
Abstract
Recent investigations have been suggesting that some neuronal subpopulations may die via programmed cell death after focal ischemic injury. To clarify the possible roles of the genes involved in the cell-death program, this study examined the expression of three members of the interleukin-1β converting enzyme (Ice) gene family (Ice, Nedd2, and Yama/CPP32) and two members of the bcl-2 gene family (bcl-2 and bcl-x) in the rat brain after permanent occlusion of the middle cerebral artery. Northern blot analysis revealed a transient induction of Nedd2 mRNA 8 h after the ischemic insult (3.8-fold) and an increase in Yama/CPP32 mRNA 16 to 24 h after the insult (5.8-fold at 24 h), whereas the expression of Ice remained constant. The expression of bcl-2 and bcl-x remained constant after the ischemic insult. Taking into account the key role of the Ice gene family in the execution of programmed cell death, the induction of Ice gene family might play a causative role in apoptotic cell death.
During focal ischemia, neurons in a densely ischemic focus are usually doomed unless reperfusion is quickly instituted. In contrast, neurons in a less densely ischemic penumbral zone remain viable for at least 4–8 h and then die (Siesjö, 1992). Recent studies have provided evidence that some neuronal subpopulations may die after permanent or transient cerebral ischemia via apoptosis or programmed cell death following the activation of an endogenous cell death program (Linnik et al., 1993; Li et al., 1995).
The best characterized genetic system of programmed cell death (PCD) is the worm Caenorhabditis elegans (C. elegans), in which 14 genes regulating PCD during development have been identified (Ellis et al., 1991). Two of these genes, ced-3 and ced-4, play essential roles in either the initiation or execution of the cell death program (Ellis and Horvitz, 1986). Several mammalian homologues of the ced-3 gene have been discovered as an executioner of PCD or apoptosis. These genes include the interleukin-1β converting enzyme (Ice) (Yuan et al., 1993), Nedd2/Ich-1 (Kumar et al., 1994; Wang et al., 1994), Yama/CPP32 (Fernandes-Alnemri et al., 1994; Tewari et al., 1995), Tx/Ich-2 (Faucheu et al., 1995; Kamens et al., 1995), Mch-2 (Fernandes-Alnemri et al., 1995). On the other hand, another gene of C. elegans, ced-9 protects cells from PCD by antagonizing the function of ced-3 and ced-4 (Hengartner et al., 1992). The ced-9 gene are found to be homologous with the bcl-2 proto-oncogene (Hengartner and Horvitz, 1994) and bcl-x (Boise et al., 1993), of which overexpression prevents neurons from apoptosis (Farlie et al., 1995; González-García et al., 1995). To elucidate the possible involvement of the Ice and bcl-2 gene families in apoptotic neuronal cell death which may occur in the ischemic brain, expression of Ice, Nedd2, Yama/CPP32, bcl-2, and bcl-x was investigated in the rat brain following intraluminal permanent occlusion of the middle cerebral artery (MCA) using Northern blot analysis.
MATERIALS AND METHODS
Surgical preparation
Male Sprague–Dawley rats (Shimizu Laboratory Supplies Co. Ltd., Kyoto, Japan) weighing 280 to 320 g were used for the experiments. Focal cerebral ischemia was induced according to a method of intraluminal MCA occlusion (Zea Longa et al., 1989). After an overnight fast of 12 h, the rats were anesthetized with 4.0% halothane and maintained on 1.0% halothane in a mixture of 70% N2/30% O2 using a face mask. The rectal temperature of the rats was maintained at 37.5 ± 0.5°C during surgery with a heating lamp and a pad connected to a rectal thermistor (Animal Blanket Controller ATB 1100, Nihon Kohden, Tokyo, Japan). The right femoral artery was cannulated to monitor mean arterial blood pressure and to obtain blood samples for measuring pH, Pa
Quantitative analysis of infarct volume
Selected rats (n = 8) were subjected to quantitative image analysis of infarct volume 24 h after the ischemic treatment. Coronal sections of the brain (2-mm thickness) were prepared and stained with 2.3.5-triphenyltetrazolium chloride (TTC, Sigma, St. Louis, MO, U.S.A.). The volume of an infarct was calculated using NIH Image Version 1.55.
In situ detection of DNA fragmentation
The rat brains were fixed by transcardial perfusion with 10% buffered formalin phosphate (pH 7.4) at 2, 4, 8, 16, 24, and 48 h after the ischemic treatment. Coronal sections (50 μm thickness) were prepared and subjected to the terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) described previously (Gavrieli et al., 1992) with slight modifications. In brief, sections were immersed in 3% H2O2 at room temperature for 5 min and dehydrated. After rehydration, the sections were immersed in the terminal deoxynucleotidyl transferase (TdT) buffer (30 mM Trizma base, 140 mM sodium cacodylate, 1 mM cobalt chloride) at room temperature for 15 min, and then incubated with 12.5 μM biotinylated dUTP (Boehringer Mannheim, Mannheim, Germany) and 0.15 U/μl TdT (Takara, Co. Ltd., Kyoto, Japan) at 37°C for 70 min. The reaction was terminated by TB buffer (300 mM sodium chloride, 30 mM sodium citrate). The sections were then subjected to the avidin–biotin procedure using ABC kit (Vector Laboratories, Burlingame, CA, U.S.A.).
Gel electrophoresis for DNA fragmentation
The rats were killed by decapitation at 4, 8, 16, and 24 h after the ischemic treatment (n = 2 at each time point). Sham-operated control rats were sacrificed 8 h after treatment. The brains were removed, and each cerebral hemisphere was quickly frozen in liquid nitrogen. The DNA was prepared according to the method of Linnik et al. (1993). The brains were minced, and cells were lysed on ice in 5 mM Tris-HCl (pH 8.0) containing 5 mM EDTA acid and 0.5% Triton X-100 for 30 min. Genomic DNA was pelleted by centrifugation at 13,000 g for 20 min. DNA that did not sediment during centrifugation was purified by phenol/chloroform/isoamyl alcohol (25:24: 1) extraction and ethanol precipitation before RNase A digestion (100 μg/ml) for 30 min at 37°C. Samples were then reextracted with chloroform/isoamyl alcohol (24:1) and reprecipitated in ethanol; DNA was separated on 2% agarose gel and visualized with ethidium bromide.
RNA isolation and Northern blot analysis
Rats were killed by decapitation at 1, 2, 4, 8, 16, and 24 h after the ischemic treatment (n = 3 at each time point). Sham-operated control rats were killed 8 h after treatment. Each cerebral hemisphere was frozen quickly in liquid nitrogen. Total RNA was prepared with the RNA isolation reagent (TRIzol Reagent, Life Technologies, Inc., Gaithersburg, MD, U.S.A.), and poly(A)+ RNA was isolated by Oligotex –dT30 Super (Nihon Roche, Co. Ltd., Tokyo, Japan). Twenty micrograms of total RNA or 3 μg of poly(A)+ RNA was separated on 1% agarose–formaldehyde gel and transferred to a nylon membrane (Pall Biodyne transfer membrane, Pall Biosupport Division, East Hills, NY, U.S.A.). Hybridization and subsequent washes were carried out as described previously (Takahashi et al., 1993). Probes for Northern blot analysis were cDNA of murine Ice (Miura et al., 1993), murine Nedd2 (Kumar et al., 1994), human Yama/CPP32, human bcl-2 (Tsujimoto et al., 1985), murine bcl-x (Kamesaki et al., unpublished data). All cDNAs used here except for Yama/CPP32 cDNA are gifts from the original researchers. Yama/CPP32 cDNA was obtained using polymerase chain reaction (PCR). The images were established by scanning with a bioimage analyzer (Bas 2000, Fujix, Tokyo, Japan). The radioactivity of each sample was quantified using the bioimage analyzer and normalized by the radioactivity detected with a cDNA probe of glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Reverse transcription PCR analysis for Nedd2
One microgram of total RNA samples of sham-operated controls and rats killed 8 and 24 h after treatment were subjected to a reverse transcription (RT)-PCR analysis for mapping of Nedd2 mRNA. The RT reaction and PCR were carried out using RT-PCR kit (Takara, Co. Ltd., Kyoto, Japan). The following conditions were used for the RT reaction: 50°C for 20 min, 99°C for 5 min, and 5°C for 5 min. The following conditions were used for the PCR amplification: cDNA products of the RT reaction were denatured for 2 min at 94°C before 40 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 1 min 30 s. The RT-PCR product was separated on 1.8% agarose gel and visualized with ethidium bromide. Control experiments using primers for Nedd2 showed that the amount of amplified PCR products was nearly proportional to the amount of input RNA within the range of 0.5 to 4 μg after 40 cycles of amplification (Fig. 2B, bottom). The following oligonucleotides were used as primers: murine Nedd2 primers (Wang et al., 1994), 5′ primer (5′-ATGCTAACTGTCCAAGTCTA-3′), 3′ primer (5′-GTCTCATCTTCATCAACTCC-3′); rat ribosomal protein L27a primers used as an internal control (Hoshimaru et al., 1996), 5′ primer (5′-ATCGGTAAGCACCGCAAGCA-3′), 3′ primer (5′-GGGAGCAACTCCATTCTTGT-3′).
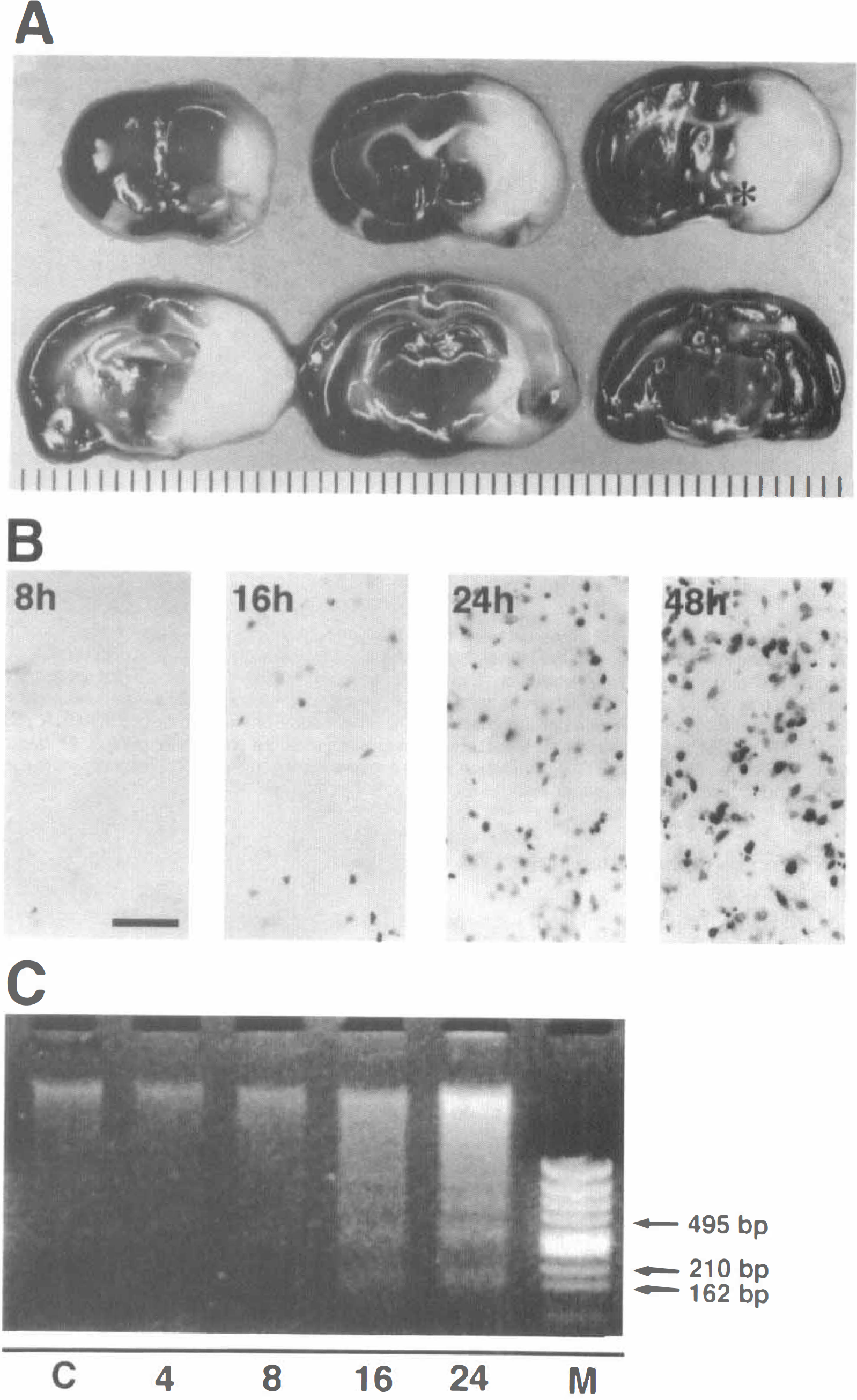
Brain sections showing an ischemic infarct 24 h after permanent middle cerebral artery (MCA) occlusion stained with 2.3.5-triphenyltetrazolium chloride.
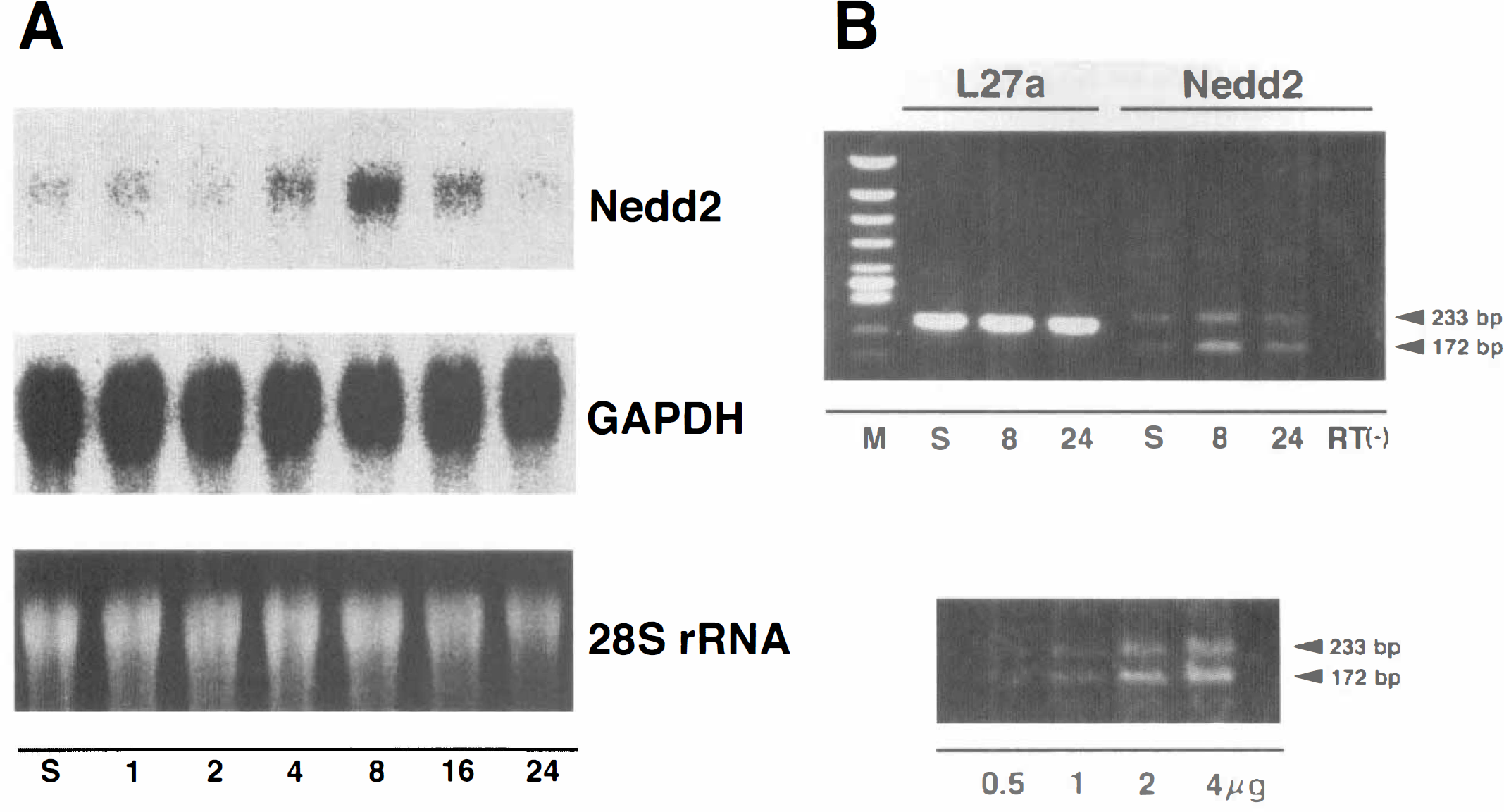
Expression of Nedd2 in the rat brain following permanent middle cerebral artery (MCA) occlusion.
RESULTS
Reproducible cerebral infarction after permanent occlusion of the MCA
Physiological variables during surgical preparations are shown in Table 1. During surgical manipulations, mean arterial blood pressure, Pa
Physiological variables during surgical manipulation (n = 8)

Data are presented as mean ± SD.
MABP, mean arterial blood pressure.
DNA fragmentation after permanent occlusion of the MCA
Cells with fragmented DNA were not observed 4 h after the ischemic insult but appeared 8 h thereafter, and subsequently the number of apoptotic cells progressively increased (Fig. 1B). These cells were localized mainly to the inner boundary zone of the infarct, although some scattered apoptotic cells were observed throughout the ischemic core, including the caudolenticular nucleus and the adjacent neocortex. No cells with fragmented DNA were observed in the contralateral nonischemic hemisphere.
Agarose gel electrophoresis of purified DNA from the ischemic zone showed random DNA degradation 8 h after the ischemic insult, which was observed as a continuous smear. The amount of random DNA degradation progressively increased thereafter. Although dense smearing was predominant, a ladder of multiples of about 180-bp DNA fragments appeared 16 h after the ischemic insult and increased thereafter (Fig. 1C).
Induction of Nedd2 and Yama/CPP32 mRNA in the ischemic brain after MCA occlusion
Nedd2 mRNA was detected as a single 3.5-kb band by Northern blot analysis using total RNA. The amount of Nedd2 mRNA increased to 3.8-fold higher expression relative to the sham-operated control 8 h after the ischemic insult and then decreased almost to the basal level within 24 h (Figs. 2A and 4). On the contralateral side, little increase of expression was observed (data not shown). On the other hand, the expression of Ice, Yama/CPP32, and bcl-2 was too low to be detected by Northern blot analysis using total RNA and was analyzed using poly(A)+ RNA. Yama/CPP32 mRNA was detected as a single band 2.5 kb long as reported previously, and significant induction was observed 16 to 24 h after the ischemic insult (3.1-fold and 5.8-fold higher expression at 16 h and 24 h, respectively) (Figs. 3 and 4). On the other hand, the expression of Ice (1.6 kb long), bcl-2 (3.5 kb and 7.5 kb long), and bcl-x (2.7 kb long) remained constant after the ischemic insult (Figs. 3 and 4).
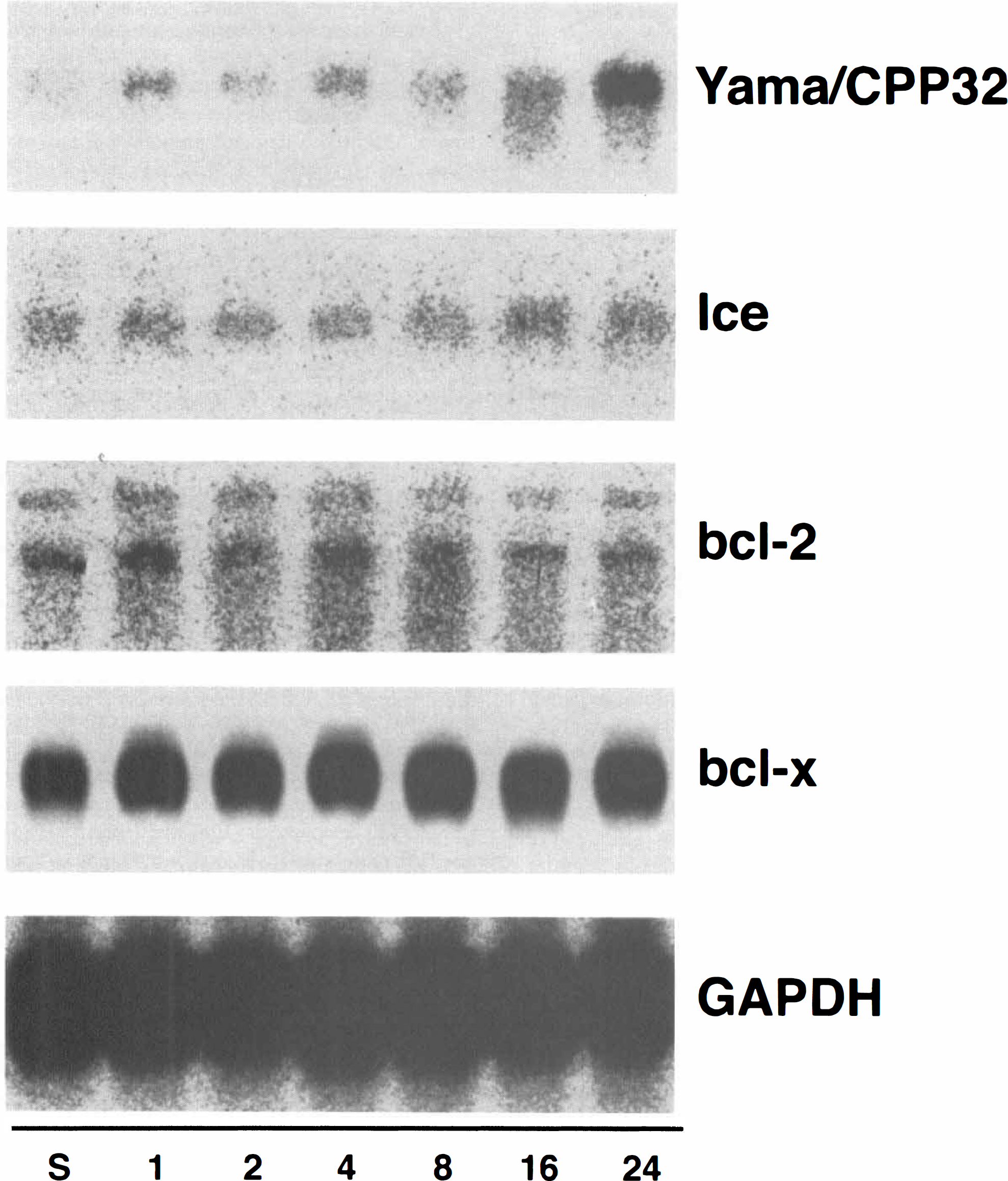
Expression of Yama/CPP32, Ice, bcl-2, and bcl-x in the rat brain following permanent middle cerebral artery (MCA) occlusion. Northern blot analysis of 3 μg of poly(A)+ RNA isolated from the ischemic cerebral hemisphere with a human Ice, human Yama/CPP32, human bcl-2, and murine bcl-x cDNA probes.
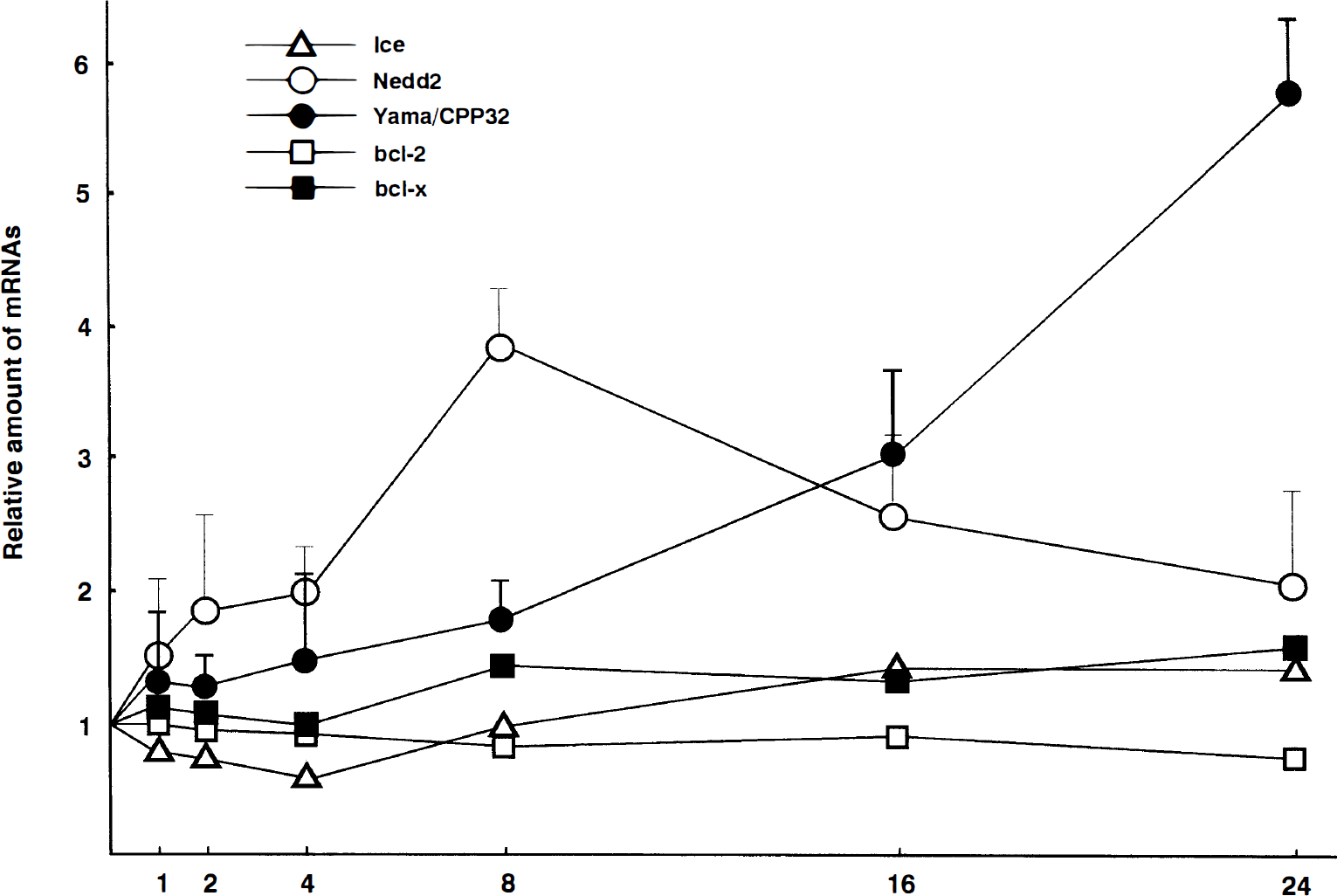
Time course of Ice, Nedd2, Yama/Cpp32, bcl-2, and bcl-x mRNA expression in the rat brain following permanent middle cerebral artery (MCA) occlusion. Data are given as relative amount to sham-operated controls, and are plotted as the mean (±SD) of three independent analyses.
Discrimination between an active and inactive form of Nedd2 mRNA by RT-PCR
An alternatively spliced form of murine Nedd2 mRNA, Nedd2s mRNA has been suggested to encode an inactive form of Nedd2 (Kumar et al., 1995). To discriminate between Nedd2 and Nedd2s mRNA, specific primers that produce the PCR products of different sizes between Nedd2 (172 bp) and Nedd2s (233 bp) were used for quantitative RT-PCR analysis. Both Nedd2 and Nedd2s mRNA can be detected in sham-operated control animals. Eight hours after the ischemic treatment, both transcripts were obviously induced, and the induction of Nedd2 was higher than that of Nedd2s (Fig. 2B, top).
DISCUSSION
Recent investigations have suggested that apoptosis has an important role in the development and extension of an ischemic infarct. Internucleosomal DNA fragmentation, which indicates apoptotic cell death, has been demonstrated by gel electrophoresis in purified DNA from the ischemic rat brain following permanent or transient MCA occlusion (Linnik et al., 1993; Tominaga et al., 1993; MacManus et al., 1994). Dead cells with DNA fragmentation have been observed in the ischemic zone after transient MCA occlusion (Li et al., 1995; Charriaut-Marlangue et al., 1996). In addition, protein synthesis inhibitors and bcl-2 have been demonstrated to protect neuronal cells partially against permanent focal ischemia (Linnik et al., 1993; Martinou et al., 1994). These data have suggested that apoptosis occurs concurrently with necrosis in focal ischemia. In our present permanent MCA occlusion model, DNA fragmentation was demonstrated by TUNEL and gel electrophoresis. Cells exhibiting DNA fragmentation were rarely observed 8 h after the ischemic insult but progressively increased in number thereafter. Accordingly, the DNA ladder that is typical of apoptosis appeared 16 h after the ischemic insult and increased in intensity after that, although the amount of random DNA degradation, which reflects necrosis, progressively increased after 8 h of ischemia. The time course of the appearance of DNA fragmentation is almost similar to that demonstrated in previous studies of rat focal ischemia (Tominaga et al., 1993; Li et al., 1995). Therefore, our permanent MCA occlusion model is appropriate for the study of the apoptosis-related genes.
The bcl-2 proto-oncogene and bcl-x are mammalian homologues of the ced-9 gene. The bcl-2 proto-oncogene is expressed in neuronal tissues during embryonic development but is downregulated in the adult CNS. In contrast, bcl-x expression is retained in neurons of the adult CNS and functions to prevent neuronal cell death (González-García et al., 1995). It is suggested that suppression of bcl-2 mRNA production may mediate apoptosis in some human cell lines (Chen M et al., 1995). Furthermore, the volume of the infarct produced by permanent MCA occluson was significantly reduced in bcl-2 transgenic mice (Martinou et al., 1994). On the other hand, some recent reports have indicated by immunohistochemistry that bcl-2 proteins may be induced in neuronal cells after an ischemic insult (Shimazaki et al., 1994; Chen J et al., 1995); however, our data demonstrated no significant change in the expression of bcl-2 and bcl-x following focal brain ischemia.
Specific mammalian cysteine proteases that are homologous with ced-3 have been discovered as executioners of PCD or apoptosis (Ellis and Horvitz, 1986). Ice is the first protein discovered as a mammalian homologue of the ced-3 gene (Yuan et al., 1993). Some recent reports have revealed the induction of the Ice mRNA in mice mammary gland epithelium undergoing apoptosis (Boudreau et al., 1995) and in the rat brain after an injection of the immunostimulant, lipopolysaccharide (Keane et al., 1995). Our present study, however, demonstrated that the expression of Ice, which is weak in the CNS, remained constant after permanent occlusion of the MCA. Therefore, Ice may not contribute to apoptotic neuronal cell death following focal ischemic injury in the rat brain.
Yama/CPP32:
Another mammalian homologue of the ced-3 gene, which was isolated on the basis of its ability to cleave poly(ADP-ribose) polymerase (PARP) during the induction of apoptosis, has recently drawn attention (Tewari et al., 1995) because PARP is considered one of target proteins for apoptosis triggered by the ced-3 gene family (Martin and Green, 1995; Whyte and Evan, 1995). In our present study, 3.1- and 5.8-fold higher expression of Yama/CPP32 were observed 16 and 24 h after the ischemic insult, respectively. This enzyme mRNA was induced almost at the same time that the internucleosomal DNA fragmentation occurred. A recent study indicated that the preparatory events that partially cleave genomic DNA occur several hours before the appearance of the oligonucleosomal DNA cleavage at the late stage (18–24 h) (Charriaut-Marlangue et al., 1995). Therefore, the induction of Yama/CPP32 mRNA seems too late to have a crucial role in apoptotic cell death pathway in the ischemic brain after permanent MCA occlusion. In the rat brain after permanent MCA occlusion, neutrophils infiltrate and accumulate in the ischemic zone at the late stage of the ischemia (Zhang et al., 1994); and Yama/CPP32 mRNA is known to be highly expressed in cell lines of hematopoietic lineage (Fernandes-Alnemri et al., 1994). Therefore, Yama/CPP32 mRNA induction may be representative of this neutrophil infiltration. However, further investigations on the localization of Yama/CPP32 mRNA or protein are required to clarify this point.
Nedd2 is highly expressed in the CNS during embryonic development, when massive programmed neuronal cell death occurs, and it is also slightly expressed in the adult brain (Kumar et al., 1994). Furthermore, it is reported that overexpression of Nedd2 induces apoptosis and that expression of antisense Nedd2 inhibits apoptosis (Kumar, 1995). In our present study, 3.8-fold higher expression of Nedd2 was observed 8 h after ischemic insult. In addition, RNA mapping by RT-PCR revealed that an active form of Nedd2 was prominently induced. The significance of the Nedd2 mRNA induction cannot be discussed clearly here because the induction of Nedd2 protein or its biochemical activity was not investigated in this study; however, the time course of the induction of Nedd2 mRNA seems compatible with the idea that the increased expression of this enzyme is involved in the initiation or execution of apoptosis in the ischemic brain following permanent MCA occlusion because oligonucleosomal DNA fragmentation, which is considered to be a late stage of apoptosis, occurred several hours after the induction of Nedd2 mRNA.
Footnotes
Acknowledgment:
We thank Dr. M. Miura (University of Tsukuba, Japan) for murine Ice cDNA, Dr. M. Noda (Kyoto University, Japan) for murine Nedd2 cDNA, Dr. Y. Tsujimoto (Osaka University Medical School, Japan) for human bcl-2 cDNA, and Dr. H. Kamesaki (Aichi Cancer Center Research Institute, Japan) for murine bcl-x cDNA.