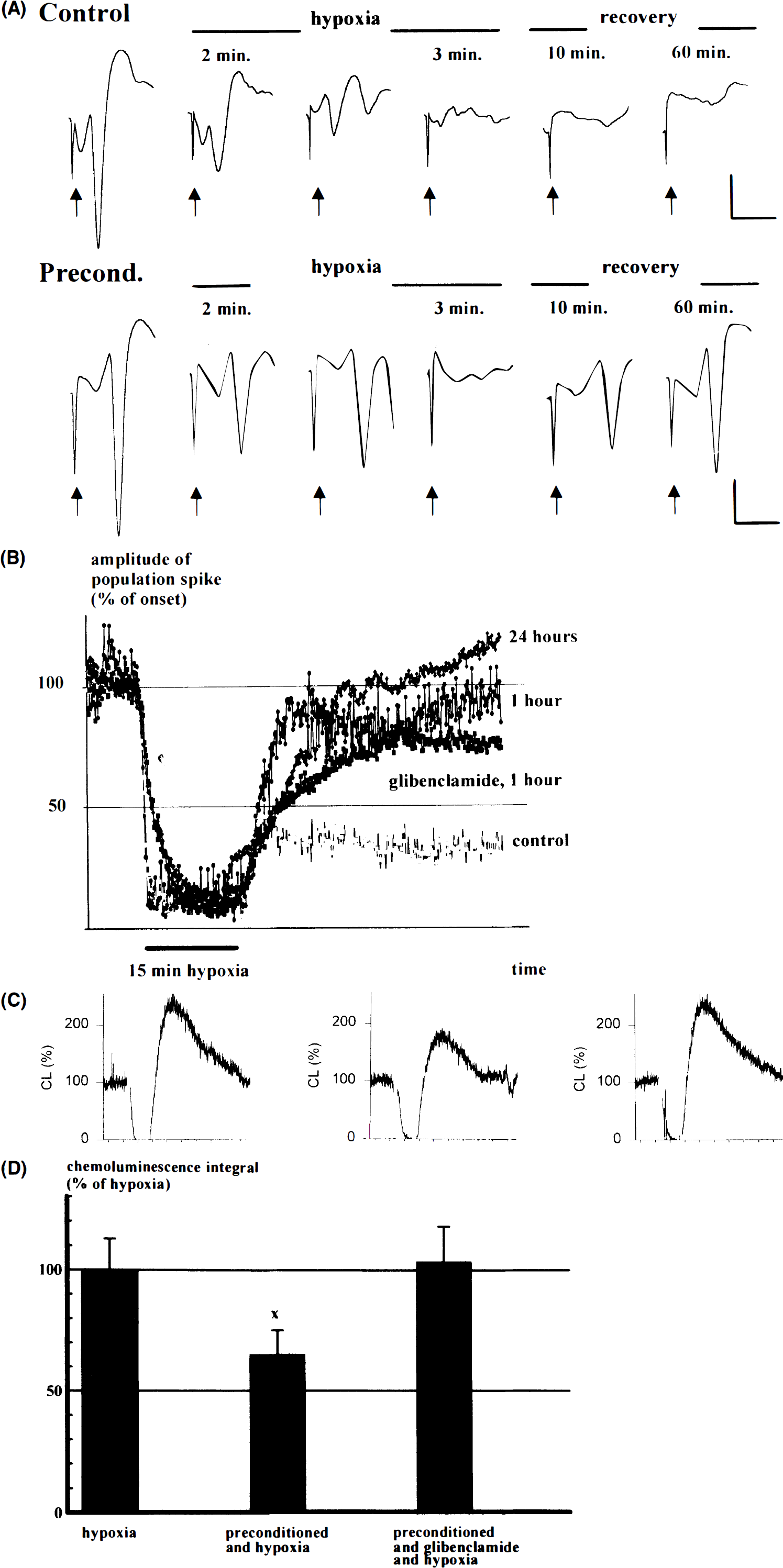
The psap in hippocampal region CA1 upon 15-min hypoxia in vitro was preserved better in preconditioned slices (Fig. 3A). A detailed time course of psap is shown in Fig. 3B. Ten minutes into hypoxia, the amplitude of the population spike already had decreased to 14 ± 7% (mean ± SD; n = 9) in control slices, while it was 14 ± 7% (n = 5) in preconditioned slices (time interval between in vivo treatment and slice preparation: 1 h). At the end of hypoxia (at 15 min), psap was 11 ± 4% in control slices vs. 14 ± 7% in preconditioned slices. After 15 min of recovery, psap was 37 ± 31% in control slices and 91 ± 20% in preconditioned slices. A similar difference of psap was observed after 45 min of recovery (31 ± 27% in control slices vs. 90 ± 15% in preconditioned slices). With a time interval of 24 h between in vivo pretreatment, psap 10 min into hypoxia was 18 ± 16% (n = 5). At the end of hypoxia, psap was 14 ± 12%. After 15 min of recovery, psap was 62 ± 28%, and after 45 min it was 107 ± 16%.
Oxygen free radicals upon in vitro anoxia
One possible toxic mechanism associated with postischemic tissue damage is increase of production of oxygen free radicals (Siesjö and Smith, 1991). Lucigenin-enhanced CL, an indicator of oxygen free radical production, was reduced in preconditioned slices during the initial 60 min of reoxygenation after 15 min of hypoxia to 65 ± 10% (mean ± SD; n = 5) of control slices (p < 0.05) (Fig. 3C and D).
Antagonism to preconditioning by glibenclamide
When the preconditioned slices were perfused with Ringer solution containing 5 μM glibenclamide 10 min prior to onset of hypoxia, no change in psap was observed. psap amounted to 9 ± 5% (mean ± SE; n = 7) at the end of hypoxia. Recovery was delayed. psap was 54 ± 31% after 15 min of recovery (p < 0.05 vs. preconditioned slices without additional treatment with glibenclamide) and 77 ± 25% after 45 min of posthypoxic recovery (Fig. 3B). Similarly, reversal of the decrease in posthypoxic oxygen free radical production was observed (104 ± 14%, mean ± SD, NS from control, p < 0.05 vs. preconditioning alone; n = 5). A similar trend was observed for the protective effect on morphologic lesions. However, while neuronal density was lower in preconditioned slices treated additionally with glibenclamide (88 ± 18%, mean ± SD; n = 10), this result did not reach levels of significance. ATP content of preconditioned slices upon hypoxia was not different with and without additional treatment with glibenclamide (data not shown).
Preconditioning against selective chemical inhibition of mitochondrial complex IV
Preconditioning following in vivo administration of 3-np could also be shown to protect against subsequent severe chemical hypoxia by cyanide in vitro. By fluorometric measurement of NADH (Riepe et al., 1996b), a preconditioning effect on energy metabolism could be observed in both hippocampal region CA1 and hippocampal region CA3 (Fig. 4). After 10 min of perfusion with 1 mM cyanide in vitro, NADH fluorescence increased to 111 ± 9% (mean ± SD; n = 10) in hippocampal region CA3 of preconditioned slices and to 127 ± 23% in control slices (p < 0.05; n = 10). In hippocampal region CA1, an increase to 123 ± 17% in control slices and to 108 ± 6% in preconditioned slices was observed (p < 0.05). In control slices, NADH fluorescence after 10 min of 1 mM cyanide was already maximal, while in preconditioned slices the maximum was reached not before 20 min of continued application of cyanide. At this time, the fluorescence was 120 ± 13% in CA1 and 114 ± 6% in CA3.
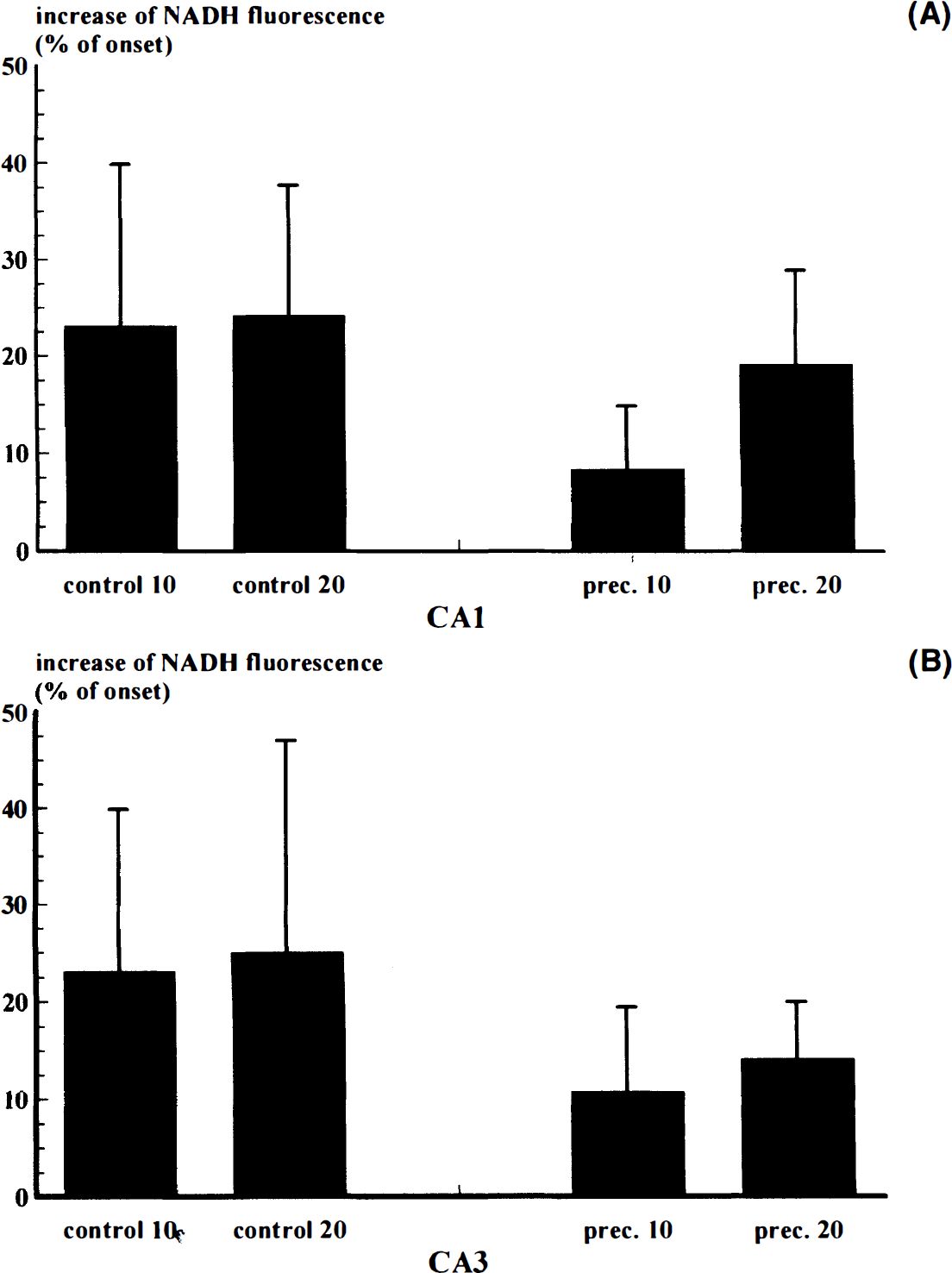
Increase of NADH fluorescence (% of onset) in the pyramidal cell layer of hippocampal regions CA1 (
DISCUSSION
A short ischemic or hypoxic episode is known to increase tolerance toward subsequent severe ischemia in heart and brain (Murry et al., 1986; Kitagawa et al., 1990). However, intermittent occlusion of cerebral arteries is an unlikely perspective for a practical neuroprotective strategy in humans. This study shows that a single intraperitoneal injection of a selective inhibitor of SDH (mitochondrial complex II) increases tolerance against subsequent hypoxia. Increased tolerance can be shown functionally and histologically. Similar to previous observations at an even higher dosage (Simpson and Isacson, 1993), the treatment itself does not impair pathological or gross clinical parameters in vivo. In analogy to ischemic preconditioning, we suggest the term “chemical preconditioning” for the induction of tolerance to massive disruption of energy metabolism by preceding inhibition of oxidative phosphorylation. With this paradigm, a protective effect of early onset and long duration can be shown. Previously, it was demonstrated by Heurteux et al. (1995) that preconditioning ischemia protects against lethal ischemia when applied with a time interval of 3 days, but worsens the damage when applied with a time interval of 1 h. Additionally, it was shown that pharmacologic induction of preconditioning with agonists at adenosine receptors, activating KATP-channels, has a protective effect when applied with a time interval of 15–30 min preceding lethal ischemia (Heurteaux et al., 1995). The present results indicate that chemical inhibition of oxidative phosphorylation with 3-np includes the characteristic of protection of long duration as observed after ischemic preconditioning in brain as well as the early-onset characteristic of pharmacologic preconditioning with adenosine agonists, activating KATP-channels. This is in good harmony with the observation that KATP-channels are activated upon application of 3-np (Riepe et al., 1992, 1996a). Increased tolerance by chemical preconditioning therefore has an early onset and is long-lasting.
Chemical preconditioning by selective inhibition of mitochondrial complex II does not act only by a change in succinate-related oxidation. As is shown by a delayed increase of NADH upon application of cyanide, chemical preconditioning also influences NADH-related oxidation [as expected, NADH does not increase upon prolonged application of 3-np in hippocampal slices (Riepe et al., 1996b)]. This could indicate facilitated adaptation to increased demand on preservation of energy production during disruption of oxidative metabolism in general. Alternatively, this could indicate decreased demand for maintenance of energy-requiring processes, in particular for restoration of ionic homeostasis upon synaptic activity. One mechanism that may participate in the latter is hyperpolarization due to activation of KATP-channels. The mechanism by which antagonists at KATP-channels interfere with preconditioning could be an accelerated neuronal depolarization upon inhibition of oxidative phosphorylation (Riepe et al., 1992). Indeed, it has been suggested previously that KATP-channels are involved in induction of chemical (Riepe et al., 1996a) and ischemic (Heurteaux et al., 1995) preconditioning in brain. However, incomplete reversal of increased tolerance by glibenclamide, an antagonist at KATP-channels, indicates that other mechanisms are involved in addition. Further, the present study gives additional evidence that protection against neuronal damage upon hypoxia is associated with a reduction of posthypoxic free oxygen radicals. This reduction in posthypoxic free oxygen radicals is reversed by additional treatment of slices with glibenclamide prior to hypoxia.
Further understanding of the mechanism(s) involved in chemical preconditioning is of great interest not only for development of a neuroprotective strategy, but also for understanding of the pathophysiologic cascade of excitatory agents in ischemic/hypoxic cell death. While aggravation of morphological damage is observed upon direct application of excitants after systemic pretreatment with 3-np (Simpson and Isacson, 1993), we demonstrated that neuronal death upon ischemia is reduced after chemical preconditioning. Possibly this again indicates that neurotoxicity of endogenous excitatory agents is not potentiated in situations of severely inhibited energy metabolism the more gradual or prolonged the onset of metabolic impairment is (Riepe et al., 1995). However, excitants may become toxic when the recovery mechanisms are impaired or overly stressed, e.g., by continuous or repeated application at short intervals of mitochondrial inhibitors (Brouillet et al., 1993) or repeated ischemic episodes without sufficient time in-between (Kato et al., 1991). A detailed understanding of beneficial and adverse conditions is necessary since several drugs widely applied in clinical practice interfere with mitochondrial function and energy metabolism. Last, clinical applicability of ischemic preconditioning has been demonstrated in coronary angioplasty (Tomai et al., 1994). While ischemic preconditioning by ligation of cerebral arteries supplying the brain is not possible for practical reasons, in vivo chemical preconditioning is practicable experimentally in animals and might also be in humans. Due to its early onset and long duration, chemical preconditioning might provide an opportunity for a new neuroprotective strategy in high-risk situations for ischemic stroke. In analogy to ischemic preconditioning (Chen et al., 1994), it also might be inducible repeatedly. It might even be speculated that chemical preconditioning unknowingly is in use already, since some pharmaceuticals with widespread therapeutic applications are inhibitors of mitochondrial energy metabolism.
We conclude that chemical preconditioning is a novel and practical strategy of neuroprotection against otherwise fatal inhibition of energy metabolism with early onset and long duration. Chemical preconditioning reduces posthypoxic oxygen free radicals. Activation of KATP-channels participates in mediating the protective effect by mechanisms other than interference with intracellular ATP content. Ultimately, chemical preconditioning may be a novel useful neuroprotective strategy in diseases with increased risk for cerebral ischemia.
Footnotes
Acknowledgment:
The work was supported by grants from the Deutsche Forschungsgemeinschaft to M.W.R and A.C.L. We thank Ms. A. Raupach for excellent technical assistance.