
Abstract
We recently demonstrated that closed head injury (CHI) in the rat triggers the production of tumor necrosis factor alpha (TNFα) in the contused hemisphere. Other investigations have shown that this cytokine plays a role in the inflammatory response following trauma. The present study was designed to determine whether inhibition of TNFα production or activity affects the development of cerebral edema as well as neurological dysfunction and hippocampal cell loss after CHI. To this end, we used two pharmacological agents, each acting via a different mechanism: pentoxifylline (PTX), which attenuates the production of TNFα, and tumor necrosis factor binding protein (TBP), a physiological inhibitor of TNFα activity. Both agents significantly lessened peak edema formation at 24 h and facilitated the recovery of motor function for ≤14 days postinjury. In addition, TBP attenuated disruption of the blood-brain barrier and protected hippocampal cells. PTX significantly lowered the brain TNFα level (by ∼80%), and TBP completely abolished the activity of recombinant human TNF when they were added at the same time in the in vitro bioassay. We suggest, therefore, that a decrease in TNFα level or the inhibition of its activity is accompanied by reduced brain damage.
Keywords
Tumor necrosis factor alpha (TNFα), a potent cytokine implicated in inflammation and immunity (Dinarello, 1988), is produced by activated macrophages (Gallily et al., 1989; Semenzato, 1990), as well as by astrocytes (Brenner et al., 1993). The role of cytokines in the cascade of injury has not been fully elucidated, although a recent review (Ott et al., 1994) describes changes in cytokine production and metabolic dysfunction after severe head injury. Cytokine levels appear to be augmented after head injury (McClain et al., 1991); the elevation is related to morbidity and mortality in a variety of diseases. Tracy and Cerami, in their review of the biology of TNFα, emphasize its potential as a therapeutic target due to its broad scope of injurious and beneficial effects (1994). A rat model of closed head injury (CHI) has been developed in our laboratory (Shapira et al., 1988), and we have described the activation of an inflammatory pathway, namely, phospholipase A2, and increased eicosanoid production (Shohami et al., 1987, 1989). We have demonstrated that following CHI in the rat, there is an increase in TNFα and interleukin-6 (IL-6) production in the cortical tissue adjacent to the site of injury (Shohami et al., 1994).
Since cytokine activity was identified as early as 1 h post-CHI, before infiltration of inflammatory cells is detectable (Shapira et al., 1988), the rise in cytokines has been attributed to increased cytokine production by resident brain cells (astrocytes and microglia) rather than to its release from invading monocytes. Assuming that elevated TNFa levels play a detrimental role in the pathophysiology of CHI, the present study was designed to determine the effects of TNFα inhibition on the development of cerebral edema as well as on neurological dysfunction and hippocampal cell loss after CHI. To this end, two pharmacological agents acting via different mechanisms were used.
Pentoxifylline (PTX), a methylxanthine, has been known for many years for its hemorrheological properties. This drug has been shown to prevent or attenuate the production of TNFα induced by bacterial lipopolysaccharides (LPS) both in vitro (Strieter et al., 1988) and in vivo (Zabel et al., 1989) at the transcriptional level (Doherty et al., 1991). TNFα binding protein (TBP) is a cysteine-rich glycoprotein identified and purified from human urine (Peetre et al., 1988; Seckinger et al., 1988; Engelmann et al., 1989). TBP corresponds to a soluble fragment of the cell-surface TNFα receptor (TNFα-R) generated by proteolytic cleavage of the extracellular binding domain (Nophar et al., 1990). It displays a sequence homology to the extracellular portions of the TNF-R p80 chain and to nerve growth factor. TBP is a physiological inhibitor of TNFα, which acts in vitro by competing with the cell-surface TNFα receptor. We hypothesized that if TNFα plays a part in the pathophysiology of CHI, TBP might counteract its effect and find potential use as a therapeutic agent in our CHI. Indeed, our results support the notion that TNFα is a harmful mediator in CHI, and its suppression by both agents correlates with improved recovery following brain injury.
MATERIALS AND METHODS
Closed head injury model
Male Sabra rats (Hebrew University strain) weighing 200–220 g were used. CHI was induced under ether anesthesia by a calibrated weight-drop device that falls over the exposed skull, covering the left hemisphere 1–2 mm lateral to the midline in the midcoronal plane, as described previously (Shapira et al., 1988). Rats were kept according to the institutional regulations of the Animal Care Committee.
Neurological evaluation
Rats were evaluated on the basis of clinical criteria, which include reflexes and motor functions, and were graded according to the neurological severity score (NSS) 1 h after CHI (Table 1). Rats with a score of 15–20 (>90% of the injured animals) were considered severely injured and were included in the study. At various times post-CHI (24 h to 2 weeks) the animals were reevaluated. The difference between the new score and that at 1 h was defined as the extent of recovery (ΔNSS) and was used to assess the neuroprotective properties of the drugs. The greater the difference, the better the recovery.
Neurological severity score

Evaluation of edema
At 24 h post-CHI, the rats were decapitated, and their brains were removed. Since we had shown that the region adjacent to the core of injury develops the highest degree of edema, samples of this region were taken to assess drug effectiveness. The water content in a ∼20-mg segment of cortical tissue adjacent to the site of injury was measured, using the dry/wet weight ratio, as described by Shapira et al. (1988).
Evaluation of BBB integrity
Blood-brain barrier (BBB) integrity was tested using the Evans Blue dye extravasation method. Evans Blue was injected i.v. 3 or 23 h after CHI, and 1 h later the animals were perfused with saline, their brains were removed, the dye was extracted, and its fluorescence was determined (Uyama et al., 1988).
TNFα determination
TNFα titer was determined by a cytotoxicity assay. Brain extracts were bioassayed as described previously (Sher et al., 1990, Shohami et al., 1994), using the BALB/c CL.7 line as target cells. Briefly, cells were plated in Dulbecco's Modified Eagle's Medium containing 5% fetal calf serum. After 24 h, threefold dilutions of the brain extracts were added to the target cells, followed by the addition of actinomycin D (Sigma, St. Louis, MO, U.S.A.). The cultures were incubated for 20 h, stained with 2% crystal violet, rinsed, and dried. Destruction of CL.7 cells was assessed by measuring the absorbance of the stained cells (550 nm) with an MR 700 microplate reader (Dynatech, Farmingdale, NY, U.S.A.).
The TNFα titer (S50), defined as the reciprocal of the dilution of test extract required to destroy 50% of the target cell monolayer, was determined. Various dilutions of a known concentration of recombinant TNFα were added to the target cells to quantify the S50, and the data were converted into picograms per milligram of tissue protein. The specificity of TNFα cytotoxicity was confirmed using anti-TNFα antibodies in the same assay, as has been described (Shohami et al., 1994). The capacity of TBP to neutralize TNFα activity was demonstrated in vitro by coadministration of recombinant human TNFα (rh-TNFα) and TBP in the bioassay.
Neuropathological analysis
Fourteen days post-CHI rats were reanesthetized with ether, and their brains were perfused transcardially with 4% formaldehyde. The brains were removed for histopathological evaluation, and 6-μm-thick coronal sections were cut using a microtome and stained with hematoxylin and eosin. Hippocampal damage was quantified under a light microscope (Nikon) by an uninformed observer by counting the number of viable and dead cells in the CA1–3 fields of the hippocampus.
Agents
PTX (Sigma) was used as a stock solution of 4 mg/ml in saline. Traumatized rats were injected i.v. with PTX (20 mg/kg) immediately after induction of CHI. TBP was the generous gift of Interpharm Laboratories Ltd. (Rehovot, Israel). A stock solution of 1 mg/ml saline was used, and a dose of 1 mg/kg was administered i.v. within 5 min of CHI. Recombinant human TNFα was obtained from Genetech Inc. (San Francisco, CA, U.S.A.).
Experimental protocol
Rats were subjected to CHI and treated with the tested agent or its vehicle immediately after induction of trauma. NSS was assessed 1 h later, and the animals were returned to their cages. NSS was reevaluated at 24, 48, and 72 h and at 7 and 14 days, and ΔNSS was calculated at each time point. Other groups of rats were decapitated 24 h post-CHI for determination of water content or at 4 or 24 h for evaluation of BBB integrity. For measuring TNFα levels, groups of rats were traumatized in parallel, treated with the different agents, and decapitated 4 h post-CHI, when TNFα accumulation peaks (Shohami et al., 1994).
To further assess the role of endogenous TNFα in the pathophysiology of CHI, a group of rats were injected intracerebrally, into the right lateral ventricle, with 24 μg of rh-TNFα (in 30 μl of saline) 5 min before CHI. Within 5 min of CHI the rats were injected i.v. with PTX, and their NSS and water content were evaluated 24 h later.
Statistical analysis
Percentage water content, TNFα levels, and percentage cell loss are presented as the mean ± SD and compared using the two-tailed Student's t test. Clinical recovery, expressed as ΔNSS, is shown as the median (range) and compared by the two tailed nonparametric Mann Whitney test. The level of significance was p < 0.05.
RESULTS
Effect of PTX
We first determined whether PTX attenuates TNFα production in our model of CHI. Cortical tissue of the contused hemisphere 4 h post-CHI contained 2.16 ± 1.89 ng TNFα/mg protein in the control traumatized rats, whereas the nontraumatized rats had undetectable levels of the cytokine. The TNFα level was significantly suppressed after PTX treatment: 0.44 ± 0.39 ng/mg protein (p = 0.025, n = 8 rats per group).
Brain tissue water content was determined at 24 h, the time of peak edema formation in our CHI model. As can be seen in Fig. 1A, the PTX-treated rats had a significantly lower water content than did the controls (83.1 ± 1.13 versus 84.7 ± 1.33%, p = 0.0125). We then addressed the functional state of the traumatized rats and compared the ΔNSS of control and PTX-treated rats for 14 days postinjury. NSS was assessed at 24, 48, and 72 h as well as 1 and 2 weeks post-CHI. At each of these time points, the ΔNSS of the PTX-treated rats was significantly higher than that of the control, untreated CHI rats, indicating better recovery (Fig. 1B).
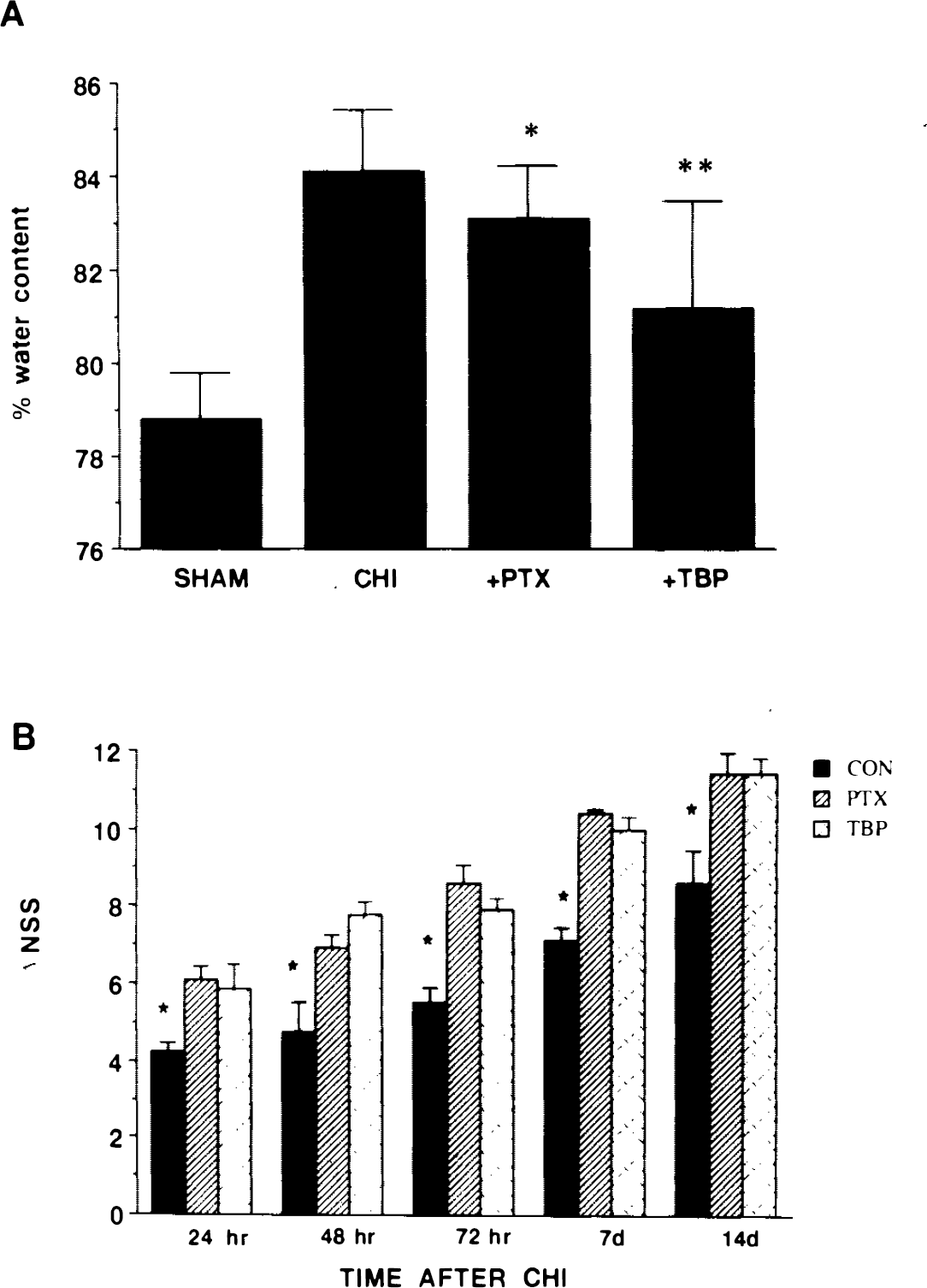
Effect of drug therapy on edema formation
The effect of PTX on BBB integrity was evaluated at 4 h and 24 h, according to the amount of Evans Blue accumulated in the brain. At 4 h post-CHI, in the contused hemisphere of the PTX-treated rats, there was 75 ± 17.9 ng dye/mg tissue (n = 7), compared with 117 ± 22.6 (n = 7) in the controls (this difference did not reach significance). At 24 h post-CHI the level of dye was identical in the two groups (229 ± 131 and 233 ± 90 ng dye/mg tissue in the PTX-treated and untreated rats, respectively).
We next tested whether rh-TNFα injected before CHI would reverse the protective effect of PTX given 5 min after injury. Indeed, the ΔNSS of the PTX-treated rats at 24 h was 7 (range 5–9, n = 8), while that of the traumatized rats treated with rh-TNFα and PTX was 6 (range 5–7, n = 9), p = 0.02, Mann Whitney test. This value was not significantly different from that obtained for the control (traumatized, untreated) rats, indicating that rh-TNFα reversed the beneficial effect of PTX on the clinical outcome. In contrast, the water content of these two groups was similar and significantly lower than that of the control, indicating that rh-TNFα did not abolish the effect of PTX on edema.
The effect exerted by PTX on neurons in the hippocampal CA2 and CA3 regions was assessed by counting the number of viable cells following treatment with this agent. In CA2: 23 ± 16.7% dead cells were found in the controls vs 10.8 ± 18.3% in the PTX-treated rats. In CA3: cell death was 32.7 ± 18.9% in the control non-treated rats (n = 5), vs 20.5 ± 14.5% in the PTX-treated rats (n = 8). The differences were not significant.
Effect of TBP
The effect of TBP on edema formation and functional recovery (ΔNSS) after CHI is evident in Fig. 1. Less water accumulated at the site of injury in the TBP-treated rats compared with that in untreated rats after a similar degree of injury (81.16 ± 2.32, n = 11, versus 84.17 ± 1.33%, n = 9; p = 0.007, two-tailed t test; Fig. 1A). In addition, recovery of motor functions was significantly facilitated in the TBP-treated rats during the 2-week follow-up (Fig. 1B). At all time points, the ΔNSS of the TBP-treated rats was significantly higher (p < 0.05) than that of the controls, indicating better recovery of motor functions. The effect of TBP on the disruption of the BBB was examined at 4 and at 24 h post-CHI. Whereas at 4 h the levels of Evans Blue in the TBP-treated rats were similar to those in the control, at 24 h, they were significantly lower: 76.6 ± 42.8 (n = 6) versus 233.4 ± 117.9 (n = 9, p < 0.01).
Examining cell viability in the hippocampus showed that there was significantly less cell damage in the CA2 and CA3 regions of the TBP-treated rats (n = 7) compared with that in the untreated rats (n = 5). In the CA2 it was 3.42 ± 1.44 versus 23.02 ± 16.7% (p < 0.05), and in the CA3 it was 14.5 ± 10.4% versus 32.7 ± 18.9% (p = 0.056, marginally significant). The protective effect of TBP on hippocampal cells is shown in Fig. 2.
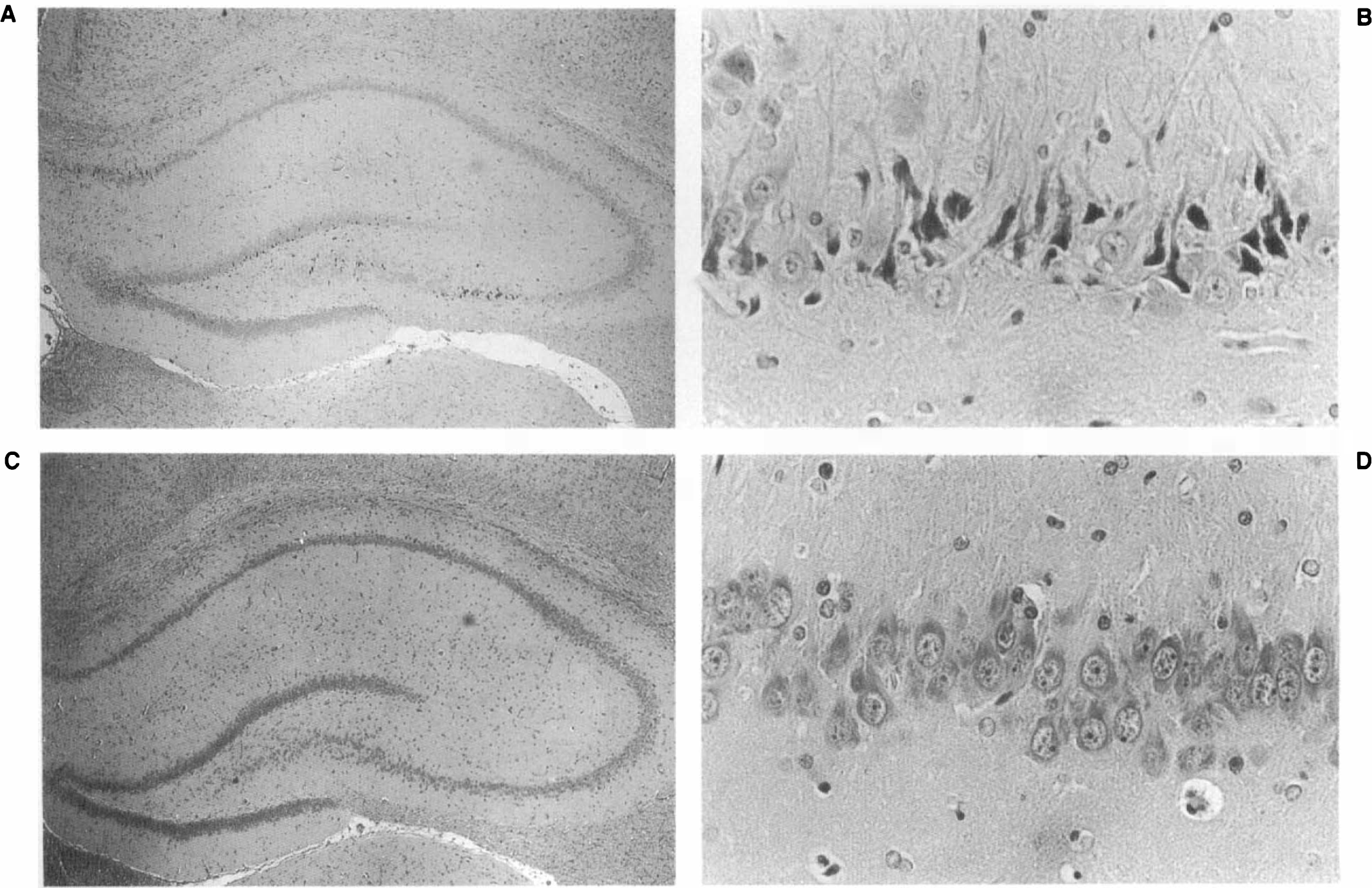
Illustration of neuroprotection of TBP (1 mg/kg i.v., administered within 5 min of CHI) in the hippocampus. Rats were killed 14 days post-CHI, and brain sections (5 μm thick) were stained with H&E. Vehicle-treated rats:
In an attempt to verify that TBP treatment indeed reduced brain TNFα activity, brain extracts were prepared from the traumatized, TBP-treated rats and tested in the bioassay. The results show that there was no reduction in TNFα activity in the brain homogenates of the treated rats (S50 of 61 ± 35, n = 12, in the control, untreated rats, versus 151 ± 106, n = 7, in the TBP-treated rats, which was not significant). However, the activity was totally abolished when TBP was added in vitro along with rh-TNFα (S50 of 1,919 for the rh-TNFα, and 0 for the rh-TNFα added together with TBP).
DISCUSSION
The results of the present study support the notion that TNFα mediates at least some of the harmful events occurring following traumatic brain injury. Indeed, PTX reduced the early and transient surge of TNFα activity in the brain following CHI. This finding concurs with earlier reports by a number of investigators regarding the drug's mechanism of action (Strieter et al., 1988; Doherty et al., 1991). We showed that PTX also ameliorated motor dysfunction, even as late as 14 days postinjury, and reduced brain edema at 24 h, the time of maximal edema (Shapira et al., 1988). We, therefore, suggest that the low level of TNFα is associated with the cerebroprotection observed under these conditions. We previously showed that there is a transient increase in TNFα production after CHI, starting at 1 h, peaking at 4 h, and terminating at ∼12 h postinjury (Shohami et al. 1994).
In the present study, PTX was administered only once, immediately after CHI, whereas the protective effect was evident even as late as 2 weeks postinjury. It is conceivable that the drug, by interfering with the production of a harmful mediator, prevents progressive damage. TNFα may affect a cascade of other mediators (e.g., IL-6) during the very early post-CHI stages (≤6 h). Reversal of the PTX effect on clinical recovery, achieved by coadministration of rh-TNFα, strongly supports the role of TNFα in mediating motor impairment occurring after injury. Although TNFα reportedly damages cerebral endothelial cells (Terada et al., 1992) and increases BBB permeability (Megyeri et al., 1992), i.c.v. administration of rh-TNFα did not abolish the effect of PTX on brain edema in our model. This finding implies that edema formation may be mediated by various mechanisms in which other mediators (cytokines, free radicals, eicosanoids, etc.) are involved. This notion is supported by our present finding that PTX is not a potent inhibitor of BBB impairment, i.e., its effect on water content may be related to cytotoxic rather than to vasogenic edema.
One may also speculate that the effect of PTX is not specific to TNFα suppression and that it can affect other cytokines. Parhar et al. (1993) have found that LPS-induced cytokine (IL-1α, IL-2, IL-6, TNFα, and gamma interferon) as well as surface receptor expression (IL-1α, IL-2, and TNFα) were affected by PTX. Our results agree with those of Quagliarello et al. (1991), who studied the influence of IL-1 and TNFα on BBB permeability. They showed that the two cytokines acted synergistically to disrupt BBB function, whereas TNFα alone was less effective as an independent inducer of BBB injury. One could, therefore, speculate that in our model of CHI, the production of various cytokines is triggered, leading to a deleterious additive effect in the traumatized rat.
A more direct pharmacological approach to overcoming the negative effects of various cytokines is neutralization of these properties by specific antibodies or by soluble receptors. This strategy has been successfully applied by Rothwell and colleagues, who showed that the antagonist of the IL-1 receptor inhibited neuronal death in a model of focal cerebral ischemia (Relton and Rothwell, 1992) and fluid percussion (Toulmond and Rothwell, 1995). In animal models of endotoxic and septic shock, several investigators have shown that development of shock and tissue injury could be prevented by anti-TNF antibodies (Tracy et al., 1987; Hinshaw et al., 1989; Emerson et al., 1992).
In the present study we treated the traumatized rats with TBP within 1 h of CHI and found reduced edema and BBB impairment at 24 h, a significant amelioration of neurological dysfunction =≤14 days post-CHI, and protection of hippocampal neurons. TBP, a specific anti-TNFα compound, acts as a soluble fragment of the cell-surface TNFα receptor and binds this cytokine, thus neutralizing its activity. TBP, a natural molecule that interferes with TNFα activity, prevents the interaction of the latter with the target cell. The apparent ineffectiveness of TBP in the bioassay of brain extracts of TBP-treated rats may be attributed to the fact that the brain homogenates were prepared by sonicating the brain tissue. This procedure may have dissociated the complex and liberated the free TNFα that had accumulated in the brain after injury, yielding the high activity observed in the bioassay.
In contrast to the PTX-treated brains, where TNFα production was inhibited, in the TBP-treated rats, TNFα was produced in response to injury, and was probably neutralized thereafter, resulting in the improved in vivo outcome. However, the complex TNF-TBP may be unstable under the extraction conditions, leading to the release of free TNFα and the expression of its activity in the in vitro assay system. Indeed, upon co-administration of rh-TNFα and TBP to the target cells, there was a loss of activity. We therefore assume that TBP acts to neutralize the direct or indirect actions of TNFα in the brain.
In their study of the pharmacokinetics and tissue distribution of TBP in mice, Gascon et al. (1992) found that only a minor fraction of the i.v.-injected TBP was present in the brain, probably owing to its inability to cross the BBB. In our model of CHI, there is early impairment of the BBB, which may facilitate the penetration of nonpermeable molecules and could account for the observed effect of TBP on brain function. Our results, showing the ameliorative effects of PTX and TBP on CHI outcome, could be interpreted as showing the involvement of TNFα, and probably of other cytokines activated by TNFα, in the pathophysiology of brain injury. If so, neutralization of its deleterious effects could be a novel approach to the treatment of CHI patients.
Footnotes
Acknowledgment:
This study was partly supported by the Israel Ministry of Absorption and by the David R. Bloom Center for Pharmacy. We gratefully acknowledge the support of the Friends of the Lautenberg Center and the Concern Foundations of Los Angeles. We appreciate the skillful assistance of Semion Breiterman Ph.D., from the Department of Experimental Surgery, Hadassah Medical Center.