
Abstract
DNA nick end-labeling (TUNEL) and heat shock protein (HSP)70 immunocytochemistry were performed on the same brain sections 1 (n = 6), 3 (n = 12), and 7 (n = 7) days following permanent middle cerebral artery (MCA) occlusions produced in adult rats using the endovascular carotid suture method. In the cortex at 1 and 3 days following MCA occlusions, HSP70 immunoreactive neurons were located outside areas of infarction and showed little evidence of DNA fragmentation. HSP70-stained cortical neurons were intermingled with TUNEL cells near the infarct, but extended for greater distances away from the infarct. DNA fragmentation occurred in CA1 hippocampal neurons in 39% of the animals at 1 and 3 days following ipsilateral MCA occlusion. Bilateral DNA fragmentation occurred in CA1 neurons in one subject. HSP70 protein was expressed in CA1 hippocampal neurons in nine of 18 (50%) animals at 1 and 3 days following MCA occlusions, including all animals that exhibited hippocampal DNA fragmentation. Three animals had bilateral expression of HSP70 in CA1 neurons. Cells that stained for either HSP70 protein or DNA fragmentation existed in close proximity to one another. Approximately 5–7% of HSP70-stained cells were TUNEL stained and 3% of TUNEL-positive cells also stained for HSP70. There was no HSP70 staining or DNA fragmentation in the brains of sham-operated controls (n = 4) or in the brains of animals 7 days following MCA occlusions. These data suggest that ischemic cells capable of translating HSP70 protein generally do not undergo DNA fragmentation. These data support the concept that most HSP70 protein-containing neurons in the cortical “penumbra” and hippocampus survive ischemic injury and are “reversibly injured.” It is shown that CA1 hippocampal pyramidal neurons die or are reversibly injured in ˜50% of animals following permanent MCA occlusions. Although the mechanism of this hippocampal injury is unknown, it could relate to transynaptic activation of N-methyl-
Keywords
The hsp70 heat shock gene is induced in brain following ischemia (Gonzalez et al., 1989, 1991; Kinouchi et al., 1993a,b, 1994a,b; Li et al., 1992; Nowak, 1991; Nowak and Jacewicz, 1994; Sharp et al., 1992; Vass et al., 1988). Denatured proteins (Anathan et al., 1986) and other signals activate heat shock factors, which bind to the heat shock elements in the hsp70 promoter to initiate hsp70 transcription (reviewed in Nowak and Jacewicz, 1994; Massa et al, 1996). In severely ischemic areas of the brain, cells destined to die may transcribe hsp70 mRNA but may not translate it into heat shock protein (HSP)70 (Nowak and Jacewicz, 1994; Kinouchi et al, 1993b). hsp70 mRNA and HSP70 protein are both induced in neurons outside areas of infarction (Kinouchi et al., 1993a,b; 1994a). The extent of this neuronal HSP70 protein induction can be used to describe a “penumbra” of stress gene induction surrounding areas of infarction (Kinouchi et al., 1993b; Massa et al., 1995). Although it was suggested that HSP70 immunostained neurons may be “reversibly injured” and survive the injury (Massa et al., 1996; Nowak and Jacewicz, 1994; Sloviter and Lowenstein, 1992), the fate of such HSP70 protein-expressing neurons is unclear.
HSP70 is also induced in regions that are some distance from areas of infarction. Middle cerebral artery (MCA) occlusions induce hsp70 mRNA in hippocampus, substantia nigra, and thalamus (Kinouchi et al., 1994a,b; Ferriero et al., 1990; Kamii et al., 1994b; Welsh et al., 1992; Yamada et al., 1994). The immediate early genes (IEGs), including c-fos, c-jun, jun-B, and NGFIA, are also induced in regions remote from areas of infarction including cortex, hippocampus, substantia nigra, and thalamus (An et al., 1993; Collaco-Moraes et al., 1994; Gass et al., 1992; Kamii et al., 1994a; Kinouchi et al., 1994; Sagar et al., 1988; Welsh et al., 1992; Uemura et al., 1991). hsp70 and JEG gene-inductions in these regions are notable because they lie outside the MCA distribution, suggesting that such inductions may be caused by secondary responses to ischemia such as spreading depression (Nedergaard and Astrup, 1986), diaschisis (Feeney and Barron, 1986; Ginsberg et al., 1977), deafferentation (Yamada et al., 1994), or transynaptic activation caused by ischemia-induced spreading depression (Kinouchi et al., 1994b).
It is unclear whether the remote induction of hsp70 or IEGs following focal cerebral ischemia is associated with cell death. Neuronal death in brain regions remote from sites of focal ischemic injury has been well described. The thalamus is known to undergo a slow, long-term atrophy following large MCA occlusions in humans (Tamura et al., 1991) and in animal models (Ciricillo et al., 1994; Fujie et al., 1990; Hara et al., 1993; Iizuka et al., 1990; Kataoka et al., 1989; Myers et al., 1991; Nagasawa et al., 1994a,b; Tamura et al., 1991; Tokuno et al., 1992; Yamada et al., 1991). Following large MCA strokes that infarct striatum, the ipsilateral substantia nigra slowly atrophies in humans (Forno et al., 1983; Inagaki et al., 1992) and in experimental animal models (Nakane et al., 1992; Saji et al., 1994; Tamura et al., 1990). Although there are conflicting data on whether damage occurs in the hippocampus following MCA occlusions (Benyo et al., 1995; Bereczki and Csiba, 1993; Czurko and Nishino, 1993; Okada et al., 1995), a recent study by Zhu and Auer (1995) demonstrated bilateral hippocampal neuronal cell death following MCA infarctions.
The present study was undertaken to determine whether HSP70 protein was induced and whether DNA fragmentation occurred in cells in the remote regions described above. In situ DNA nick end-labeling (TUNEL) was performed at various times following MCA occlusions. This method detects fragmented genomic DNA occurring during apoptotic and necrotic cell death (Gavrieli et al., 1992; Gold et al., 1994; Grasl-Kraupp et al., 1995; Li et al., 1995a,b; MacManus et al., 1993, 1994, 1995). HSP70 immunocytochemistry was performed on the same sections. Since we have suggested that HSP70-stained cells survive ischemic injury (Massa et al., 1996), it was anticipated that HSP70 and TUNEL would stain different populations of cells following MCA occlusions. It was also anticipated that HSP70 protein would be induced in regions remote from the infarction where hsp70 mRNA is induced following focal ischemia (Kinouchi et al, 1993a,b; 1994a,b).
MATERIALS AND METHODS
Animal preparation
The MCA of male Sprague-Dawley rats (Bantin and Kingman, Freemont, CA, U.S.A.) (280–320 g) was permanently occluded using the endovascular internal carotid artery suture method of Longa et al. (1989) with minor modifications. Anesthesia was induced using 3% isoflurane in a mixture of 70% N2O:30% O2 (vol/vol). Rats were intubated and anesthesia maintained with mechanical ventilation using 1.5% isoflurane in the above N2O:O2 mixture. The right common carotid artery (CCA) was exposed at its bifurcation through a midline cervical incision. The various branches of the external carotid artery (ECA) were coagulated. The pterygopalatine artery was ligated with a 4–0 silk suture. The ECA was transected and a 3–0 nylon monofilament suture, its tip rounded by heating, was inserted into the ECA stump. It was advanced into the internal carotid artery (ICA) for a distance of 22 mm beyond the CCA bifurcation to occlude the origins of the MCA and proximal anterior cerebral artery (ACA). The suture was secured in place with a ligature and the wound closed. The animals were allowed to survive for 1 (n = 6), 3 (n = 12), or 7 (n = 7) days with food and water available ad libitum. Seizures were not observed in the current experiments at any time following MCA occlusions.
Surgery required 15–20 min. Rectal temperature was maintained in the normal range before, during, and for 10 min after ischemia using a thermistor-controlled heating blanket. Postischemia hyperthermia, which occurs with the suture model (Zhao et al., 1994), could not be controlled. However, since the body temperature never goes >39.5°C following MCA occlusions, this would not be high enough to induce HSP70 by itself. In addition, hyperthermia only induces HSP70 in endothelial cells and glial cells, so that HSP70 induction in neurons in this study cannot be accounted for by hyperthermia alone (Brown, 1990). Following MCA occlusions, there was no mortality in the 1-day group (0/6, six survivors), 33% mortality in the 3-day group (6/18, 12 survivors), and 36% mortality in the 7-day group (4/11, seven survivors). Brains of the animals that died showed swelling of the infarcted hemisphere. Isoflurane was chosen as the anesthetic agent since recovery is rapid and it has no reported harmful effects that might induce HSP70 (Sharp et al., 1991).
Controls (n = 4) were subjected to an identical sham surgery. Control animals were killed 3 days following surgery since this was the time of maximal HSP70 induction in the MCA occlusion groups. At the appropriate time following the surgeries, ischemic and control animals were given a lethal overdose of chloral hydrate. Once deeply anesthetized, they were perfused through the ascending aorta with 200 cc of 10% glycerol in phosphate-buffered saline (PBS). After perfusion, the brains were rapidly removed and frozen in 2-methyl butane at −25°C. Brains were embedded in Lipshaw matrix, wrapped in foil, and kept at −70°C until sectioning was performed.
TUNEL
A modification of the TUNEL technique described by Gavrieli et al. (1992) was used. Coronal 20 μm sections were cut in a cryostat (Hacker Instruments, Fairfield, New Jersey, U.S.A.) at −20°C and collected on Fisherbrand Superfrost/Plus slides (Fisher Scientific, Pittsburgh, PA, U.S.A.). Sections were fixed for 10 min in 4% paraformaldehyde and placed in 0.1 M PBS, (pH7.4)2 × 15 min, 0.3% Triton X-100 in PBS 2 × 15 min, and PBS 2 × 15 min. IX terminal deoxynucleotidyl transferase (TdT) buffer (Gibco BRL, Gaithersburg, MD, U.S.A.) was applied for 15 min. A mixture of TdT (Gibco BRL) (300 U/ml) and biotin 14-dATP (40 μM) in IX TdT buffer was applied on the sections and then were incubated at 37°C for 60 min. Slides were washed in 2X SSC, pH 7.0, (2 × 15 min), 2% bovine serum albumin (BSA) in PBS, and PBS 2 × 15 min. Avidin-biotin-horseradish peroxidase solution (Elite Vectastain, Burlingame, CA, U.S.A.) was applied for 30 min followed by 0.175 M sodium acetate solution 2 × 15 min. Staining was visualized using 0.015% diaminobenzidine (DAB), 0.001% H2O2, and 1% nickel sulfate in 0.175 M sodium acetate. After staining, slides were rinsed with water. Positively-stained cells had dark blue to black, round nuclei.
HSP70 immunocytochemistry
After processing for TUNEL, HSP70 immunocytochemistry was carried out on the same sections. HSP70 mouse monoclonal antibody C92 (Amersham, Arlington IL, U.S.A.) (1:4000 in 1% BS A, vol/vol) was applied to sections and incubated at 4°C overnight. Sections were washed in 1% BS A 2 × 15 min and incubated in second antibody (biotinylated sheep antimouse IgG) (Amersham) (1:200 in 1% BS A) for 30 min. After washes in PBS 2 × 15 min, avidin-biotin-horseradish peroxidase solution was applied as above. Sections were washed in PBS and placed in 0.015% DAB and 0.001% H2O2 in PBS for 2–8 min. Slides were washed with water, dehydrated in ethanol, and cover slipped. HSP70-stained cells had brown staining of the cell body and dendrites. Nissl staining was performed on adjacent sections. The C92 monoclonal antibody to HSP70 was raised to human HSP70 protein (Welch and Suhan, 1986). It detects two bands on Western blots of ischemic gerbil brain (Vass et al., 1988) and detects HSP70 protein expressed from the rat hsp70 cDNA (Longo et al., 1993; Narasimhan et al., 1996).
Controls and cell counting
Positive controls for HSP70 immunostaining and TUNEL included brains of rats that had kainic acid (10 mg/kg, i.p.) seizures induced 24 h previously. Positive controls for the TUNEL staining also included control brain sections treated with DNAase. Negative controls for the HSP70 immunostaining included deletion of the primary or secondary antibody. Negative controls for TUNEL staining included normal rat brain frozen sections from rats that received no treatment of any kind.
Counting was performed to determine what proportion of HSP70 stained neurons also had evidence of TUNEL. Counting was performed on animals (n = 6) that demonstrated HSP70 staining in hippocampus at 3 days following MCA occlusions. HSP70-stained neurons were counted in CA1 of the hippocampus and in the dorsal neocortex adjacent to the infarction in regions showing the highest density of stained cells of both types. At least 50 HSP70 stained cells per animal were counted; cells that were also TUNEL-positive were recorded.
RESULTS
DNA fragmentation
There was no TUNEL evidence of infarction or any edema in any brain region of sham-operated control subjects (Fig. 1F; Table 1). Every brain at 1, 3, and (most at) 7 days following MCA occlusions demonstrated evidence of a midline shift and edema in the infarcted hemisphere. In brains of ischemic animals, TUNEL staining showed labeled cell nuclei exhibiting a dark blue to black color (Figs. 1A,C,E; 2A-F). In some cells, labeled DNA appeared to aggregate in more densely-stained punctate masses, which had the appearance of being compacted against the nuclear membrane (Fig. 2A). Other cells, however, exhibited homogeneous staining of labeled nuclei, which could be consistent with either necrosis or apoptosis (Fig. 2A).
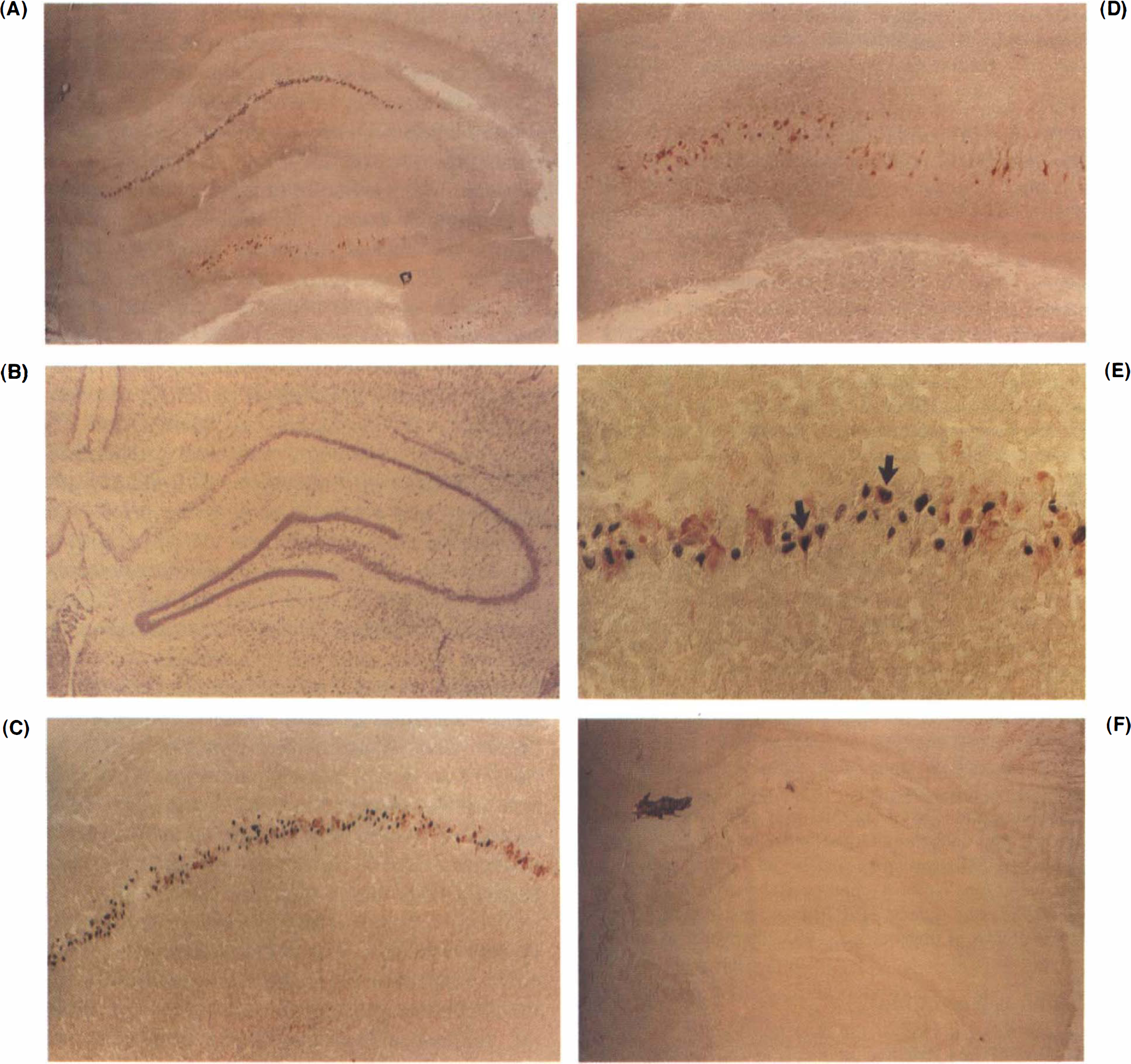
Photomicrographs of the hippocampus (
TUNEL in the cortex was detected by 1 day, was greatly increased at 3 days (Fig. 2E,F), and had disappeared in cortex by 7 days (not shown). Areas of infarction, mainly in the cortex and basal ganglia, had many stained cells at all time points, with the amygdala and adjacent basal cortex exhibiting the greatest number. Outside the areas of infarction, TUNEL cells were observed in the ipsilateral hippocampus in two of six 1-day animals, and in five of 12 3-day animals (Table 1). Hippocampal DNA fragmentation was most pronounced in the CA1 region, particularly the medial aspect (Fig. 1A). In one animal at 3 days following ischemia, the contralateral CA1 had numerous stained cells (Table 1; Fig. 2C,D). DNA fragmentation was rare in cells in other fields of Ammon's horn. Hippocampal DNA fragmentation was not observed in the 7-day group (Table 1). No stained cells were observed in the substantia nigra or thalamus except in the few cases where these areas were part of a large infarct (data not shown).
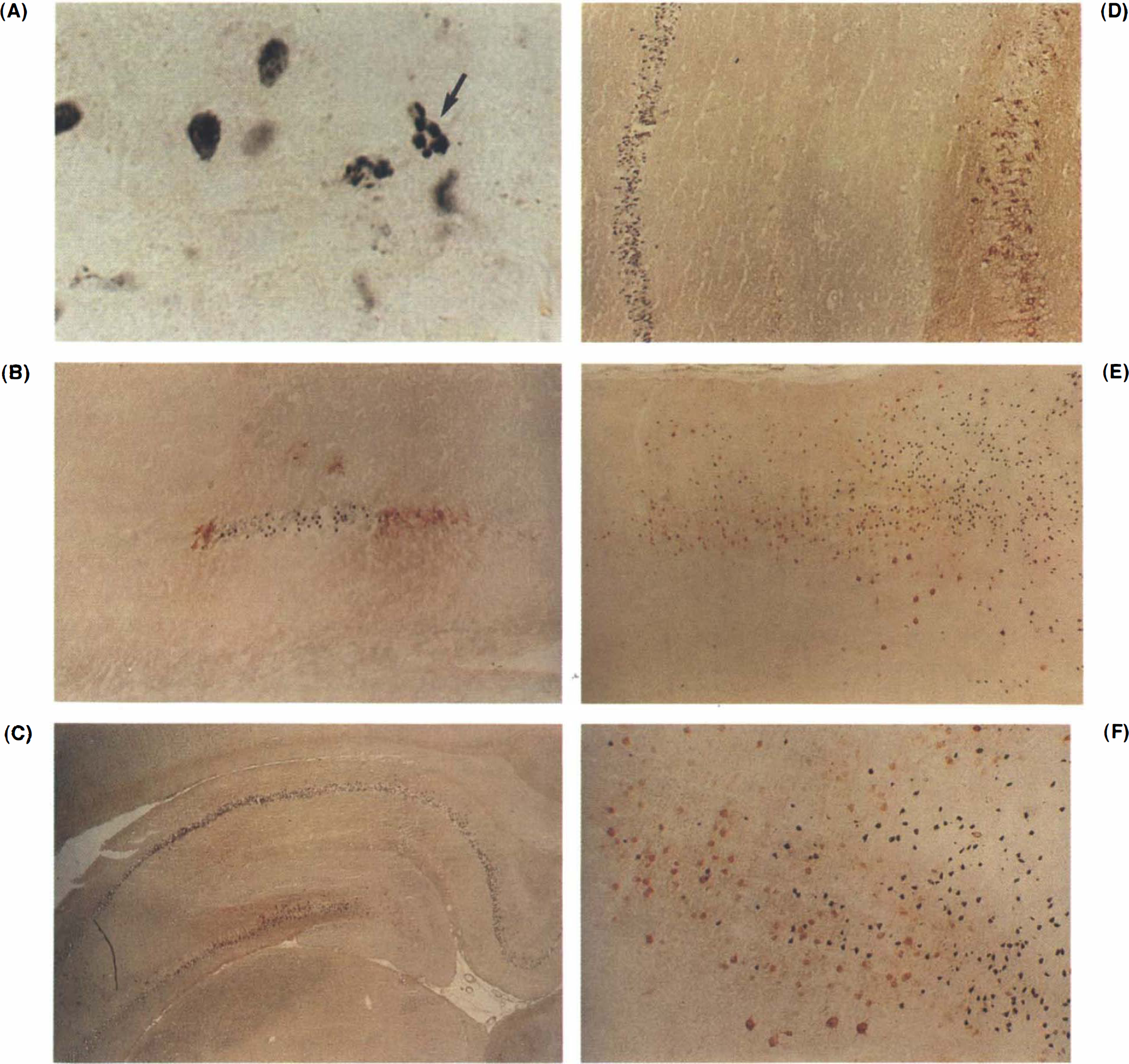
A TUNEL of hippocampal pyramidal neurons 72 h following permanent MCA occlusion with two cells demonstrating apoptotic bodies, with discrete balls of stained DNA, adjacent to many more stained cells that demonstrate varying degrees of chromatin condensation.
HSP70 induction
Use of peroxidase as the sole amplification substrate for double staining of molecules compartmentalized to the nucleus and cytoplasm has been previously described. Although the Nickel/DAB precipitate from the TUNEL reaction would have masked any HSP70 antigenic sites in the nucleus of these cells, the cytoplasm of these cells was free of TUNEL label. Reliable detection of HSP70 protein in the cytoplasm of nuclear stained TUNEL cells was, therefore, possible (Fig. 1C,E).
HSP70-immunostained neurons were not detected in the brains of sham-operated, control animals. In animals subjected to permanent MCA occlusions, HSP70 protein induction within areas of infarction was confined mainly to endothelial cells, which may survive the ischemia (Kinouchi et al., 1993a,b; Dietrich et al., 1984). In some animals, a rim of HSP70 immunostaining occurred in the dorsolateral cortex, a border zone between MCA and AC A territories. In these areas, many, but not all, neurons stained intensely for HSP70 protein, as did variable numbers of glial cells.
In areas remote from the infarction, 50% of animals in the 1-day group and 50% in the 3-day group expressed HSP70 in the hippocampus (Table 1). HSP70 was expressed in the contralateral hippocampi of three animals (two in the 1-day group, one in the 3-day group). HSP70 induction in hippocampus at 1 and 3 days following ischemia was greatest in neurons in the CA1 region (Figs. 1A,C,E; 2B), but also occurred in CA3 and the dentate hilus (Figs. 1A,D; 2C,D). The dentate gyrus rarely expressed HSP70. HSP70 was not induced in any contralateral area except the hippocampus (Fig. 2C, D). There was no evidence of HSP70 protein induction in the 7-day group (Table 1) and no induction in the substantia nigra or thalamus in any animal at any time.
HSP70 induction in cells in relation to DNA fragmentation in cells
At the edge of the infarcted area in dorsomedial cortex, as the MCA territory extended medially towards ACA territory, the number of TUNEL cells diminished while the number of HSP70-positive neurons increased (Fig. 2E,F). The border between these two territories corresponded to the area of greatest regional co-mingling of cells that stained positively for either HSP70 induction or DNA fragmentation (Fig. 2E,F). This area was located immediately adjacent to the infarction. In this area, a few double-stained cells could be located on most cortical sections (not shown for cortex; see Fig. 1E for double-labeled cells in CA1 of the hippocampus). Double-labeled cells in cortex represented a minority of the total population of cells that were positively stained for either DNA fragmentation or HSP70 alone. Of 590 counted HSP70 -stained cells, 5.5% showed DNA fragmentation in the dorsal cortex 3 days following infarction.
Summary of TUNEL (DNA fragmentation) and HSP70 immunostaining in ipsilateral CA1 (ipsi) and contralateral CA1 (contra) pyramidal neurons of hippocampus of control animals (n = 4), and of animals 1 n = 6), 3 (n = 12), and 7 (n= 7) days following permanent MCA occlusion

Values represent the number of animals.
In the cases where hippocampal DNA fragmentation was observed in most of the CA1 pyramidal neurons, CA3 pyramidal neurons and dentate hilar neurons stained intensely for HSP70 (Figs. 1A,D; 2C,D). As the CA1 field coursed laterally in these animals, the number of TUNEL cells diminished while the number of HSP70-positive cells increased (Fig. 1A,C). In animals that had lesser degrees of DNA fragmentation in CA1, HSP70 induction occurred in neurons at the margins of small zones of CA1, where only TUNEL cells were detected (Fig. 2B). As in cortex, hippocampal cells that stained for HSP70 protein were observed adjacent to cells that demonstrated TUNEL (Fig. 1C,E). Cells that stained for HSP70 protein and double stained for TUNEL were observed in the ipsilateral hippocampus (Fig. 1E), but accounted for only 6.8% of 680 counted HSP70-stained cells in the CA1 region of animals 3 days following MCA occlusions. Of TUNEL-stained cells, 3% also stained for HSP70 in the hippocampus.
DISCUSSION
This study confirms that hippocampal neurons can be damaged and die following permanent MCA occlusions in adult rats. It is proposed that the HSP70 immunoreactive pyramidal neurons detected in CA1 of hippocampus and in the penumbra in cortex following MCA occlusions are “stressed” ischemic neurons that survive the injury.
Previous evidence for hippocampal injury and cell death following focal ischemia
Histological changes in hippocampus have not always been observed following MCA occlusions. For example, although long-term spatial memory impairment has been noted following MCA occlusion in rats in a recent study (Okada et al., 1995), no histological changes were noted in the hippocampus. Others have described defects of memory following MCA occlusion (Markgraf et al, 1992; Okada et al., 1995, Wahl et al, 1992), although it was not always clear whether these deficits correlated with damage to hippocampus (Ott and Saver, 1993). In spontaneously hypertensive rats (SHR), defects in memory have been recorded after MCA occlusions without any apparent effects on hippocampus (Yamaguchi et al, 1994).
Studies of gene expression, however, have shown that hsp70, c-fos, and other genes may be induced throughout hippocampus following MCA occlusions (An et al., 1993; Li et al., 1992; Kinouchi et al., 1994a,b; Welsh et al., 1992), although this may not occur in all models (Collaco-Moraes et al., 1994). Induction of these genes in the hippocampus appears to be dependent, in part, on the oxidative stress of the hippocampus (Kamii et al., 1994a,b).
Biochemical changes have also been detected in the hippocampus following MCA and carotid occlusions. In the Levine model of combined hypoxia and carotid occlusion in the 7-day old rat, changes in ATP levels and induction of interleukin-1β and HSP70 have been detected in the hippocampus as well as the distribution of the MCA (Ferriero et al., 1992; Szaflarski et al., 1995). Transient MCA ischemia decreases ATP levels and produces acidosis in the hippocampus as well as in the cortex and thalamus (Bereczki and Csiba, 1993). Unilateral carotid occlusion elevates extracellular amino acids in the hippocampus in SHR and normal rats (Gemba et al., 1992). Bilateral carotid occlusion elevates glutamate and taurine in hippocampus in SHR and normal rats, and can produce bilateral CA1 neuronal cell death with upregulation of endothelins ET1 and ET3 in SHR rats (Yamashita et al., 1993). Mild focal ischemia can protect the gerbil hippocampus against subsequent global ischemia (Miyashita et al., 1994). MCA occlusion in normal adult Sprague Dawley rats produces moderate decreases of blood flow and glucose metabolic rate in the hippocampus (Shiraishi et al., 1989).
Histological changes in hippocampus have been documented in a few focal MCA ischemia studies. Embolic occlusion of the MCA may decrease hippocampal volume (Lyden and Lonzo, 1994). MCA occlusions result in loss of hippocampal CA1 parvalbumin immunoreactivity, which can be prevented by nimodipine (Benyo et al., 1995). Unilateral carotid occlusion combined with systemic hypotension produced damage mainly in the ipsilateral CA1 region of Wistar rats in 21 of 161 animals and, in four of these, contralateral hippocampal damage was also observed (Zhu and Auer, 1995). A recent study by Czurko and Nishino (1993) has demonstrated argyrophyllic, dead CA1 pyramidal neurons in the hippocampus at 3 days following 2 h of MCA occlusion using the endovascular suture model employed in the present study. Results of this previous study, combined with the present results, confirm that CA1 pyramidal neurons can be injured following MCA occlusions in rats, although only ˜50% of the animals sustained hippocampal damage following permanent MCA occlusions in the present study.
Possible mechanisms of neuronal cell injury in hippocampus following MCA occlusion
Even though the hippocampus is outside the distribution of the middle cerebral artery, the mechanism of hippocampal cell injury might be due in part to ischemia. Moderate decreases (50%) of glucose metabolism and blood flow (Shiraishi et al., 1989), as well as increases of metabolism (Levy and Duffy, 1977; Lockwood et al., 1989) have been observed in hippocampus following MCA occlusions. In addition, all animals in this study demonstrated evidence of a marked midline shift at death, indicative of edema, which could affect blood flow to hippocampus. The small zones of HSP70 induction and DNA fragmentation in CA1 of hippocampus following MCA occlusion in some animals are similar to the small zones of HSP70 protein induction observed in the hippocampus following global ischemia using the rat two-vessel occlusion, systemic hypotension model (Gonzalez et al., 1991). In that study, it was suggested that the ischemia around individual penetrating arteries to the hippocampus that run perpendicular to CA1 might account for small zones of injured cells in CA1. The present findings would be consistent with this hypothesis as well, since a region of CA1 pyramidal neurons with DNA fragmentation was usually bounded on either side by HSP70 immunoreactive neurons (Fig. 2B). Ischemia around a single penetrating vessel might create a “microcore” surrounded by a “micropenumbra.” Lastly, the combination of unilateral carotid artery occlusion and elevated intracranial pressure (Busto and Ginsberg, 1985) produced marked decreases of blood flow in the hippocampus as well as the cortex and striatum, markedly decreased ATP and phosphocreatine, and increased lactate in these same structures. The magnitude of the changes in the hippocampus were similar to those observed in the core of the infarction. Therefore, it seems possible that elevated intracranial pressure due to edema following MCA occlusion could have decreased cerebral blood flow in the hippocampus and at least contributed to the death of CA1 neurons in the hippocampus in this study.
Ischemia alone, however, probably does not explain the selective vulnerability of CA1 (compared to other hippocampal neurons) to injury, demonstrated in this and other studies. An alternative hypothesis for cellular injury in the hippocampus following MCA occlusion could be related to trophic support. The thalamus slowly atrophies and neurons die slowly following cortical lesions and infarctions (Iizuka et al., 1990) presumably due to loss of trophic support (Ciricillo et al., 1994). The rather rapid death of CA1 neurons following MCA occlusion in this study, however, might be more consistent with a rapid, transynaptic death of cells rather than a loss of trophic support.
Overexcitation of hippocampal neurons, resulting from loss of inhibitory input or overactivation of excitatory input from the ischemic core or penumbra projecting to hippocampus, is another mechanism that might account for the hippocampal cell death observed in this study. The glucose metabolic rate of the substantia nigra increases following ischemic destruction of inhibitory afferents projecting from the striatum (Tamura et al., 1981). Furthermore, delayed cell death in the substantia nigra following destruction of the striatum can be prevented by intraventricular infusion of the GABAergic agonist muscimol (Saji et al, 1994). Direct hippocampal overexcitation by electrically-deregulated cells that project to the hippocampus from the ischemic core or penumbra is possible. The amygdala is an injured region in this model that shares afferent and efferent connections with CA1. It is notable that administration of MK801 prior to MCA occlusion blocks induction of hsp70 mRNA and immediate early gene mRNAs (c-fos, jun-B, others) in the hippocampus and thalamus (Kinouchi et al., 1994b). These data support the possibility that MCA infarctions produce overactivation of N-methyl-
An overlooked mechanism of injury could be related to primary opening of the blood-brain barrier (BBB). Following unilateral MCA occlusion, bilateral opening of the BBB has been demonstrated by diffuse, bilateral leakage of horseradish peroxidase (Dietrich et al., 1988). It is possible that opening of BBB produced by MCA occlusion resulted in exposure of both hippocampi to blood-borne molecules, including iron-containing heme proteins, which could mediate induction of hsp-70, c-fos, and other genes, and possibly contribute to the cellular injury observed.
Apoptosis versus necrosis of hippocampal neurons following MCA occlusion
Two morphologically distinct forms of cell death have been defined—necrosis and apoptosis (Schwartz et al., 1993; Searle et al., 1982). Apoptotic morphology is defined by blebbing of the plasma membrane, chromatin condensation, and, ultimately, formation of nuclear membrane-bound “apoptotic bodies” (Searl et al., 1992). Apoptosis is characterized biochemically by endonuclease-mediated internucleosomal DNA cleavage that results in the appearance of “laddered” DNA of 185–200 bp multiples when DNA is electrophoresed on gels (Bursch et al., 1990; Charriaut-Marlangue et al., 1995; Kure et al, 1991; MacManus et al., 1993, 1994, 1995). There is growing evidence that global and focal ischemia, as well as other types of brain injury, can produce cell death via apoptosis, as evidenced by DNA laddering and TUNEL staining typical of that seen in apoptosis (Charriaut-Marlangue et al., 1995; Heron et al, 1993; Kihara et al, 1994; Li et al., 1995a,b; Linnik et al., 1993; MacManus et al., 1993, 1994, 1995; Nitatori et al., 1995; Okamoto et al., 1993; Schreiber et al, 1993; Sci et al., 1994).
The TUNEL method has been used to identify apoptotic cells in situ, although the method has been questioned, since it can stain DNA that is cleaved during necrosis as well. Gold et al. (1994) suggested that DNA damage after necrosis occurs at random sites and involves single-stranded nicks, in contrast to the internucleosomal double-stranded breaks observed in apoptosis. In that study, the specificity of DNA nick translation by DNA polymerase I, which adds nucleotides only to single-stranded DNA breaks, was compared to that of TUNEL or “tailing” by TdT, which labels double-stranded breaks in DNA. The nick end labeling appeared to have greater specificity for apoptotic compared to necrotic cell death, as detected by gel electrophoresis, flow cytometry, and morphological analysis, whereas the reverse was true for nick translation. Kihara et al. (1994) found that necrotic tissue in glioblastoma cells was not stained by nick end labeling. However, McManus et al. (1995) demonstrated diffuse TUNEL staining, including staining in CA1 neurons, in brain following decapitation ischemia, whereas there was little evidence of DNA laddering and, hence, no apoptosis following this type of cell death. Therefore, TUNEL alone cannot be used as evidence of apoptotic cell death.
It is notable that the cellular morphology observed using TUNEL in this study was very similar to that described by Li et al. (1995a,b), who used the same surgical model to produce focal ischemia. The majority of nuclei positively labeled by this technique contained, within them, more densely-stained punctate masses. However, only a small percentage of positively-labeled cells exhibited discrete and perfectly separate (i.e., no labeled chromatin between them) apoptotic bodies, possibly because once chromatin condensation is complete, apoptotic bodies are rapidly phagocytosed and degraded; thus, the time window in which they are likely to be visualized might be short. The presence of apparent apoptotic bodies in at least some cells in the hippocampus and cortex in this study supports the idea that at least some cells undergo an apoptosis-like death.
Whatever the mechanism of cell death leading to TUNEL in the present study, be it apoptosis or necrosis, it seems reasonable to conclude that the cells stained using this method are dead. This is supported by the fact that the DNA would have to be cleaved into many pieces for the cell to be stained, suggesting that this is a terminal event.
Relationship Between cell death and reversible cell injury
The pattern of HSP70 expression and DNA fragmentation at the margin of the infarcts in the dorsomedial cortex suggests a gradient in the severity of the ischemic injury. Although HSP70-stained neurons were intermixed with nick end-labeled neurons, HSP70-positive neurons extended well beyond the area of infarction and the region defined by the TUNEL cells. It has been suggested (reviewed by Massa et al., 1996; Vass et al, 1988) that HSP70 immunoreactive neurons survive ischemic injury. This is based on the observations that (a) HSP70 protein-expressing neurons appear morphologically normal, (b) HSP70 protein-containing neurons are only seen in areas outside the infarction, (c) neurons destined to die in CA1 in the gerbil global ischemia model do not synthesize HSP70 protein, (d) and prolonged stimulation of the perforant pathway induces HSP70 protein in cells that will survive from the stimulation (Massa et al, 1995; Nowak, 1991; Nowak and Jacewicz, 1994; Sloviter and Lowenstein, 1992). The present data are consistent with this hypothesis since HSP70 immunoreactive neurons in this study generally did not demonstrate evidence of DNA fragmentation. It is proposed, therefore, that the zone of HSP70 protein-immunoreactive neurons defines a “penumbra” of reversible neuronal injury wherein the bulk of the injured neurons will survive the ischemic insult. This “penumbra” of reversible neuronal injury is generally larger than the “penumbra” region of selective neuronal cell death that occurs within a small rim just outside an area of infarction (Nedergaard and Astrup, 1986; Li et al., 1995a,b; present study).
The finding that HSP70 protein-containing neurons generally do not have evidence of DNA fragmentation also has other implications. Both types of cells can be found in the zone immediately adjacent to infarctions; this suggests that cells very close to one another appear to sustain variable degrees of injury, although the further away a cell is from the ischemic core, the less likely it is to undergo selective cell death. In addition, since HSP70 protein and DNA fragmentation are infrequently observed within the same cell, the idea is supported that cells that are lethally injured may not be capable of synthesizing HSP70 protein (Kinouchi et al, 1993a,b; Massa et al., 1995). This is consistent with our previous data showing that neurons within an infarct may or may not make hsp70 mRNA, but do not synthesize HSP70 protein (Kinouchi et al., 1993a,b). One could also speculate that cells that synthesize HSP70 protein may be protected from undergoing apoptosis (Massa et al., 1996) since HSP70 protein can protect cells against injury produced by heat shock, light, and other types of insults (Nowak and Jacewicz, 1994; Massa et al., 1995).
The finding that a small percentage of cells have evidence of DNA fragmentation in spite of expressing HSP70 protein suggests that HSP70 expression does not invariably protect against cell death. This may help explain results of studies in which many of the CA1 cells, which express HSP70 protein, may die following global ischemia in the rat (Simon et al., 1991), and in which many of the hippocampal CA1 and CA3 neurons that express HSP70 protein following kainic-induced seizures may go on to die (Gonzalez et al., 1989; Vass et al., 1989; Planas et al., 1995).
Although MCA occlusions produce atrophy of the thalamus and substantia nigra in a number of models—and HSP70 protein has been noted in the substantia nigra following MCA occlusions (Yamada et al., 1994)—no HSP70 protein was detected in these regions in this study. The failure to detect HSP70 in the substantia nigra might be related to the absence of hyperglycemia and postischemic seizures (Nevander et al., 1985; Inamura et al., 1988). The failure to detect HSP70 in the thalamus might be related to slow, delayed atrophy of the thalamus not being detected within the 1-week period in this study.
It is concluded that cell death can occur in the hippocampus following MCA occlusion in adult rats. Neuronal HSP70 induction occurs in brain areas in which selective neuronal cell death also occurs. However, HSP70 and DNA fragmentation occur infrequently in the same cells, suggesting that cells that are dead do not make HSP70 protein and that HSP70-stained cells are reversibly injured and, likely, survive the ischemic injury. Lastly, “penumbras” of different size can be defined on the basis of the distribution of cells with fragmented DNA and cells that stain immunocytochemically for HSP70 protein.
Footnotes
Abbreviations used
Acknowledgment:
This work was supported by NIH grants NS28167, NS14543, HL53040 (F.R.S. and Dr. Scott Panter), the Merit Review Program of the Department of Veterans Affairs (F.R.S., P.R.W.), and the Finnish Academy of Sciences and the Neurology Foundation of Finland (J.H.).