
Abstract
The interaction between ATP-sensitive K+ channels (KATP) and nitric oxide (NO) was studied in pial arterioles of piglets. We examined the effects of N-nitro-
Recent studies have revealed that ATP-dependent potassium channels (KATP) are present in cerebral arterioles (Ksoll et al., 1991; Faraci and Heistad, 1993) and are involved in cerebral blood flow regulation in both physiological and pathophysiological conditions (Faraci et al., 1994; Taguchi et al., 1994). Due to potential therapeutic possibilities, the mechanisms through which exogenous KATP regulators influence vascular tone are currently under intense study (Cook and Chapman, 1993; Challinor-Rogers and McPherson, 1994).
Several fundamental dilator responses of pial arterioles [e.g., to acetylcholine, glutamate, or N-methyl-
It is generally accepted that activation of KATP results in hyperpolarization and, thus, relaxation of vascular smooth muscle. However, recent experiments indicate that compounds that open KATP can induce vasorelaxation by mechanisms that are partly independent of hyperpolarization (Quast et al., 1994). Despite definite advances in the understanding of potassium channel-induced vasorelaxation, an important question remains unanswered: does NO contribute to pial arteriolar dilatation induced by potassium channel openers?
The finding that glibenclamide, an inhibitor of KATP, inhibits hyperpolarization to NO is a novel observation in rabbit and rat mesenteric arteries (Garland and McPherson, 1992; Murphy and Bray den, 1995). It suggests that a reciprocal relationship between KATP and NO may exist and that K+ channel activation may be a mechanism for the vasorelaxant effect of NO.
The first goal of this study was to determine whether vasorelaxation induced by aprikalim, through activation of KATP, is dependent on production of NO. We compared the effects of Nω-nitro-
MATERIALS AND METHODS
Newborn (1–7 days old) piglets of either sex with a body weight of 0.9–1.4 kg, were used in this study. Anesthesia was induced with sodium thiopental (30 mg/kg) followed by intravenous injection of α-chloralose (75 mg/kg). Supplemental doses of α-chloralose were given as needed to maintain a stable level of anesthesia. Animals were intubated via tracheotomy and artificially ventilated with room air. Rectal temperature was maintained at 37–38°C by a heating pad. Systemic arterial blood pressure was recorded via a right femoral artery cannula connected to a pressure transducer. The right femoral vein was cannulated for drug administration. The heads of the piglets were fixed in a stereotaxis frame. The scalp was incised and connective tissue over the parietal bone was removed. A 19-mm craniectomy for a stainless steel-glass cranial window was made in the left parietal bone, 10 mm rostral to the coronal suture. The dura was exposed, cut, and reflected over the skull. The cranial window with three needle ports was placed into the hole, sealed by bone wax and cemented with SuperGlue and dental acrylic. The closed window was filled with artificial cerebrospinal fluid (aCSF), which was warmed to 37°C and equilibrated with 6% O2 and 6.5% CO2 in N2 gas mixture. The composition of the aCSF was as follows (millimolar): KCl 2.9, MgCl2 1.4, CaCl2 1.2, NaCl 132, NaHCO3 24.6, urea 6.7, and glucose 3.7. Diameters of pial arterioles were measured using a microscope (Wild M36, Switzerland) equipped with a video camera (Panasonic, Japan) and video micro scaler (IV-550, For-A Co. Newton, MA, U.S.A.). Following the surgical procedure the cranial window was gently infused with aCSF several times until a stable baseline arteriolar measurement was obtained.
Protocols
In the first group (n = 6), we tested whether aprikalim elicits reproducible dilatation of pial arterioles over time. Arteriolar responses were determined to topically applied aprikalim of 10−6 and 10−8 M during control conditions and 45 min after the vessel diameter returned to baseline.
To examine the effect of inhibition of NOS on the aprikalim-evoked vasodilatation, we used two experimental groups. In one group (n = 8), we measured the change in diameter of pial arterioles (˜100 μm in diameter) to topical aprikalim (10−8 and 10−6 M) before, and 45 min after
In a separate group of animals (n = 4), we examined vascular responses to topical sodium nitroprusside (SNP) at doses of 10−6 and 10−5 M prior to and 45 min after
In another group (n = 7) arteriolar responses were determined in response to 10−4 M glutamate and to 10−4 M NMDA before and 45 min after the 7-NI treatment. In the final group of piglets (n = 9), we examined whether dilator responses of the pial arterioles to NO were mediated by KATP. We determined changes in vessel diameter to topical SNP at doses of 10−6 and 10−5 M before and in the presence of glibenclamide (10−5 M), an inhibitor of KATP. In a previous study we have shown that dilatation to aprikalim was completely inhibited when pretreated and coadministered with 10−5 M glibenclamide (Busija and Louis, 1995).
Nitric oxide synthase activity ex vivo was measured by quantification of the conversion of [l4C]-citrulline from
Drugs
Statistics
Data are expressed as mean ± standard deviation (SD). A paired t test was used for comparing data between two groups. For repeated measurement analysis, analysis of variance (ANOVA) was used, and the Student-Newman-Keuls test was then performed. Data analyses were performed on absolute and percent change data. A p value <0.05 was regarded as statistically significant.
RESULTS
Under control conditions, mean arterial blood pressure was similar in the various groups of animals (57 ± 4 mm Hg, n = 40). Arterial blood gases and pH were monitored regularly throughout the experiment and were kept in the physiological range (Pco2 = 33 ± 3 mm Hg, Po2 = 87 ± 7 mm Hg and pH = 7.44 ± 0.03, n = 40).
Administration of aprikalim resulted in dose-dependent arteriolar dilatation (Table 1). In the time-control group, 10−8 and 10−6 M aprikalim dilated pial arterioles by 10 ± 3% and 21 ± 4%, respectively, at the first application. Arteriolar dilatation to aprikalim did not change over time. It dilated by 10 ± 5% and 20 ± 5% with the second application (Table 1).
Arteriolar responses to repeated drug application in groups investigated
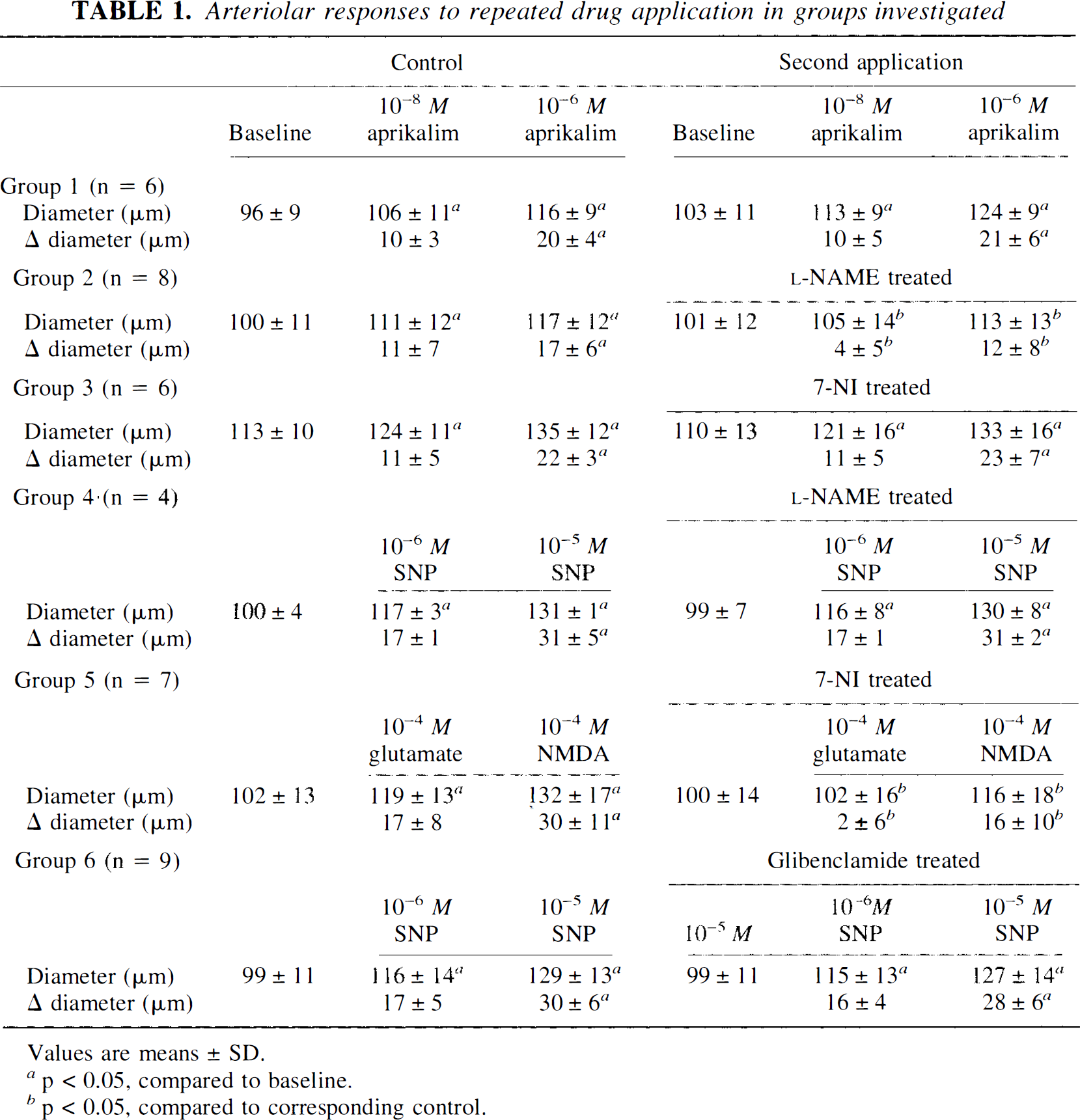
Values are means + SD.
p < 0.05, compared to baseline.
p < 0.05, compared to corresponding control.
Intravenous administration of
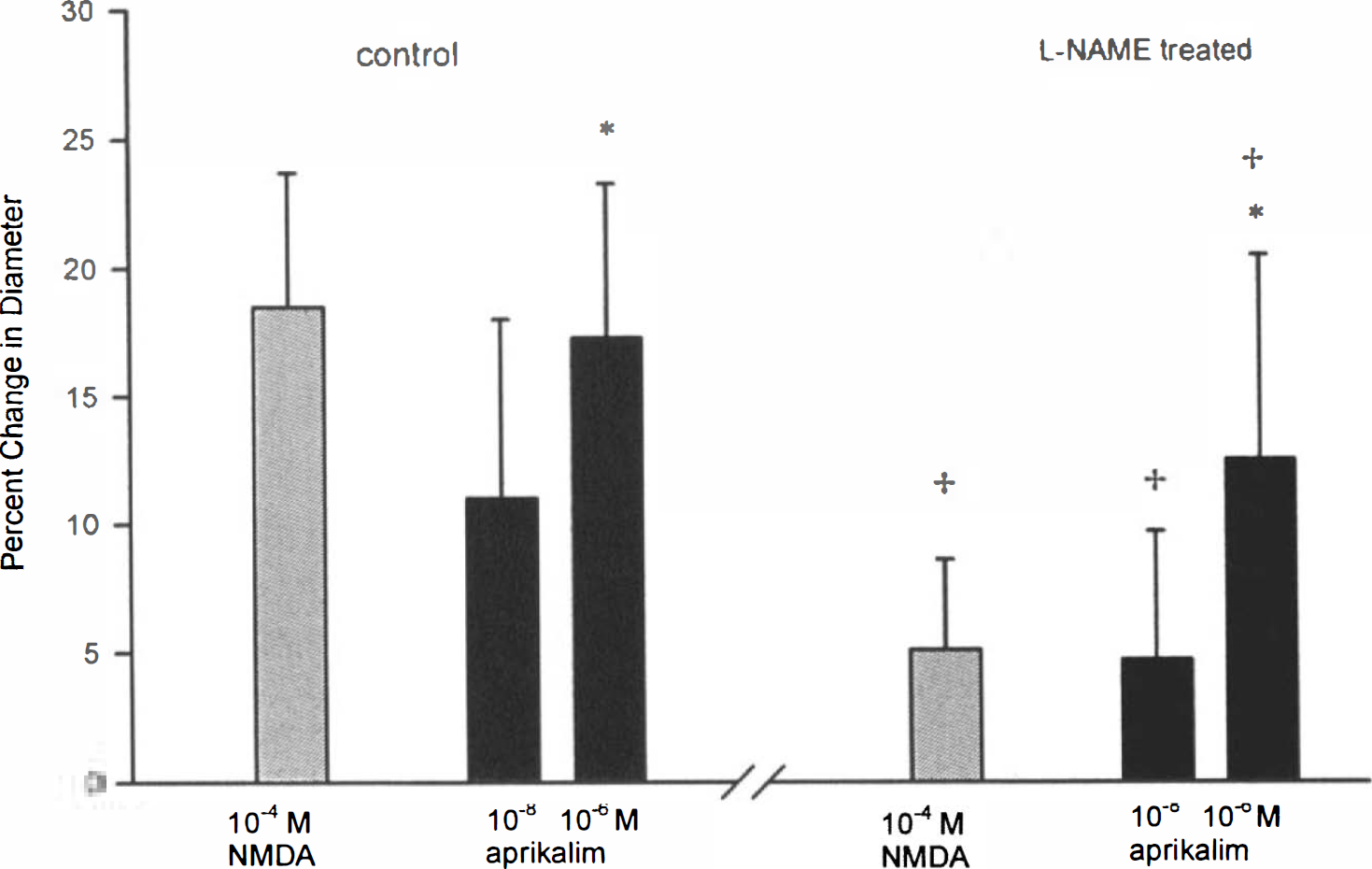
Percent change from control arteriolar diameter during application of N-methyl-
Intraperitoneal administration of 7-NI (50 mg/kg) had no effect on systemic blood pressure and on pial arterial diameters. Administration of 7-NI reduced the vasodilator activity of glutamate (10−4 M) and NMDA (10−4 M). The dilatory responses were 17 ± 8% and 29 ± 10% in control circumstances and 2 ± 6% and 16 ± 10% after drug application, respectively. However, the vasodilator responses to 10−8 and 10−6 M aprikalim were not reduced (Table 1, Fig. 2).
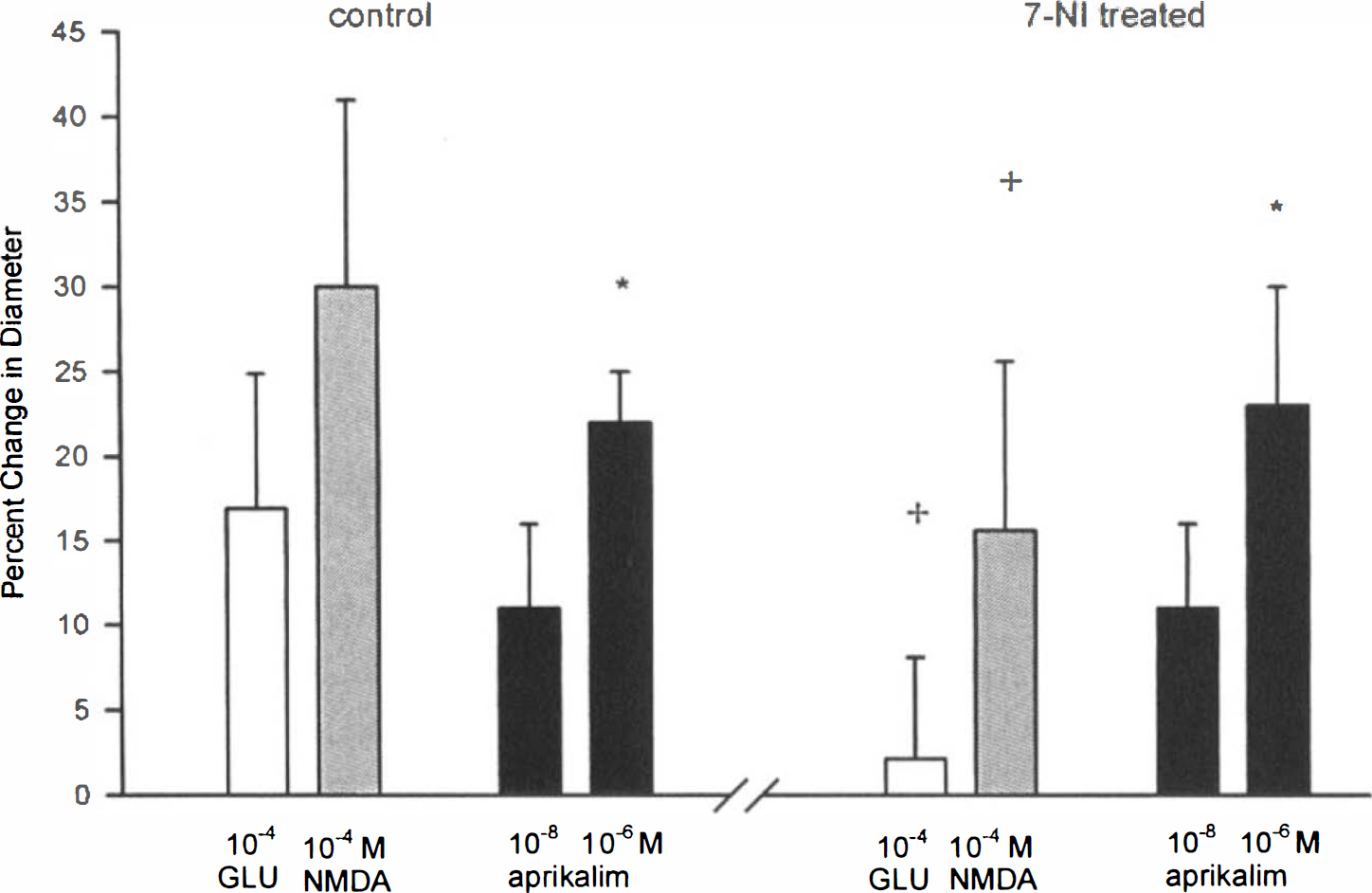
Percent change from control arteriolar diameter in response to 10−4 M glutamate (GLU), 10−4 M N-methyl-
After intravenous administration of
Dilatation of cerebral arterioles in response to SNP was not affected by
Pretreatment and coadministration of 10−5 M glibenclamide, an inhibitor of ATP-sensitive potassium channels, did not change the response to SNP (Table 1, Fig. 3). In this group (n = 9) 10−6 and 10−5 M SNP dilated pial arterioles by 17 ± 5 and 30 ± 7% prior to and by 16 ± 4% and 28 ± 6% after glibenclamide, respectively. Administration of glibenclamide did not alter resting arteriolar diameter (101 ± 12 μm in the absence and 102 ± 13 μm in the presence of glibenclamide, n = 9).
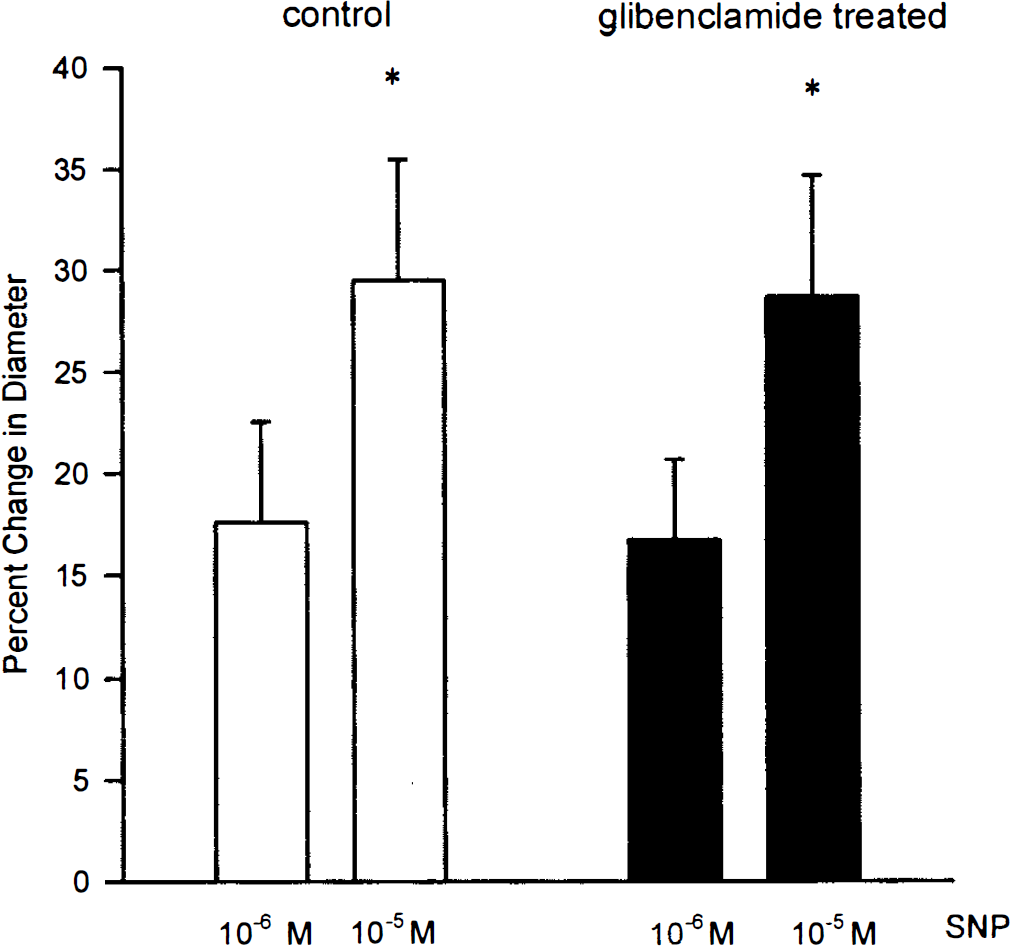
Percent change from baseline arteriolar diameter in response to 10−6 and 10−5 M sodium nitroprusside (SNP) in absence (open bars) and in presence of 10−5 M glibenclamide (filled bars), a selective inhibitor of ATP-sensitive potassium channels (n = 9, *p < 0.05 between the two doses of SNP).
DISCUSSION
The major findings in the present study are the following: (1) inhibition of NOS by
Compounds that open K+ channels have a variety of potential therapeutic actions, and they are under intensive investigation (Quast, 1992; Challinor-Rogers and McPherson, 1994). The detailed molecular basis of the mechanism of action of KATP openers is unknown at the present time (Quast et al., 1994). The binding site of these compounds has been difficult to identify. There is also a discrepancy between the concentrations required to cause vasodilatation and those needed to observe 42K+ efflux in vascular smooth muscle. Usually a 10–30 times higher concentration is needed to observe tracer efflux than to elicit vasodilatation (Atwal, 1994). These facts point out that additional mechanisms may also be involved in vasodilatation caused by KATP openers (Quast et al., 1994).
In our study 10−8 and 10−6 M aprikalim dilated pial arterioles by 10–21% in control conditions. These magnitudes of dilatation are similar to those found by Mayhan and Faraci (1993) and to our previous results (Busija and Louis, 1995). We do not know why the dose-response curve for aprikalim is relatively flat compared to other stimuli. Similar response profiles have been found for other agents such as glutamate (Meng et al., 1995), acetylcholine (Rosenblum et al., 1992), and bradykinin (Baumbach et al., 1994). In addition, it is worth noting that pial arteriolar dilation by 20% would reduce vascular resistance by one half.
We hypothesized that NO may also play a role in the vasorelaxation response to aprikalim. This view is supported by the results of some, but not all, investigations. For example, Mayhan and Faraci, 1993 showed a slight, but not significant impairment in dilatation to aprikalim after NG-monomethyl-
We found that intravenous
Increasing evidence suggests that NO participates in the maintenance of resting cerebral blood flow but the sources of NO remain to be elucidated. Both endothelial-and neuronal-derived NO may participate, but their relative contribution differs depending on animal species, brain region, and segment of cerebral vasculature (Iadecola et al., 1994). Data obtained from experiments using various NOS inhibitors suggest that the route of administration and timing of drug treatment influences pial arteriolar tone. Experiments by Armstead et al (1994) and Armstead (1995) showed that topically applied
In the present study, we evaluated how two different inhibitors of NO synthase affect pial arteriolar diameter. Intravenous administration of
Because
Our present data support previous findings that dilatation of pial arterioles in response to glutamate and NMDA are sensitive to NOS inhibition (Meng et al., 1995; Faraci and Brian, 1995). Furthermore, our results give the first direct evidence that glutamate-induced vasodilatation could be blocked by inhibition of neuronal NOS. The observation that NMDA-induced vasodilatation was less sensitive to the selective inhibition of neuronal NOS suggests that a reduced NOS activity is still enough to mediate vasodilation when NMDA receptors are activated.
The selective neuronal NOS inhibitor, 7-NI, may open novel therapeutic strategies in the neurodegenerative diseases. Several studies suggest that NO of neuronal origin contributes to cell death following cerebral ischemia or anoxia, but the mechanisms are unknown (Dalkara et al., 1994). Some evidence suggests that neurotoxicity includes the interaction of NO with superoxide radical-forming peroxynitrite. A novel observation of Schulz et al. (1995) indicated that 7-NI protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MTPT) -induced neurotoxicity. In another study (Yoshida et al., 1994), it has been shown that 7-NI decreased infarction size when given 5 min after occlusion of the middle cerebral artery in rats. On the other hand, hemodynamic responses to glutamate or NMDA may play a role in excitotoxic brain injury (Dalkara et al., 1994). We conclude that selective neuronal NOS inhibition can reduce the glutamate-induced and NO-mediated cerebral injury.
In our experiments large doses (50 mg/kg) of 7-NI had no effect on blood pressure and did not change pial arteriolar diameter, whereas NOS activity in the cerebral cortex decreased by 44%. In a previous study by Kelly et al. (1995), inhibition of neuronal NOS by 7-NI decreased cerebral blood flow, which was measured by an autoradiographic technique. Their result is in conflict with our present finding and other reports of Yoshida et al. (1994) and Faraci and Brian (1995). These authors, using a cranial window preparation, have reported that 7-NI had no effect on the diameter of pial arterioles. The discrepancy between the different results probably reflects species and methodological differences.
NO is thought to relax vascular smooth muscle by stimulation of guanylate cyclase, increasing levels of cGMP, cGMP-dependent phosphorylation of a protein kinase, and kinase-dependent phosphorylation of various targets. The end result is a decreased calcium sensitivity of the contractile apparatus. In a recent report by Kubo et al. (1994), patch clamp analyses demonstrated that the open probability of individual KATP channels could be increased by cGMP. Thus we considered the possibility that inhibition of KATP would influence the pial arteriolar dilatation to SNP. In the current study KATP was blocked by 10−5 M glibenclamide. This dose of glibenclamide has completely blocked pial arteriolar dilatation activated directly by aprikalim (Busija and Louis, 1995) and inhibited CGRP-induced vasodilatation as well (Louis et al., 1996). The finding that glibenclamide had no effect on SNP-evoked pial arteriolar dilatation matches similar findings in rabbits and rats. In those studies even a higher dose (10−3 M) of glibenclamide did not affect vasodilatation evoked by nitroglycerin or SNP (Faraci and Heistad, 1993; Faraci et al., 1994). Conversely, Armstead (1996) reports that 10−6 M glibenclamide attenuates SNP-induced dilatation of small pial arteries and arterioles. We do not know why these differences are present. Although the exact reason for this discrepancy remains unclear, it may be due to different timing in drug administration and differences in the basal diameter of the vessels under investigation.
In the present study, topical application of 10−5 glibenclamide had no influence on baseline diameter of the pial arterioles. This finding is in agreement with the observations of Faraci and Heistad (1993) and provide further evidence that activity of KATP had no effect on the basal tone of pial arterioles. However, our result differs from results of Jackson (1993), who reported that glibenclamide produced concentration-dependent vasoconstriction in hamster cheek arterioles. Similarly, Clapp and Gurney (1992) found that KATP channels regulate resting potential of pulmonary arteriolar smooth muscle cells. The disparate results reveal regional and species differences in the physiological role of arteriolar KATP channels.
In summary, we found that inhibition aprikalim-induced dilatation of pial arterioles is mediated partly by NO. The source of NO is likely the vascular endothelium.
Footnotes
Abbreviations used
Acknowledgment:
Supported by grants HL-30260, HL-46558, and HL-50587 from the National Institutes of Health. We thank Lisa Moore for assisting in the NOS activity assay.