
Abstract
Cerebral blood flow (CBF) rises when the glucose supply to the brain is limited by hypoglycemia or glucose metabolism is inhibited by pharmacological doses of 2-deoxyglucose (DG). The present studies in unanesthetized rats with insulin-induced hypoglycemia show that the increases in CBF, measured with the [14C]iodoantipyrine method, are relatively small until arterial plasma glucose levels fall to 2.5 to 3.0 mM, at which point CBF rises sharply. A direct effect of insulin on CBF was excluded; insulin administered under euglycemic conditions maintained by glucose injections had no effects on CBF. Insulin administration raised plasma lactate levels and decreased plasma K+ and HCO3– concentrations and arterial pH. These could not, however, be related to the increased CBF because insulin under euglycemic conditions had similar effects without affecting CBF; furthermore, the inhibition of brain glucose metabolism with pharmacological doses (200 mg/kg intravenously) of DG increased CBF, just like insulin hypoglycemia, without altering plasma lactate and K+ levels and arterial blood gas tensions and pH. Nitric oxide also does not appear to mediate the increases in CBF. Chronic blockade of nitric oxide synthase activity by twice daily i.p. injections of NG-nitro-L-arginine methyl ester for 4 days or acutely by a single i.v. injection raised arterial blood pressure and lowered CBF in normoglycemic, hypoglycemic, and DG-treated rats but did not significantly reduce the increases in CBF due to insulin-induced hypoglycemia (arterial plasma glucose levels, 2.5-3 mM) or pharmacological doses of deoxyglucose.
A number of studies have examined the effects of insulin-induced hypoglycemia on cerebral blood flow (CBF) with somewhat disparate results. Some reported small or no changes (Kety et al., 1948; Eisenberg and Seltzer, 1962; Hernandez et al., 1980; Ghajar et al., 1982), but most found significant increases in CBF (Della Porta et al., 1964; Norberg and Siesjö, 1976; Abdul-Rahman et al., 1980; Hollinger and Bryan, 1987; Bryan et al., 1987; Ichord et al., 1994). The discrepant results may have been due to species differences, the presence or absence of anesthesia, the nutritional state of the animals, the level and duration of hypoglycemia, or differences in the methods employed to determine CBF. In all studies in which it was determined, cerebral energy metabolism, for example, cerebral oxygen consumption (CMRO2) or glucose utilization (CMRglc), was reduced during insulin-induced hypoglycemia, the number of structures affected, and the extent of reductions in metabolism varying with the severity of the hypoglycemia (Kety et al., 1948; Eisenberg and Seltzer, 1962; Norbert and Siesjö, 1976; Ratcheson et al., 1981; Ghajar et al., 1982; Suda et al., 1990).
The mechanism responsible for the increased blood flow is unknown, but it is of interest that pharmacological doses of 2-deoxyglucose, which produce clinical effects like those of insulin-induced hypoglycemia despite elevated blood glucose levels (Landau et al., 1958), also cause marked increases in local CBF throughout the brain in unanesthetized animals (Breier et al., 1993). This would suggest that neither direct action of insulin nor the blood glucose level per se is responsible.
A vasodilator agent derived from vascular endothelium, the endothelium-derived relaxing factor (EDRF), has been shown to mediate the vasodilator effects of acetylcholine (Furchgott and Zawadzki, 1980). This factor has been identified as nitric oxide (NO) (Palmer et al., 1987; Ignarro et al., 1987). Nitric oxide is enzymatically produced in brain tissue and vascular endothelium, and it mediates the effects of a number of vasodilator drugs and plays a role in the regulation of systemic blood pressure and vascular tone (Ignarro, 1989; Moncada et al., 1991). It also provides a tonic vasodilator influence on the cerebral circulation; inhibition of its synthesis reduces cerebral blood flow throughout the brain despite an elevation in mean arterial blood pressure (MABP) (Sokoloff et al., 1992; Adachi et al., 1994). Inhibition of NO synthase activity has also recently been reported to attenuate the increases in CBF during insulin-induced hypoglycemia in anesthetized, mechanically ventilated piglets, suggesting that NO plays a role in mediating the cerebrovascular response to hypoglycemia (Ichord et al., 1994).
In the present studies, we examined the effects of insulin-induced hypoglycemia and pharmacological doses of 2-deoxyglucose on CBF. Both modes of producing cerebral glucoprivation result in increases in CBF, despite major differences in systemic effects. By comparing their effects, we have been able to eliminate several possible mechanisms responsible for the cerebral vasodilator effects, for example, a direct action of insulin as well as some of the systemic effects of insulin, including those on blood constituents. We have also examined and found no evidence to support a role for nitric oxide in the mediation of the increases in CBF during glucoprivation.
MATERIALS AND METHODS
Chemicals
Chemicals were purchased from the following sources:
Animals
All procedures performed on animals were in strict accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and were approved by the local animal care and use committee. Normal adult male Sprague-Dawley rats weighing 270 to 380 g were obtained from Taconic Farms (Germantown, NY, U.S.A.) and maintained in a climate-controlled room on a normal 12 h light/dark cycle with food and water available ad libitum. The rats were fasted but allowed free access to water for about 16 h immediately before the experiment. On the day of the experiment, the rat was anesthetized with halothane (5% for induction and 1.0–1.5% for maintenance) in 70% N2O/30% O2, and polyethylene catheters (PE 50, Clay-Adams, Parsippany, NJ, U.S.A.) were inserted into both femoral arteries and one femoral vein. One arterial catheter was used for continuous recording of arterial blood pressure, and the other was standardized at exactly 16 cm long and used for sampling of arterial blood; the venous catheter was used for injection of agents and tracers. A loose-fitting plaster cast was then applied to the lower abdomen and hips and taped to a lead brick to prevent locomotion. Body temperature was continuously monitored by a rectal probe and maintained at 37°C by thermostatically controlled heating lamps. At least 3 h were then allowed for recovery from the effects of the surgery and anesthesia before continuation of the experimental procedure.
Physiological variables
We monitored MABP with a blood-pressure analyzer (Micro-Med, Inc., Louisville, KY, U.S.A.) that had been calibrated with an air-damped mercury manometer. Arterial blood Pa
Measurement of local cerebral blood flow
Local CBF was determined by the autoradiographic [14C] iodoantipyrine method as previously described (Sakurada et al., 1978), modified only by the use of a programmed i.v. infusion to obtain a constantly rising tracer concentration in the arterial blood (i.e., ramp input function). Local CBF was computed by means of the operational equation of the [14C]IAP method with appropriate corrections for the lag and washout of the dead space in the arterial sampling catheter (Frerichs et al., 1994); arterial catheter flow rates per minute were adjusted to equal at least 40 dead space volumes per minute to minimize the magnitude of the corrections. Average rates of blood flow for the brain as a whole, weighted appropriately for the relative sizes of its component structures, were determined from the individual rates of blood flow, pixel by pixel, with the computer program developed by G. Mies (Max Planck Institut für Neurologische Forschung, Köln, FRG) for use with the Macintosh personal computer and Image 1.08 program.
Experimental procedures
The experiments were designed to characterize and examine the possible mechanisms of the effects of glucoprivation on CBF induced by insulin and also by pharmacological doses of DG. Therefore, in all the experiments, a variety of physiological variables appropriate to the condition being studied were examined 3 h after recovery from the anesthesia, at various times during the establishment of the glucoprivic state, and always immediately before the determination of local CBF. In all cases, arterial plasma glucose concentration and arterial blood gas tensions and pH were measured at 15-min intervals after administration of either insulin or DG and again immediately before the procedure for measuring CBF.
Effects of insulin-induced hypoglycemia on CBF.
Thirty rats were given i.v. injections of 0.125 to 15 U/kg insulin to achieve a range of levels of hypoglycemia; 15 control rats were administered comparable volumes of physiological saline alone. The procedure for measuring CBF was carried out 30 min after the insulin or saline administration. The dosage of insulin and the timing of the measurement of CBF after its administration were chosen on the basis of the results of preliminary experiments to establish the conditions needed to lower arterial plasma glucose concentrations into the 2 to 6 mM range without producing seizures, coma, or significant changes in arterial blood gas tensions and pH that could influence CBF.
Effects of insulin on CBF in euglycemic rats.
In seven rats, insulin (10 U/kg), immediately followed by 2.5 ml of a 10% (wt/vol) solution of glucose, was injected intravenously and this was usually sufficient to maintain euglycemia, but in a few rats it was necessary to inject an additional 1 ml of the 10% glucose solution to keep the plasma level from falling below 5.5 mM. Seven control rats for this group received no insulin but about the same volume of physiological saline in place of the insulin and 10% glucose solution. The procedure for measuring CBF was initiated 30 min after the administration of the insulin or saline.
Effects of pharmacological doses of 2-deoxyglucose on CBF.
Six rats were given 200 mg/kg of a 10% (wt/vol) aqueous solution of DG intravenously; six control rats were given equivalent volumes of physiological saline. CBF was determined 15 min after the injections.
Effects of inhibition of nitric oxide synthase on the CBF response to glucoprivation.
To examine the possibility that NO was involved in the mechanism of the cerebrovascular response to glucoprivation, the effects of acute and chronic inhibition of NO synthase activity on the changes in CBF elicited by glucoprivation were examined. For chronic inhibition, the rats were injected intraperitoneally twice daily for 4 consecutive days with 50 mg/kg of L-NAME. This regimen was reported to inhibit NO synthase activity in the brain almost completely (Dwyer et al., 1991), and we have confirmed that it inhibits brain NO synthase activity by at least 85% (Adachi et al., 1994). Twenty-five rats pretreated in this manner received i.v. injections of 0.25 to 20 U/kg of insulin 16 to 18 h after the last dose of L-NAME, and CBF was measured 30 min after the insulin injection. Twenty-two rats, pretreated similarly with saline instead of L-NAME, also made hypoglycemic with insulin as described above, served as controls for the effects of L-NAME in the presence of insulin-hypoglycemia.
Five rats chronically treated with L-NAME as described above were injected with pharmacological doses of i.v. DG (200 mg/kg) 16 to 18 h after the last dose of L-NAME, and CBF was measured 15 min after DG administration. Six rats similarly pretreated with L-NAME but given saline instead of DG were studied as controls for the effects of DG in the L-NAME-treated rats. Six rats, pretreated with saline instead of L-NAME and given DG and six rats pretreated with saline and given saline instead of DG were studied as controls for the effects of DG in the absence of L-NAME pretreatment.
For acute inhibition of NO synthase activity, six rats were infused intravenously with a dose of 30 mg/kg of L-NAME in 1.2 to 1.5 ml of normal saline over a 1- to 1.5-min period. Six control rats received similar infusions of normal saline alone. Fifteen minutes after the L-NAME infusion, 10 to 20 U of insulin were administered intravenously to three of the rats and saline to the other three, and arterial plasma glucose concentration and MABP were monitored. Similar experiments were carried out in six other rats, three administered 200 mg/kg of DG 30 min after L-NAME administration and three given saline instead of DG; CBF was measured 15 min later. In accordance with the reported time-dependence of the effect of L-NAME on NO synthase activity (Iadecola et al., 1994), the measurement of CBF was initiated 45 min after the L-NAME administration when the degree of inhibition is close to its maximum. In all cases, MABP had reached a maximum at 10 to 15 min after the L-NAME administration and remained close to that level at the time of the measurement of CBF.
Effects of insulin-induced hypoglycemia and deoxyglucose-loading on the EEG.
It was not feasible to obtain EEG tracings in the rats prepared for the determination of CBF. Therefore, EEG recordings were made in separate groups of rats. The EEG was recorded in one group of four rats made hypoglycemic with insulin like those in which CBF was determined, (i.e., 2.1 to 5.6 mM glucose in arterial plasma) and in another two rats given pharmacological doses of DG (200 mg/kg). These rats had been previously prepared by the placement of brass screws biparietally in the calvarium that served as electrodes for bipolar recording.
RESULTS
Effects of insulin-induced hypoglycemia and deoxyglucose loading on EEG
In the four animals made hypoglycemic by insulin, the EEG pattern was monitored continuously from the time of injection of the insulin until the arterial plasma glucose level had reached 2.0 mM. Thirty minutes after injection of the insulin, when the procedure for measuring CBF was ordinarily initiated, the EEG pattern was either unchanged from that before the insulin administration or displayed minimal slowing. All the rats exhibited progressive reductions in spontaneous activity with increasing hypoglycemia, but they all remained responsive to tactile and auditory stimuli. The administration of 200 mg/kg of DG intravenously also produced little if any change in the EEG; there was some reduction in spontaneous activity with preservation of responses to tactile and auditory stimulation.
Effects of insulin-induced hypoglycemia on physiological variables and CBF
Insulin-treated rats with hypoglycemia in the range of 2.6 to 5.6 mM arterial plasma glucose levels showed reduced arterial blood pH and plasma K+ concentrations and a rise in arterial plasma lactate concentration (p < 0.05, analysis of variance [ANOVA] and Dunnett's t test) (Table 1). Further reductions in plasma pH and K+ concentration and increases in lactate concentration occurred in more severe hypoglycemia (2.1–2.5 mM glucose) (p < 0.01, ANOVA and Dunnett's t test). Arterial HCO3– concentrations were statistically significantly reduced by hypoglycemia but only by about 4% from control levels (Table 1). Arterial P
Effects of insulin on physiological variables measured just before CBF measurement a
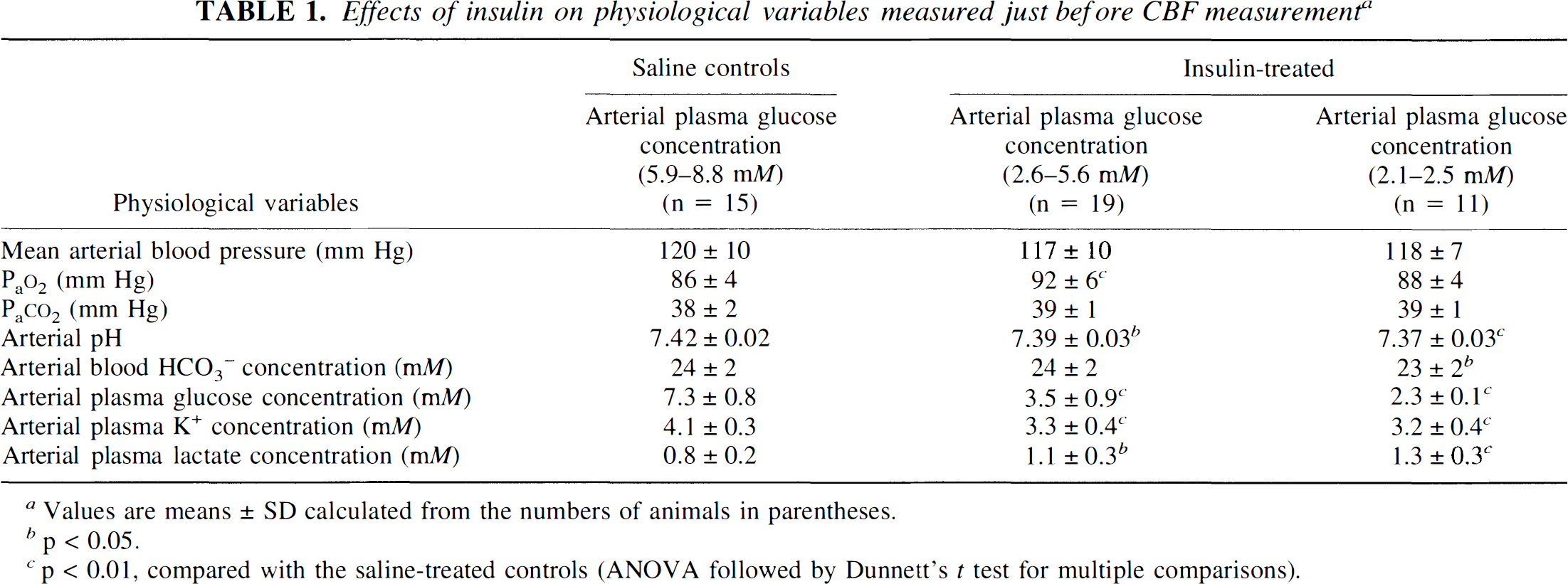
Values are means ± SD calculated from the numbers of animals in parentheses.
p < 0.05.
p < 0.01, compared with the saline-treated controls (ANOVA followed by Dunnett's t test for multiple comparisons).
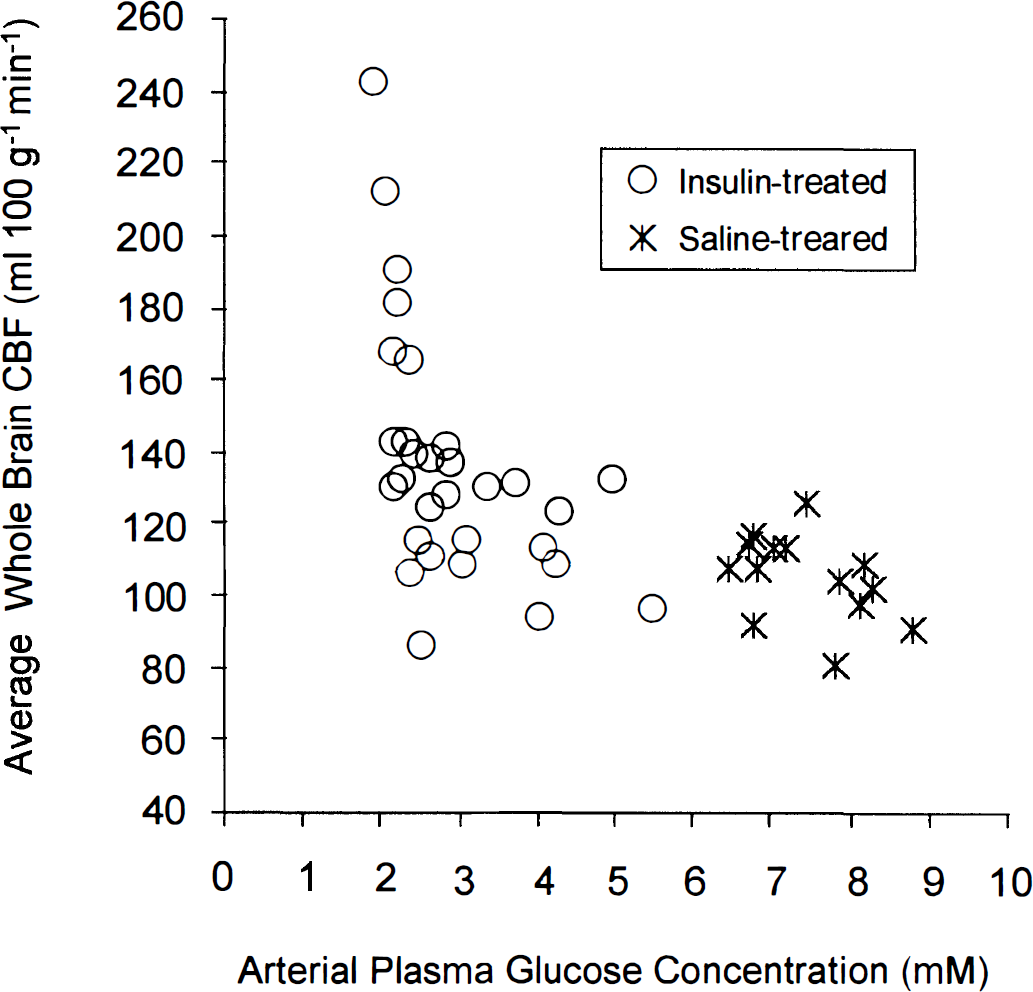
Average blood flow in the brain as a whole as a function of arterial plasma glucose concentration in rats made hypoglycemic with insulin and in saline-treated controls.
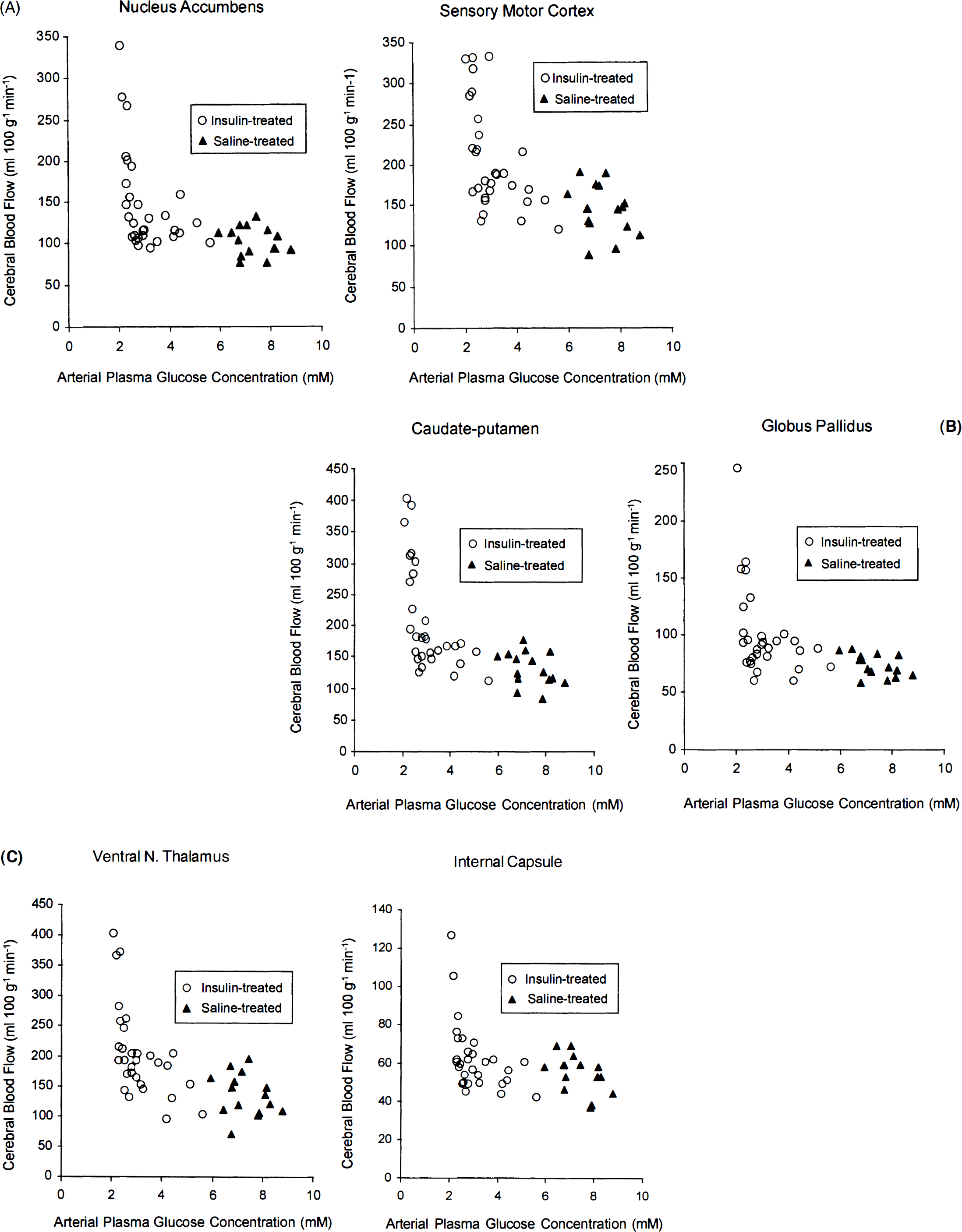
Local blood flow as a function of arterial plasma glucose level in several representative brain structures of rats with insulin-induced hypoglycemia and in saline-controls.
Effects of insulin on physiological variables and CBF in euglycemic rats
To determine whether the effects of insulin on CBF were direct or a secondary consequence of the hypoglycemia, the effects of insulin administration on CBF in rats maintained in a euglycemic state were examined. A high dose of insulin (10 U/kg) was used in these studies, but the arterial plasma glucose level was monitored and maintained more or less euglycemic by simultaneous injections of glucose as needed. In these euglycemic rats, insulin had the same effects on plasma constituents as it did in the hypoglycemic rats, for example, slight reductions in arterial blood pH and HCO3– concentration, a pronounced reduction in plasma K+ concentration, and a greater than 50% rise in plasma lactate concentration (Table 2). In the hyperinsulinemic euglycemic rats average CBF in the brain as whole was not statistically significantly different from the values in the control rats administered saline rather than insulin (Fig. 3), indicating that it was not insulin per se but the hypoglycemia that caused the increases in CBF seen in hypoglycemia. These results are in agreement with those of Duckrow (1988), who also found no effects of insulin on CBF under euglycemic conditions.
Effects of insulin on physiological rats variables in euglycemic rats a
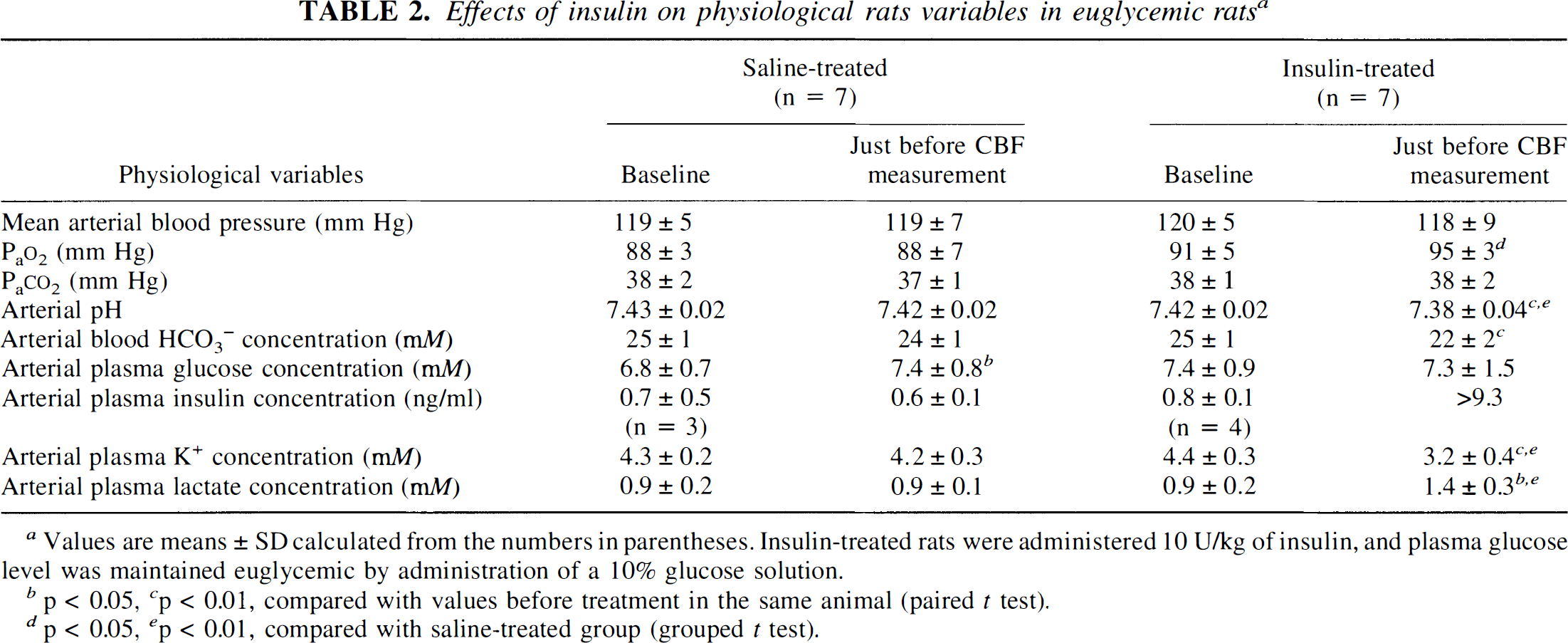
Values are means ± SD calculated from the numbers in parentheses. Insulin-treated rats were administered 10 U/kg of insulin, and plasma glucose level was maintained euglycemic by administration of a 10% glucose solution.
p < 0.05
p < 0.01, compared with values before treatment in the same animal (paired t test).
p < 0.05
p < 0.01, compared with saline-treated group (grouped t test).
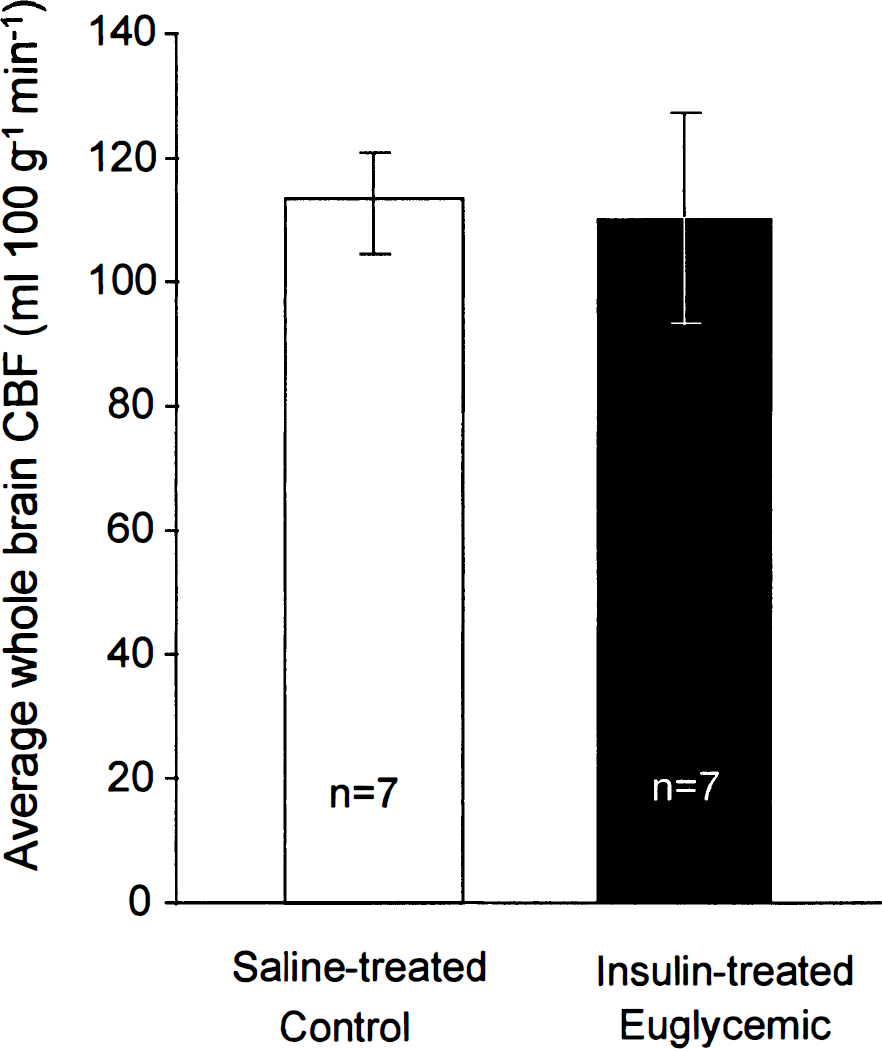
Effects of insulin on average blood flow (±SD) in the brain as a whole in saline-treated control and insulin-treated euglycemic rats. The rats that received insulin were also given i.v. injections of 10% glucose to maintain euglycemia. There was no statistically significant difference in CBF between the two groups by grouped t test.
Effects of pharmacological doses of deoxyglucose on physiological variables and CBF
Cerebral glucoprivation was also induced by the i.v. administration of 200 mg/kg of DG. Fifteen minutes after the injection, plasma glucose levels had risen significantly, arterial Pa
Effects of pharmacological doses of DG on physiological variables and CBF a
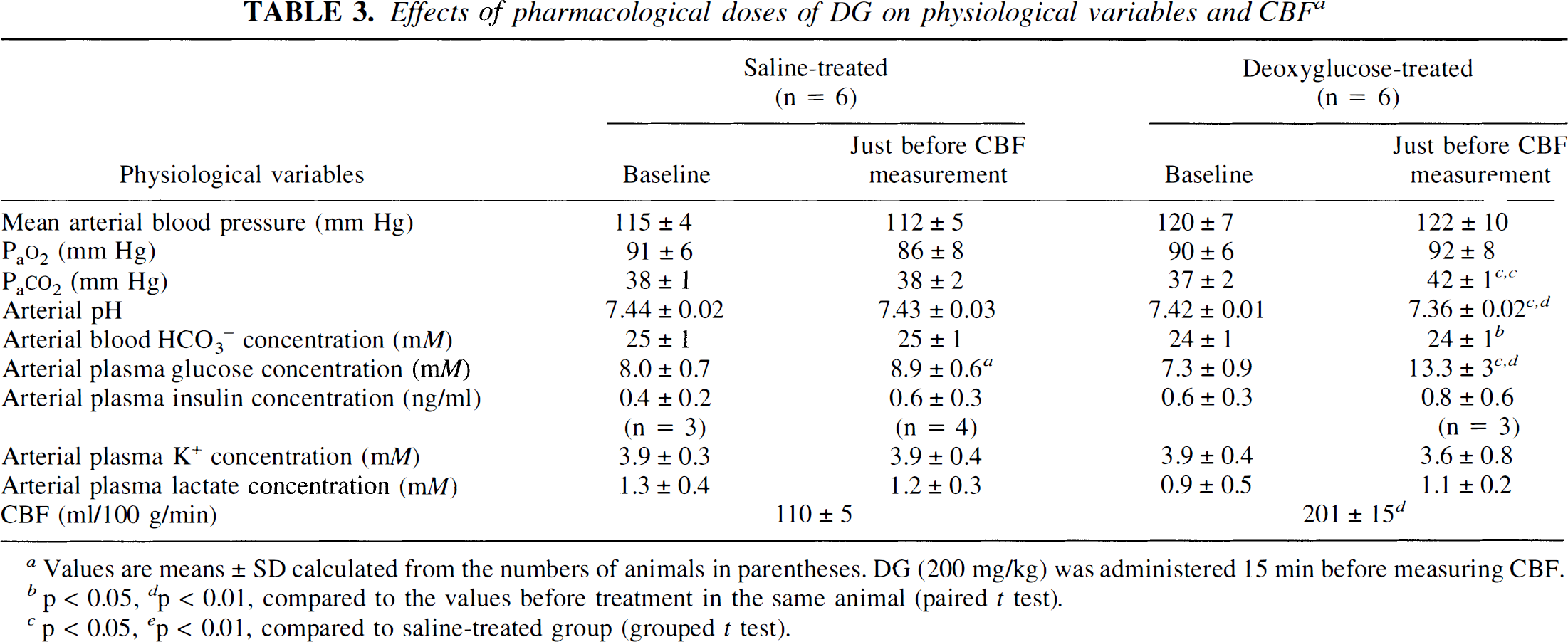
Values are means ± SD calculated from the numbers of animals in parentheses. DG (200 mg/kg) was administered 15 min before measuring CBF.
p < 0.05
p < 0.01, compared to the values before treatment in the same animal (paired t test).
p < 0.05
p < 0.01, compared to saline-treated group (grouped t test).
Systemic effects of L-NAME
Intravenously administered L-NAME (30 mg/kg) raised MABP in every rat. In L-NAME-treated rats not subsequently subjected to glucoprivation, MABP rose from a baseline level of 123 ± 6 (mean ± SD) to 145 ± 8 mm Hg (n = 6, p < 0.0002 by paired t test) at the time of the measurement of CBF. The acute L-NAME administration raised MABP from a baseline mean of 125 ± 1 to 155 ± 5 mm Hg (n = 3, p < 0.01 by paired t test) in the group subsequently treated with insulin and from 129 ± 14 to 149 ± 10 in the DG-treated rats (n = 3, p < 0.05 by paired t test). None of the other physiological variables that were measured was significantly affected by the acute L-NAME administration.
The twice-daily i.p. injections of L-NAME for 4 days also had no significant effects on the measured physiological variables, except for increases in MABP to levels like those after acute L-NAME administration. In the group subsequently treated with insulin, MABP was 158 ± 8 mm Hg (mean ± SD) in the 25 rats chronically pretreated with L-NAME compared with 133 ± 10 mm Hg in the 22 control rats that did not receive the chronic L-NAME administration (p < 0.001, grouped t test). In rats given loading doses of DG, MABP was 149 ± 7 mm Hg in those treated with L-NAME (n = 5) compared with 127 ± 8 (n = 6) in the control rats (n = 6) that did not receive L-NAME (p < 0.001, grouped t test).
Effects of chronic L-NAME pretreatment on the CBF responses to insulin-induced hypoglycemia or DG loading
Insulin-induced hypoglycemia.
In agreement with the findings of Adachi et al. (1994), chronic pretreatment with L-NAME tended to reduce CBF below the levels in the saline-pretreated control rats with arterial plasma glucose concentrations in the 3 to 6 mM range (Fig. 4). When the saline control rats (e.g., those treated with neither L-NAME nor glucoprivation) from both the insulin-hypoglycemia (Fig. 5) and DG-loading (Fig. 6) studies were pooled and the same was done with the rats chronically treated with L-NAME but not subjected to glucoprivation, the reduction of CBF by chronic L-NAME treatment was statistically significant (saline controls: mean ± SD = 106 ± 12, n = 21; L-NAME-treated: mean ± SD = 96 ± 16, n = 13; p = 0.032 by grouped t test). When the plasma glucose level was reduced to 2 to 3 mM, CBF exhibited the same sharp rises in CBF in both the L-NAME-pretreated and saline-pretreated rats; the percent increase in CBF above baseline elicited by hypoglycemia was unaffected by the pretreatment with L-NAME, and there was no statistically significant interaction between the effects of L-NAME and hypoglycemia by two-way ANOVA (Fig. 5).
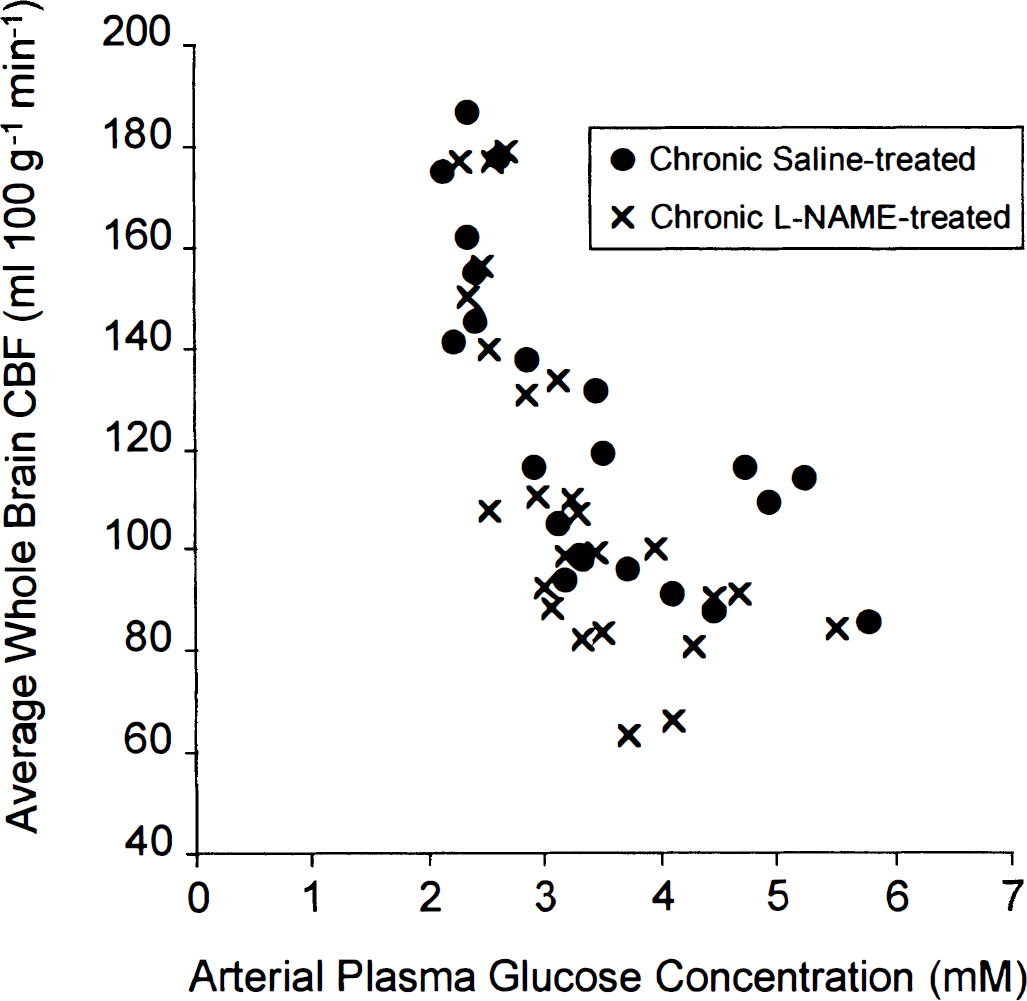
Average blood flow in the brain as a whole as a function of arterial plasma glucose concentration in insulin-treated (0.25–20 U/kg) rats after chronic pretreatment with either saline or L-NAME (50 mg/kg intraperitoneally twice daily for 4 consecutive days).
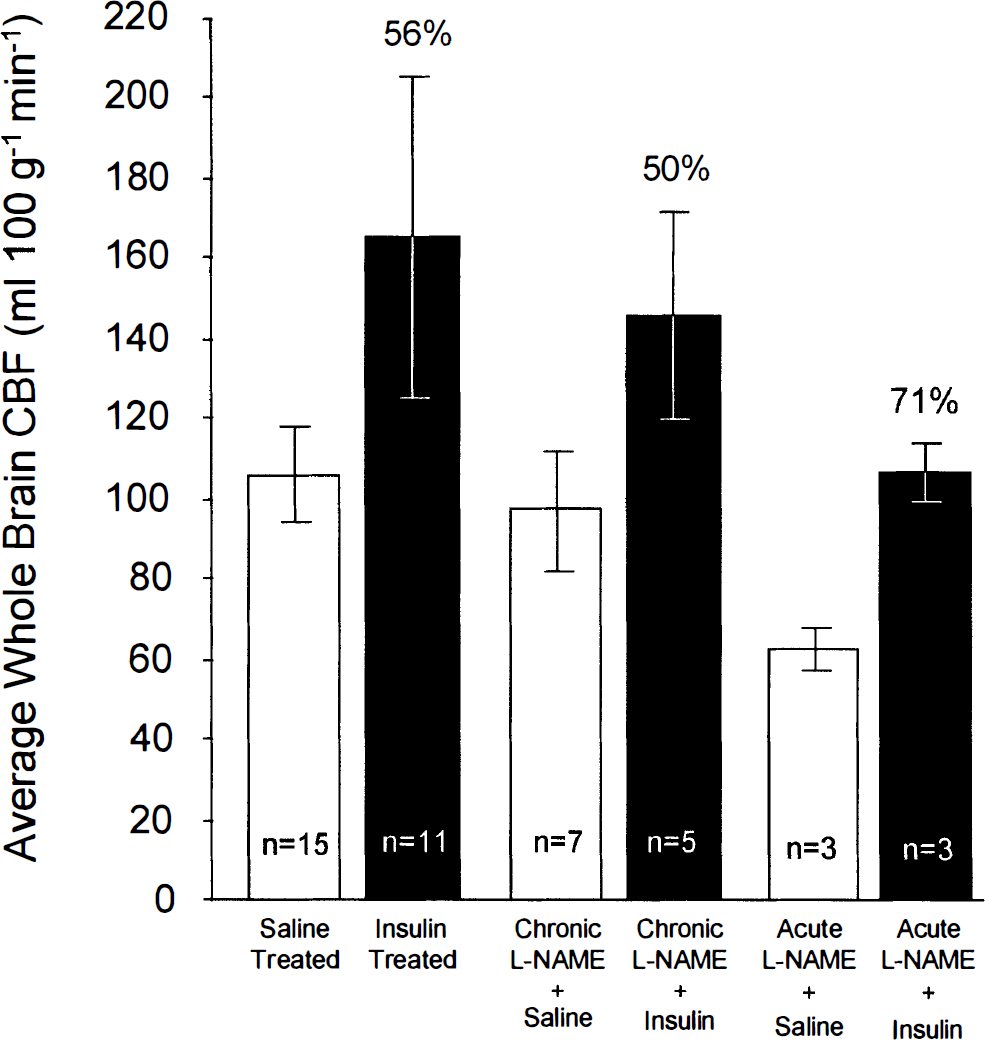
Effects of chronic (i.e., 50 mg/kg of L-NAME intraperitoneally twice daily for 4 consecutive days) and acute (i.e., 30 mg of L-NAME intravenously 45 min before measurement of CBF) inhibition of nitric oxide synthase activity on the responses of average whole brain blood flow (means ± SD) to insulin-induced hypoglycemia (arterial plasma concentration, 2.0–2.5 mM). The effects of L-NAME on the CBF responses to hypoglycemia were not statistically significant (two-way analysis of variance, no interaction between L-NAME treatments and hypoglycemia).
Pharmacologic doses of DG.
Chronic pretreatment with L-NAME raised MABP and tended to lower baseline CBF below that of the saline-pretreated control rats, but it did not prevent the marked enhancement of CBF by DG loading. The percent increases in CBF were essentially the same in both groups (Fig. 6), and there was no significant interaction between the effects of L-NAME treatment and DG loading (two-way ANOVA).
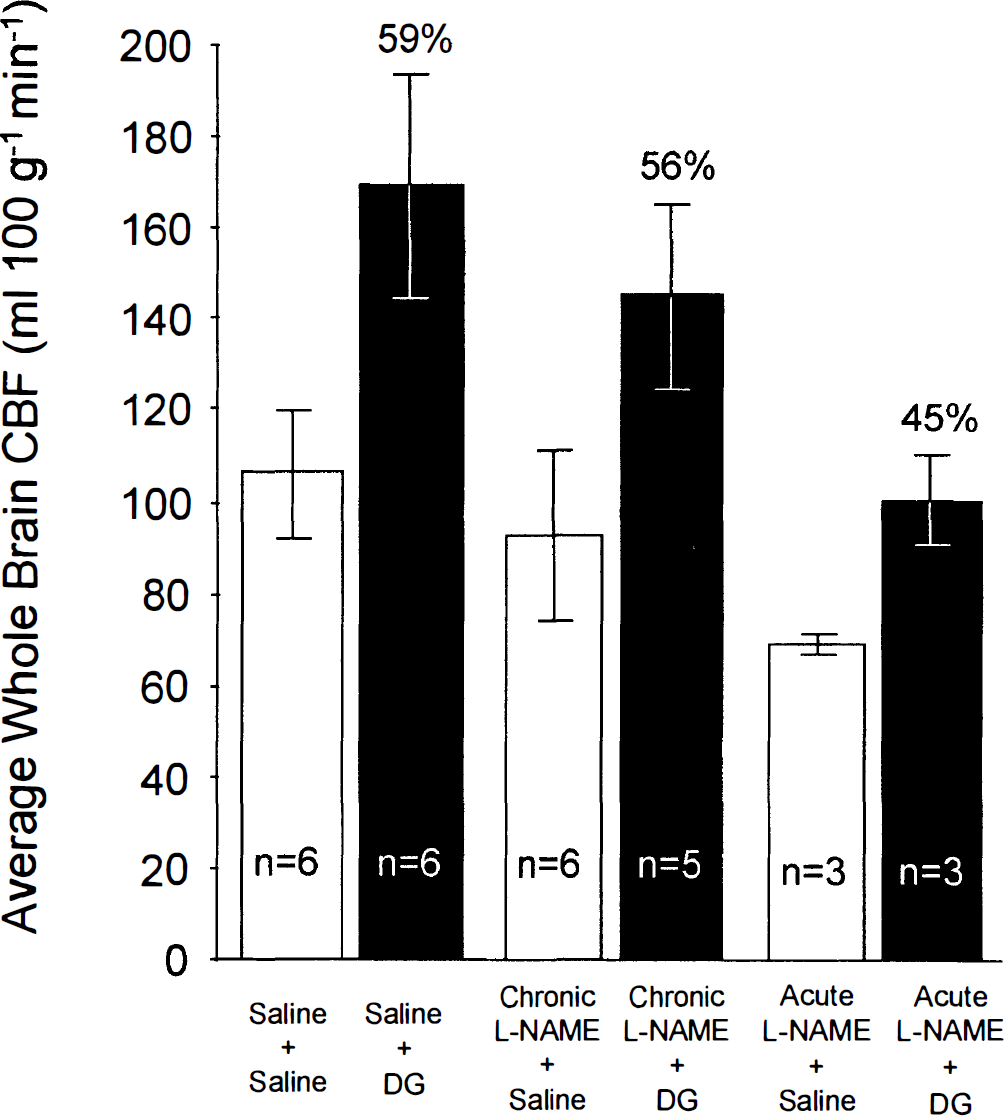
Effects of chronic (50 mg/kg of L-NAME intraperitoneally twice daily for 4 consecutive days) and acute (30 mg of L-NAME intravenously 45 min before measurement of CBF) inhibition of nitric oxide synthase activity on the responses of average whole brain blood flow (means ± SD) to pharmacological doses of 2-deoxyglucose (200 mg/kg, i.v.). The effects of L-NAME on the CBF response to DG loading was not statistically significant (two-way analysis of variance, i.e., no interaction between L-NAME treatment and DG loading).
Effects of acute L-NAME administration on CBF responses to insulin-induced hypoglycemia or DG loading
Chronic pretreatment with L-NAME has been shown to inhibit 85 to 95% of total brain NO synthase activity (Dwyer et al., 1991; Adachi et al., 1994). The increases in MABP that we observed indicated that at least endothelial NO synthase had been effectively inhibited. The results obtained with chronic L-NAME treatment therefore suggested that the cerebrovascular dilatation associated with insulin-induced hypoglycemia or blockade of cerebral glucose utilization by pharmacologic doses of DG is not mediated by NO. There have, however, been suggestions that compensatory mechanisms might come into play in the regulation of CBF during prolonged blockade of NO synthase activity (Wang et al., 1994). We therefore also examined the effects of acute administration of L-NAME on the increases in CBF associated with insulin-hypoglycemia or DG loading.
In control rats subjected to neither insulin-induced hypoglycemia nor DG loading, acute i.v. administration of L-NAME reduced CBF significantly, indeed, considerably more than did chronic pretreatment with L-NAME; CBF was 106 ± 12 (mean ± SD) in the control rats (n = 21) and 66 ± 5 in the rats given acute doses of L-NAME (n = 6) (p < 0.0001 by grouped t test). It did not, however, significantly alter the enhancement of CBF by hypoglycemia or DG loading (two-way ANOVA) (Figs. 5 and 6).
DISCUSSION
The results of previous studies on the effects of hypoglycemia on CBF have been inconsistent. The earliest quantitative studies in hypoglycemia were carried out in human subjects with the nitrous oxide method. In the first study by Kety et al. (1948) on the effects of insulin shock treatment in schizophrenia, CBF was not significantly altered when blood glucose concentrations were lowered from a control level of 74 (4.1 mM) to 19 (1.1 mM) and then to 8 mg% (0.4 mM); the reduced glucose levels were, however, sufficient to cause profound reductions in CMRO2 and CMRglc. Eisenberg and Seltzer (1962), also using the nitrous oxide method, subsequently reported no statistically significant changes in CBF in human subjects despite reduced blood glucose concentrations from a mean of 85 (4.7 mM) to 31 (1.7 mM) mg% and a 32% fall in CMRglc; surprisingly, they also observed a 16% increase in CMRO2, which they speculated was due to an epinephrine release into the blood. In another group of subjects in which blood glucose concentration was lowered from 101 (5.6 mM) to 51 mg% (2.8 mM), no significant changes occurred in CBF, CMRO2 or CMRglc. In contrast, in schizophrenic patients in insulin-induced coma studied with the same method, Della Porta et al. (1964) found a 50% increase in CBF, a 57% decrease in CMRglc, and no significant change in CMRO2.
Studies of local CBF in rats with the [14C]iodoantipyrine method have provided more uniform results. In anesthetized, paralyzed, artificially ventilated rats, local CBF was markedly increased in most regions of brain during insulin-induced hypoglycemia (e.g., blood glucose level, 1.26 mM) severe enough to produce slow-wave poly-spike activity in the EEG (Abdul-Rahman et al., 1980); CBF increased several fold above control levels when the hypoglycemia was accompanied by an isoelectric EEG (Abdul-Rahman et al., 1980; Siesjö et al., 1983). In awake, restrained rats Bryan et al. (1987) found that moderate hypoglycemia (e.g., plasma glucose level, 2.6 mM) increased local CBF in some regions of the brain (e.g., in the cerebral cortex and basal ganglia but not in the hypothalamus and cerebellum); more severe hypoglycemia (plasma glucose level, 1.5 mM) caused even greater increases in local CBF throughout the brain, including the hypothalamus and cerebellum.
The reasons for the differences in the results of these various studies are unclear, but variations in the severity of the hypoglycemia, the presence or absence of seizure activity, differences in the methods employed to determine CBF, and possibly species differences may have been involved. It is clear, however, from most studies that hypoglycemia generally is associated with cerebral vasodilator influences that are often sufficient to increase CBF.
The results of the present studies demonstrate that hypoglycemia increases CBF in almost all regions of the brain, but the effect is strongly dependent on the degree of hypoglycemia. With lesser degrees of hypoglycemia in the range of arterial plasma glucose levels between approximately 3 and 9 mM, CBF tends to rise as the plasma glucose level declines, but it is slight and of doubtful significance. When arterial plasma glucose concentration falls below 2.5 to 3 mM, however, there is an abrupt and marked increase in CBF in almost all regions of the brain. This effect occurs at a rather moderate degree of hypoglycemia because the animals, although subdued, showed no loss of consciousness or EEG abnormalities. It it of interest that the level of hypoglycemia at which the marked increase in CBF occurs is similar to the level at which glucose consumption in the brain becomes moderately depressed (Suda et al., 1990) and ATP levels in brain begin to fall (Ghajar et al., 1982).
The nature of the cerebral vasodilator influences and the mechanism of the increase in CBF provoked by hypoglycemia are still unknown. A major goal of the present study was to examine systematically the possible mechanisms that might apply. To do so, we compared the effects of two different paradigms of cerebral glucoprivation, impairment of glucose supply to the brain by insulin-induced hypoglycemia or chemical blockade of cerebral glucose metabolism. Pharmacological doses of 2-deoxyglucose are known to produce a comatose state like that of hypoglycemia while at the same time raising blood glucose levels (Landau et al., 1958). The mechanism of this effect is a blockade of glycolysis at the glucose-6-phosphate isomerase step (Wick et al., 1957; Horton et al., 1973). Short of those that produce coma or seizures, DG loading doses have also been found to produce widespread and marked increases in CBF throughout the brain (Breier et al., 1993). A reasonable assumption is that the mechanisms underlying this effect are the same as those in hypoglycemia. The comparison has led to the exclusion of a number of possible mechanisms.
Insulin administration results not only in lower blood glucose levels but also lower plasma K+ and increased lactate concentrations and a small decline in arterial pH. Insulin, administered together with glucose to maintain euglycemic conditions, produces essentially the same changes in blood and plasma without altering CBF, and DG loading raises CBF like insulin-induced hypoglycemia without those changes in blood and plasma, except for the change in blood pH. These results exclude the changes in blood or plasma lactate and K+ concentrations or blood pH and a direct action of insulin as the possible causes of the increased CBF during glucoprivation.
Nitric oxide has been identified as the EDRF which mediates acetylcholine-induced vasodilatation (Furchgott and Zawadzki, 1980; Palmer et al., 1987; Moncada et al., 1991). A possible role of NO in the increases in CBF during cerebral glucose deprivation was, therefore, investigated. Nitric oxide is produced in vascular endothelium and in neurons and glia by oxidation of arginine catalyzed by the enzyme nitric oxide synthase. The total activity of this enzyme in brain has been shown in rats to be almost completely inhibited by twice daily injections of 50 mg/kg of L-NAME for 4 days (Dwyer et al., 1991; Adachi et al., 1994). We used this regimen to prevent the formation of NO in both the endothelium and the brain cells. To avoid the influence of possible compensatory mechanisms during prolonged NO synthase inhibition, we also examined the effects of acute inhibition by a single i.v. dose of L-NAME shortly before the measurement of CBF. In both cases, the enhancement of CBF by hypoglycemia or pharmacological doses of DG was unaffected by the NO synthase inhibition. This result is partly in contrast to that of Ichord et al. (1994), who used the radiolabeled microsphere technique for determining CBF in 7 to 10-day-old piglets under anesthesia; they observed that inhibition of NO synthase activity did attenuate but did not eliminate the enhancement of CBF by hypoglycemia in forebrain, cortical gray matter, and cerebellum, but it had no effects on the responses in the brainstem, thalamus, caudate, and hippocampus. It should be noted, however, that the degree of hypoglycemia that they examined was profound enough for the EEG to become isoelectric. It is therefore difficult to compare their results with ours, which indicate that NO does not mediate the increases in CBF that we observe in unanesthetized rats with more moderate degrees of hypoglycemia.
Glucoprivation, whether induced by hypoglycemia or blockade of glucose metabolism with pharmacological doses of DG, is a stress that activates the sympathetic nervous system and raises blood epinephrine levels. Hollinger and Bryan (1987) concluded that adrenergic β-receptors mediate the increase in CBF during hypoglycemia because the effect appeared to be partially attenuated by the β-blocker propranolol. More recently, Bryan et al. (1994) found that the rise in plasma catecholamine levels was temporally dissociated from the increase in CBF and could not, therefore, be responsible for the increased CBF in hypoglycemia. Noradrenergic effects of stress are unlikely to be involved because increases in blood norepinephrine levels (King et al., 1952) and intracerebral noradrenergic mechanisms have been shown to produce cerebral vasoconstriction (Kuschinsky and Wahl, 1978) as long as the blood–brain barrier remains intact (Edvinsson et al., 1993). Studies in progress in our laboratory indicate that i.v. infusion of epinephrine that raise plasma epinephrine levels in normoglycemic rats to the same levels found in hypoglycemia have no effects on local or overall blood flow or glucose utilization in the brain.
It may be of significance that CBF begins to rise sharply in unanesthetized rats when arterial plasma glucose levels fall to about 2.5 to 3.0 mM. These are the levels at which local CMRglc begins to fall, particularly in structures with normally high rates, and brain ATP levels are not maintained (Ghajar et al., 1982; Bryan et al., 1986; Suda et al., 1990). This association suggests that the response of the cerebral vasculature may be related to limitations in the rate of glucose metabolism needed to maintain normal levels of ATP. Such a condition would result in increases in brain adenosine levels which, in fact, have been observed in hypoglycemia (Chapman et al., 1981; Van Wylen et al., 1989), and adenosine is known to have cerebral vasodilator effects (Berne et al., 1974). We are currently examining the possibility that adenosine may play an important role in the cerebral vasodilatation associated with glucoprivation. The results thus far indicate that caffeine, an adenosine receptor antagonist, attenuates in a dose-dependent fashion the increase in CBF seen in hypoglycemia.
The present studies have excluded blood pH, Pa
Footnotes
Acknowledgment:
These studies were supported in part by a grant from the National Association for Research in Schizophrenia and Affective Disorders (NARSAD).