
Abstract
White blood cells (WBCs) play vital roles in host defense. Recently, increasing interest has been directed toward the question of whether WBCs, particularly polymorphonuclear leukocytes, could also act as mediators of secondary brain damage in the setting of focal and global cerebral ischemia with and without reperfusion. Considerable insight into the importance of WBC-mediated tissue injury has been gained from studies employing antileukocyte interventions in experimental cerebral ischemia. The purpose of this article is to survey the different approaches taken to interfere with WBC inflammatory function. Emphasis is laid on a discussion of the efficacy of these interventions, their effects and side effects on cerebral and systemic parameters, and the power of evidence they provide for identification of WBCs as important factors in cerebral ischemia. The role of WBCs has been investigated in a great variety of global and focal cerebral ischemia models with and without reperfusion, leading to spmetimes contradictory results. In the light of currently available data, it seems likely that WBCs contribute to secondary brain damage in the scenario of experimental transient focal cerebral ischemia, if the insult is not too severe.
The behavior of white blood cells (WBCs) and their mobility in physiological and disease states have fascinated biologists for many decades. It was not until the recent investigation of cytoplasmic and cell membrane adhesion molecules that an understanding of the molecular mechanisms underlying margination, adhesion, and emigration of
The role of WBCs, particularly polymorphonuclear leukocytes (PMNLs) and monocytes, in the evolution of necrosis under pathophysiological conditions has been examined. Experimental studies in the 1960s indicated that reperfusion of ischemic myocardium resulted in an acceleration of tissue necrosis (Sommers and Jennings, 1964), and PMNLs were implicated as participants in “reperfusion injury.” This notion received support from studies showing that pharmacological induction of neutropenia reduced myocardial infarct size in models of coronary artery occlusion and reperfusion (Romson et al., 1983; Litt et al., 1989). As a consequence, interest in the role of WBCs in reperfusion injury has intensified dramatically over the last 10 years. Recent reports of clinical improvement with reperfusion strategies in ischemic stroke encourage the search for further therapeutic interventions. In this respect, modulation of WBC function appears as a promising candidate to improve outcome in acute stroke patients.
WBCs
WBC accumulation and tissue injury in acute cerebral lesions
In 1986 Hallenbeck et al. demonstrated significant early PMNL accumulation during the first 4 h after induction of embolic cerebral ischemia in dogs. The authors explained their finding by either increased PMNL/endothelial cell (EC) adhesion or the formation of PMNL aggregates during ischemia/reperfusion (I/R). This report stimulated a wide variety of experimental studies looking at early postishemic WBC contribution to brain damage (Tables 1–3).
Experimental studies employing antileukocyte interventions in models of global cerebral and forebrain ischemia
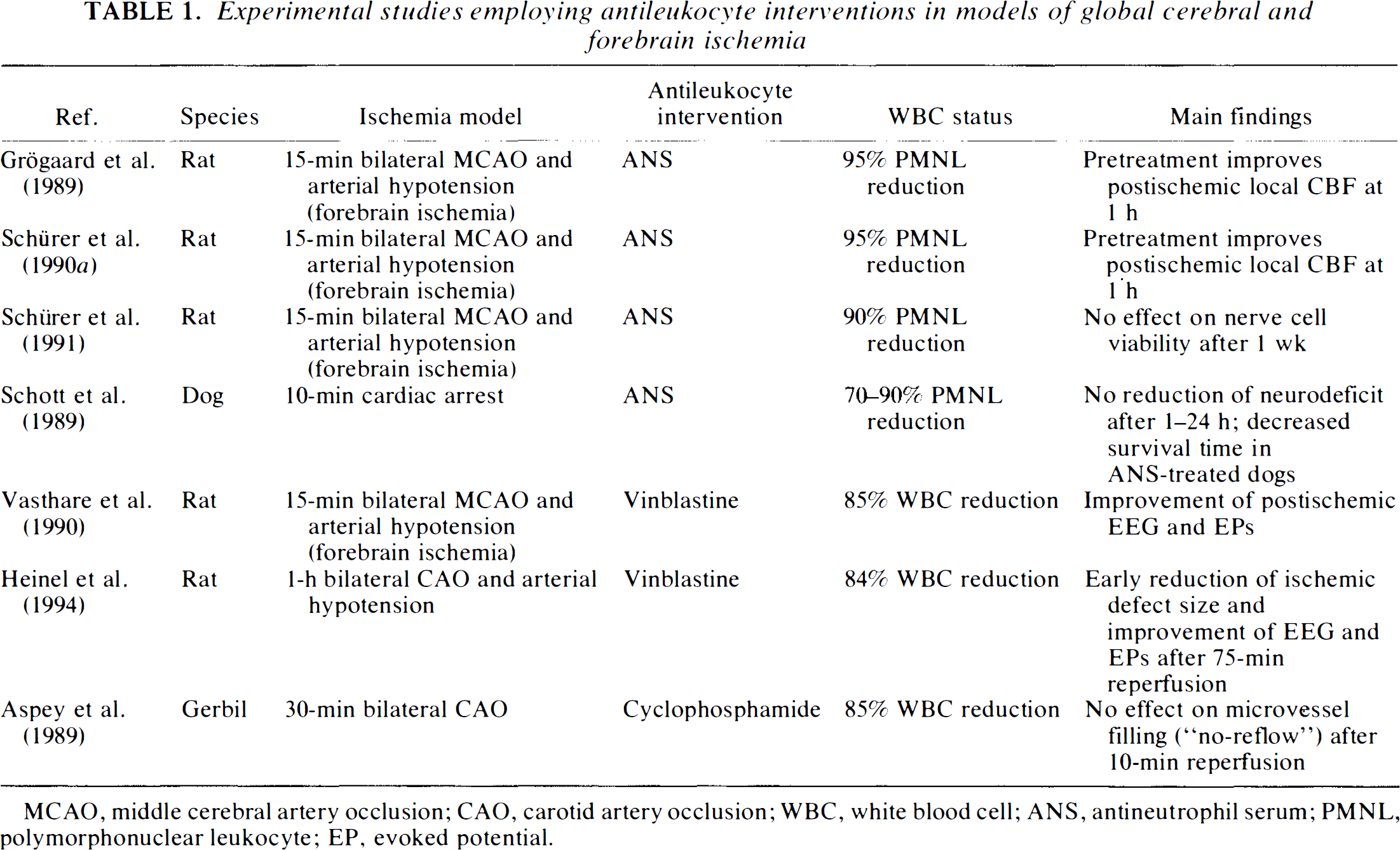
MCAO, middle cerebral artery occlusion; CAO, carotid artery occlusion; WBC, white blood cell; ANS, antineutrophil serum; PMNL, polymorphonuclear leukocyte; EP, evoked potential.
Experimental studies employing antileukocyte interventions in models of permanent focal cerebral ischemia without reperfusion

MCAO, middle cerebral artery occlusion; WBC, white blood cell; ANS, antineutrophil serum; MoAb, monoclonal antibody; PMNL, polymorphonuclear leukocyte; EP, evoked potential; ICP, intracranial pressure; TTC, triphenyltetrazolium chloride.
Experimental studies employing antileukocyte interventions in models of transient focal cerebral ischemia and reperfusion
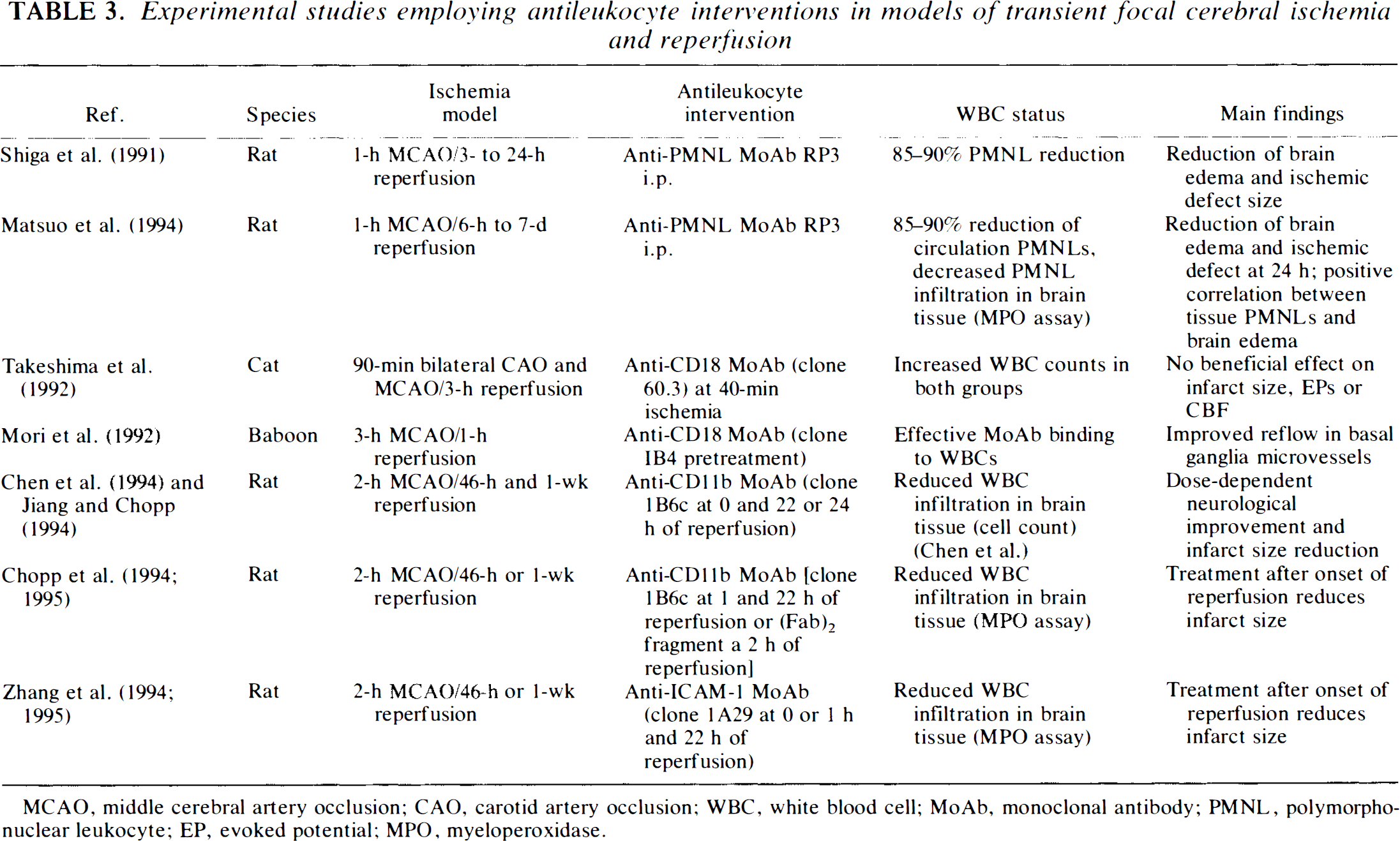
MCAO, middle cerebral artery occlusion; CAO, carotid artery occlusion; WBC, white blood cell; MoAb, monoclonal antibody; PMNL, polymorphonuclear leukocyte; EP, evoked potential; MPO, myeloperoxidase.
Mechanisms of WBC-mediated tissue injury
WBCs have been implicated in the pathogenesis of focal cerebral I/R microvascular occlusion (Del Zoppo et al., 1991; Mori et al., 1992; Del Zoppo, 1994), suggesting an element of the “no-reflow” phenomenon described after global cerebral ischemia by Ames et al. (1968), as well as in delayed postischemic hypoperfusion (Grögaard et al., 1989; Schürer et al., 1990a) first described by Snyder et al. (1975). WBCs may physically plug and obstruct microvessels (Engler et al., 1983; Schmid-Schönbein, 1987; Hatchell et al., 1994), release vasoconstrictive mediators (Lopez et al., 1989; Faraci et al., 1991; Mugge et al., 1991), cause dysfunction of the vasoreactivity of cerebral arteries (Akopov et al., 1994), injure ECs directly, or migrate into the brain tissue where they may damage parenchymal cells through the release of cytotoxic enzymes, free oxygen radicals, and products of the phospholipid cascade (Weiss, 1989; Welbourn et al., 1991). PMNLs have recently been identified as the major source of oxygen radicals in reperfusion after focal cerebral ischemia in the rat brain (Matsuo et al., 1995).
Experiments by Suematsu et al. (1993) provided the first in vivo evidence for intraendothelial oxidative stress induced by activated WBCs in the rat mesentery. The same group reported that early (<90 min) I/R-induced cell death in rat skeletal muscle was not related to WBC activation. These findings do not abrogate the potentially harmful effects of WBCs in the central nervous system because disruption of the blood-brain barrier can occur without irreversible endothelial or glial cell death (Granger and Kubes, 1994b; Kurose et al., 1994). Subsequent formation of vasogenic brain edema can be detrimental, leading to increased intracranial pressure (ICP), ischemia, and secondary cell death. This is an important difference between the brain and peripheral organs where edema formation does not have such harmful consequences.
Blocking of WBCs and/or WBC function
Probably the most valuable insight into the importance of WBC-mediated tissue injury in cerebral ischemia has been gained from studies employing antileukocyte interventions in experimental models of cerebral and noncerebral I/R. Efforts have been centered on PMNLs, since these cells are the first to arrive at the ischemic and inflammatory focus, reaching the cerebral parenchyma within hours after the insult and peaking between 24 and 72 h (Ward et al., 1988; Clark et al., 1993; Garcia et al., 1993, 1994a; Zhang et al., 1994b).
The purpose of this article is to survey the different methodological approaches directed at the inhibition of PMNL-mediated inflammatory function. It will concentrate on interventions used to either reduce the peripheral WBC count or interfere with the capability of WBCs to adhere to the vascular endothelium, assuming that this represents a key step in the subsequent events leading to enhancement of cerebral tissue injury. In an attempt to evaluate the relative scientific merit of different experimental approaches and findings, emphasis will be placed on the efficacy of these interventions and their effects and side effects on cerebral and systemic physiological parameters.
PMNL DEPLETION
Antineutrophil serum
Since the early observations of the Russian biologist Elie Metchnikoff in 1899, it has been known that “antileukocyte serum” can be prepared by immunizing animals with WBCs from another species. Intravenous administration of antineutrophil serum (ANS) leads to sequestration of PMNLs in the lungs (Sandler et al., 1987; Grögaard et al., 1989). This may be accompanied by acute hemodynamic changes (L. Schürer, personal communication) and decreased complement activity (Sandler et al., 1987). Neutropenia lasting up to 7 days can be induced by intraperitoneal administration of ANS in rodents (Simpson and Ross, 1971; Sandler et al., 1987). A reduction of circulating PMNLs ranging between 80 and 95% and monocytes up to 50% during the first hours after administration has been reported (Tables 1 and 2). ANS also activates circulating cells, ECs, and tissue macrophages, the neutropenia might therefore reflect a redistribution of PMNLs into the marginated pool (Schmid-Schönbein, 1993). Actually, an absolute reduction of all WBCs to zero does not occur owing to rapid redistribution from bone marrow stores. ANS-mediated microvascular trapping of PMNLs in the cerebral circulation might also amplify the injury process and blunt the benefits achieved by PMNL depletion.
ANS has been employed in studies of experimental global cerebral ischemia, global forebrain ischemia, and focal permanent cerebral ischemia (Tables 1 and 2). When rats were exposed to ANS prior to 15-min global forebrain ischemia and arterial hypotension, CBF improved early (1 h) after the insult (Grögaard et al., 1989; Schürer et al., 1990a). After a period of 7 days, however, quantitative histology failed to reveal any differences in nerve cell survival between neutropenic animals and untreated controls (Schürer et al., 1991). In a cardiac arrest model of global cerebral ischemia in dogs, Schott et al. (1989) reported decreased survival times in ANS-treated animals at 24 h as compared with nontreated controls. The latter study has been questioned because mortality was high even in the experimental group that did not received ANS (Kochanek and Hallenbeck, 1992). Bednar et al. (1991), after thromboembolic stroke combined with arterial hypotension in rabbits, observed improved CBF, decreased ICP, and a reduction in ischemic lesion size by 4 h as measured by 2,3,5-triphenyltetrazolium chloride (TTC), a marker of mitochondrial oxidative enzyme function. Interpretation of these studies is difficult because arterial hypotension per se may lead to PMNL activation with capillary plugging by activated cells (Bagge et al., 1980; Yamakawa et al., 1987; Barroso-Aranda et al., 1988).
A very selective depletion of PMNLs without reduction of other WBC lines in rats has been achieved by Shiga et al. (1991) and Matsuo et al. (1994) after intraperitoneal injection of RP-3, a monoclonal antibody (MoAb) developed specifically against PMNLs (Table 3). Depletion of circulating PMNLs throughout focal cerebral I/R suppressed postischemic edema formation and significantly reduced the size of the ischemic lesion by TTC at 24 h (Matsuo et al., 1994). Matsuo et al. (1994) demonstrated that tissue PMNL content correlated well with brain edema in nontreated controls: Both reached a maximum between 24 and 72 h after the insult. Garcia et al. (1994a) showed a nearly identical time course of PMNL invasion into brain parenchyma in rats with permanent middle cerebral artery occlusion (MCAO) with neutrophils peaking at 24 h.
The findings by Matsuo et al. (1994) suggest that PMNL invasion after cerebral ischemia was causally related to the development of postischemic brain injury. Unfortunately, their study is lacking in quantitative data on true neuronal and microvessel loss. TTC is associated with histochemical abnormalities that represent, under some circumstances, reversible components of cerebral injury that do not always correlate well with histology (Liszozak et al., 1984; Cole et al., 1990).
Antineoplastic agents
Antineoplastic agents such as mechlorethamine, cyclophosphamide, and vinblastine cause myelo-suppression affecting all blood cells and may cause complications such as nausea and vomiting leading to cardiovascular collapse, dehydration, and metabolic alkalosis (Dutka et al., 1989). Pretreatment of the animals is usually started 3–4 days before the ischemic insult with a 70–90% reduction in neutrophil count (Tables 1 and 2). Most authors report a concomitant decrease in hematocrit (Aspey et al., 1989; Biagas et al., 1992), platelet count (Modha et al., 1988; Aspey et al., 1989; Vasthare et al., 1990), and/or WBC count (Modha et al., 1988; Aspey et al., 1989; Vasthare et al., 1990; Helps and Gorman, 1991).
Antineoplastic agents have been used in models of experimental focal and global cerebral ischemia (Tables 1 and 2), but the observation period has generally been limited to <3 h after the insult. Within this time period, most authors reported an improvement of cerebral parameters such as CBF and evoked potentials (EPs), and in one study (Heinel et al., 1994) even ischemic injury size (by TTC). These results are partially in accordance with findings by Schürer et al. (1990a) and Grogard et al. (1989) who, in ANS-induced neutropenia, also found evidence for direct WBC involvement during ischemia and early reperfusion. However, 1 week after the insult, they were unable to detect any beneficial effect on nerve cell survival (Schürer et al., 1991). This raises the suspicion that, at least in global cerebral ischemia, leukopenia might simply delay the development of the infarct without having any effect on its final size.
All methodological approaches aiming at a reduction of WBCs have weaknesses. First, toxic side effects associated with pharmaceutical WBC depletion frequently do not permit observation periods exceeding a few hours. Second, most attempts to deplete PMNLs cause incomplete neutropenia and significant suppression of other cell lines, which makes it difficult to attribute an observation to PMNL depletion alone. It has been suggested that even small numbers of circulating neutrophils may produce postischemic microvascular dysfunction (Engler and Covell, 1987). Platelets and mononuclear cells have also been implicated in the etiology of stroke (Strachan et al., 1992). It is known, for example, that platelets accumulate in the ischemic brain (Obrenovitch and Hallenbeck, 1985; Del Zoppo et al., 1986; Okada et al., 1994) and that platelet activation can occur in experimental cerebral I/R (del Zoppo, 1994:36). Together with granulocyte activation, this in turn may trigger the release of a variety of vasoactive substances responsible for vasomotor changes and neuronal damage (Moncada and Vane, 1978; Chan et al., 1983; Vanhoutte and Houston, 1985). Lymphocytes also accumulate in ischemic brain (Barcikowska-Litwin et al., 1987), and in some of the studies cited, lymphocytes were reduced by >50% (Dutka et al., 1989; Grögaard et al., 1989; Vasthare et al., 1990; Helps and Gorman, 1991). Strachan et al. (1992) reported significant reduction of ischemic brain edema by rendering rats lympho- and thrombopenic using whole-body irradiation, without affecting PMNL counts. Finally, a reduction in circulating cell mass, through hemodilutional effects, may per se confer neuroprotection in experimental cerebral ischemia (Fischer and Ames, 1972).
MODULATION OF PMNL/EC INTERACTION
MoAbs
Adhesion of PMNLs to the endothelium of postcapillary venules is recognized as a prerequisite for subsequent emigration into surrounding tissue (Granger and Kubes, 1994a; Springer, 1994). The β2-integrin CD11/CD18 has been identified on WBCs and, together with its counterreceptor ICAM-1 on ECs, is involved in WBC adherence to stimulated endothelium. Consequently, blocking of these adhesion receptors with specific MoAbs may attenuate WBC margination and emigration (Argenbright et al., 1991; Kurose et al., 1994).
MoAbs used in experimental models of cerebral ischemia include molecules directed against the CD11b and CD18 subunit of the β2-integrin heterodimer on PMNLs and ICAM-1 on ECs (Tables 2 and 3). Adhesion molecules are not specific for a certain cell type. The CD11b subunit is shared by monocytes and PMNLs, and CD18 is found on virtually all WBCs (Springer, 1994). ICAM-1 has been detected on noncerebral vessels and on cerebrovascular ECs (Fabry et al., 1992; Wong and Dorovini-Zis, 1992; Hess et al., 1994a,b; Okada et al., 1994). It can mediate binding of all WBCs. For interpretation of the following studies, it is important to know the specificity, purity, and functional blocking characteristics of the MoAb under investigation. Unfortunately, specific epitope mapping has not been carried out in every case. A dose of 2 mg/kg i.v. of an MoAb directed against the CD18 epitope, for example, saturated rabbit or baboon PMNL adhesion sites in vivo within minutes (Wallis et al., 1986; Price et al., 1987; Mori et al., 1992) and impaired PMNL adhesion for up to 24 h (Price et al., 1987; Mileski et al., 1991).
The effects of WBC inhibition using MoAbs after focal cerebral ischemia have been studied for up to 1 week after insult (Tables 2 and 3). Support for “capillary plugging” as an important mechanism of WBC-mediated injury comes from a study by Mori et al. (1992), who investigated microvascular patency in the basal ganglia of baboons after 3 h of MCAO and 1 h of reperfusion as a measure of post-ischemic microvascular reflow. Animals receiving the anti-CD18 MoAb IB4 15 min before reperfusion revealed significantly augmented cerebral reflow in noncapillary microvessels up to a caliber of 50 μm, which the authors attributed to decreased obstruction of these vessels by PMNLs. Of note, and in contrast to other studies employing MoAbs, efforts were made to demonstrate effective binding of MoAb to CD18 adhesion sites ex vivo (83% saturation in 5 min).
Experimental studies looking at embolic stroke have yielded controversial results. In microembolic stroke produced by intracarotid injection of micro-clots in rabbits, treatment with anti-ICAM-1 MoAb was associated with neurological improvement at 24 h only when initiated within 5 min after embolization, but not if infusion was delayed until 180 min after the insult (Bowes et al., 1993). Combination with reverse transcriptase plasminogen activator was no more effective than either substance given alone. Clark et al. (1991a,b), on the other hand, reported that in irreversible embolic stroke induced by microsphere infusion in rabbits, neither anti-ICAM-1 nor anti-CD 18 MoAb pretreatment improved neurological function at 18 h.
Takeshima et al. (1992) found that in a cat model of 90-min bilateral carotid artery occlusion with unilateral MCAO and 180-min reperfusion, treatment with anti-CD 18 MoAb 60.3 40 min after ischemia did not improve the recovery of somatosensory EPs and did not decrease infarct size. The authors rejected the hypothesis that PMNLs play an important role in transient focal ischemia. However, ischemic insult in this model was severe, producing profound disturbances of evoked responses with poor recovery after reperfusion and large areas of neuronal injury. It is possible that the contribution of WBCs to the development of ischemic tissue injury in this model might have been overridden by other factors (Siesjö, 1993).
The most systematic and careful investigation of WBC-mediated injury has been conducted in rats subjected to permanent or transient (2-h) MCAO followed by 2–7 days of reperfusion (Chen et al., 1994; Chopp et al., 1994; Jiang and Chopp, 1994; Zhang et al., 1994a, 1995; Chopp, 1995). Subjects were treated with different concentrations of either anti-ICAM-1 MoAb clone 1A29 or anti-CD11b MoAb clone 1B6c upon, or 1–2 h after, reperfusion and a second dose 1 day later (Tables 2–4). The volume of ischemic injury was determined from hematoxylin/eosin-stained tissue sections. While the authors did not report neuroprotection from either antibody in their permanent ischemia model (Table 2), both antibodies significantly reduced tissue WBC infiltration and infarct size in transient ischemia, even when treatment was delayed up until 2 h after start of reperfusion. An overview of their results is given in Table 4. For example, in the group treated with 1 mg/kg anti-CD11b at reperfusion, Chen et al. (1994) observed a 39% reduction of PMNL infiltration, but no significant ischemic lesion reduction. With twice the dose of the same MoAb, however, infarct size as well as PMNL accumulation were significantly reduced (43 and 55%, respectively). This was true even when treatment with the same dose was delayed until 1 h after reperfusion (28 and 36% reduction, respectively) (Chopp et al., 1994). These results were also reflected in improved neurological performance of the animals. Measured plasma concentration of free MoAbs was sufficiently high to saturate CD11b receptors on circulating WBCs (Chen et al., 1994), which suggests that other adhesion molecules might also be involved in the PMNL migration pathway. Treatment after 2 h of reperfusion still demonstrated an >40% infarct size reduction at 1 week, indicating that treatment did not merely delay the maturation of the ischemic lesion (Chopp, 1995). Very recently, Chopp (1995) reported the successful administration of anti-CD11b (Fab)2 fragment MoAb in the same model and with comparable results. Treatment with the anti-ICAM-1 MoAb upon and at 1 h after reperfusion yielded almost identical results with infarct size reduction of 44% at 1 week postinjury (Zhang et al., 1994a, 1995). It will be important to see whether these results can be reproduced by other laboratories.
Reduction of infarct size and WBC accumulation in brain tissue of rats undergoing transient (2-h) MCAO and MoAb treatment followed by 2 or 7 days of reperfusion
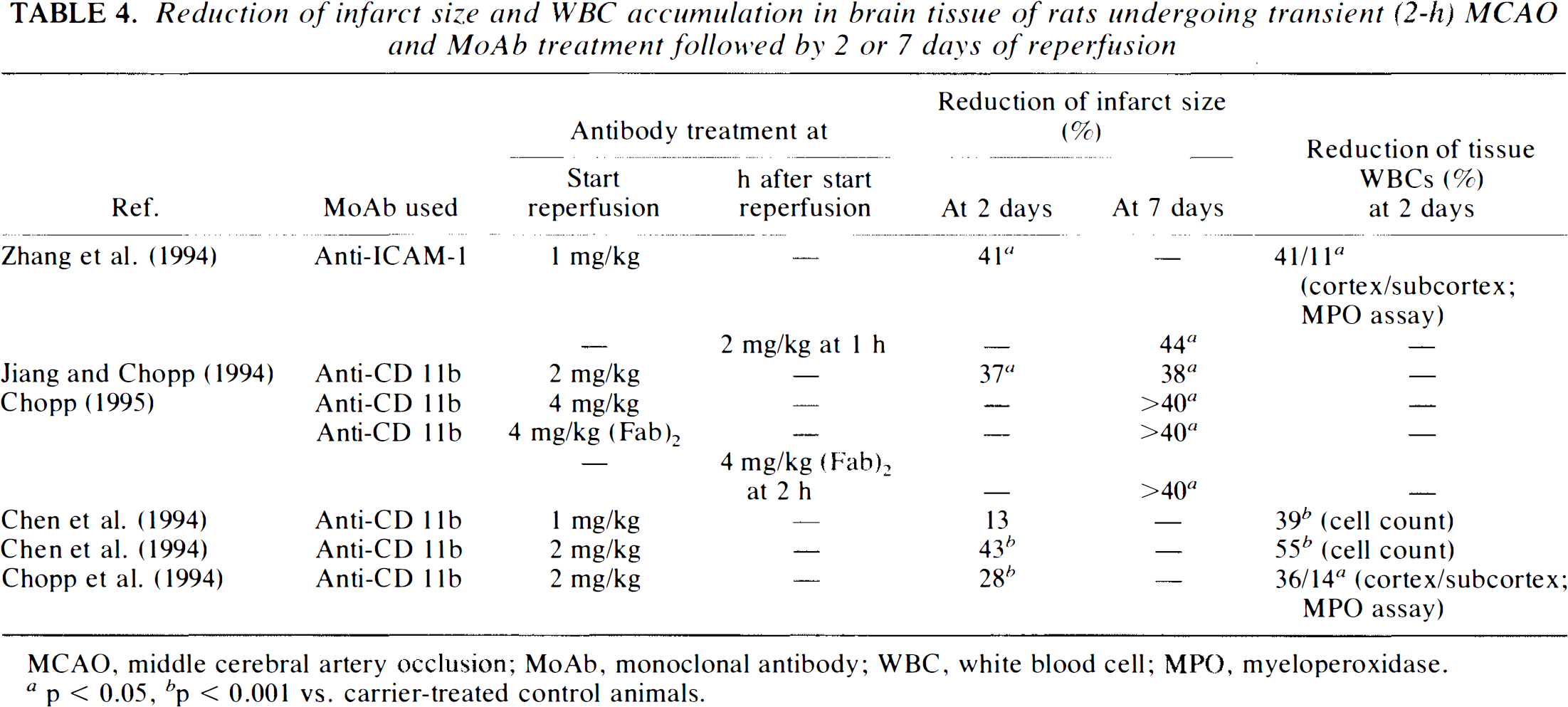
MCAO, middle cerebral artery occlusion; MoAb, monoclonal antibody; WBC, white blood cell; MPO, myeloperoxidase. a p> 0.05, b p < 0.001 vs. carrier-treated control animals.
Side effects of antileukocyte interventions with MoAbs
Of all the antineutrophil interventions discussed so far, MoAbs directed against specific adhesion receptor complexes appear to be the most appropriate candidates for clinical testing. Interference with WBC function, however, might be associated with a variety of adverse effects. Antiadhesion antibodies are not specific enough to block PMNLs selectively. The spectrum can be narrowed down to PMNLs and monocytes if the CD11b epitope is chosen as a target (Springer, 1994). The role of monocytes in the setting of cerebral ischemia, however, is still unresolved, and more work is needed to separate potentially beneficial effects on tissue repair from destructive processes (Giulian et al., 1986, 1989; Olson et al., 1987; Blight, 1992). Considering the fact that monocyte invasion into brain tissue begins only after 24 h (Ward et al., 1988; Garcia et al., 1993, 1994a), limiting antibody treatment to the first 1–2 days after the insult should avoid this potential risk. Concerns have also been raised over the application of antiadhesion molecules in patients in whom temporary paralysis of WBC function might have detrimental effects on host defense (Rosen et al., 1989; Sharar et al., 1991). In in vivo models of bacterial infection, the combination of MoAbs with antibiotics did not show any adverse effects on the severity of infection or on mortality (Tuomanen et al., 1989; Mileski et al., 1991; Saez-Llorens et al., 1991; Sharar et al., 1991; Garcia et al., 1994b). In two models of bacterial meningitis treatment with anti-CD18 MoAbs (clone IB4) decreased inflammatory blood-brain barrier damage and attenuated the formation of brain edema (Tuomanen et al., 1989; Saez-Llorens et al., 1991).
In humans, the use of mechanically WBC-depleted blood for the prevention of transfusion reactions in sensitized patients has been a well established practice for many years (Hughes and Brozovic, 1982; Brand, 1994). The first MoAb approved for human use was an immunosuppressive agent targeting lymphocyte antigens for treatment of renal transplant rejection (Russell et al., 1992). Since then, experience with MoAbs against WBC adhesion in humans has been limited to Phase I studies conducted in transplantation patients (Fischer et al., 1986; Baume et al., 1989; Le Mauff et al., 1991; Haug et al., 1993). Antibodies directed against CD18, CD11a, and ICAM-1 in varying concentrations between 7 and 40 mg/day were used in a total of 43 patients for up to 2 weeks. Minor side effects including chills, nausea, abdominal pain, headaches, and skin rash were encountered, but blood cell counts remained unaffected and no hemolysis occurred. However, one severe fungal infection (Baume et al., 1989) and one case of pneumonia (Haug et al., 1993) were observed as well. The safety of antiplatelet murine MoAbs in humans was demonstrated in coronary angioplasty patients treated with the MoAb 7E3 Fab (Ellis et al., 1993; Tcheng et al., 1994).
CONCLUSION
In their review, Kochanek and Hallenbeck (1992) concluded that, as compared to short-lasting global ischemic insults, early PMNL infiltration is “more likely to occur in focal ischemia, which is often prolonged and likely to produce severe tissue injury.” In the light of recently published experimental studies employing MoAbs to deplete (Shiga et al., 1991; Matsuo et al., 1994) or block (Chen et al., 1994; Chopp et al., 1994; Zhang et al., 1994a; Chopp, 1995; Zhang et al., 1995) PMNL adherence with high specificity, it seems justified to expand this hypothesis. WBCs clearly contribute to ischemic cell damage in the scenario of experimental focal cerebral ischemia followed by reperfusion, if the ischemic insult is not too severe. However, their role in global ischemia (Aspey et al., 1989; Schott et al., 1989; Anderson et al., 1990), permanent focal ischemia (Clark et al., 1991a,b; Strachan et al., 1992), and severe transient focal ischemia (Takeshima et al., 1992) has not yet been clarified. Based on the literature, it is not possible to identify their target and the impact they have on final pathological and behavioral outcome.
Global versus focal cerebral ischemia
A critical difference between global cerebral ischemia, on one side, and focal cerebral ischemia, on the other, is the existence of a “penumbra” in the latter, a zone of potentially salvageable cerebral tissue surrounding the ischemic focus (Astrup et al., 1981). Effects produced by antineutrophil interventions (e.g., a slight increase of CBF) may be sufficient to ameliorate or prevent cell damage in these borderline regions, whereas conditions in global or forebrain ischemia set rapidly into a state of complete energy depletion where cells die for other reasons. As reviewed recently by Siesjö et al. (1995), calcium-triggered injury might be the major determinant in global ischemia.
Transient versus permanent focal cerebral ischemia
The expression of WBC adhesion molecules on ECs appears to be triggered by reperfusion, which might explain the success of antileukocyte strategies in transient focal cerebral ischemia. In an in vitro study, hypoxia/reoxygenation, but not hypoxia alone, led to an increase of ICAM-1 in isolated brain ECs (Hess et al., 1994b). In permanent focal ischemia or very severe focal cerebral I/R, the contribution of WBC-mediated injury might be overridden by other mechanisms of secondary brain damage. These may include the loss of calcium homeostasis, leading to activation of protein kinases and new gene expression, free radical-mediated injury, and excessive acidosis (Siesjö, 1993). Reperfusion of the penumbra in focal cerebral ischemia, on the other hand, allows large numbers of WBCs to reach affected areas and induce tissue damage.
Questions concerning WBCs
The complexity of the pathophysiological problem and contradictory results obtained in a great variety of experimental approaches leave many questions unanswered (Härtl, 1995; Kochanek and Hallenbeck, 1992; Schürer et al., 1993):
Correlation between WBC accumulation and tissue injury in acute cerebral lesions. Few investigators have addressed the question of whether there exists a quantitative correlation between WBC accumulation/activation or the status of the ECs in ischemic brain parenchyma and parameters of neurological function before and after blocking/depleting WBCs. Matsuo et al. (1994) demonstrated that tissue PMNLs after transient focal cerebral ischemia correlated with brain edema in nontreated animals, both reaching a maximum between 24 and 72 h after the insult.
Blocking of WBCs and/or WBC function. If antileukocyte interventions were employed, the efficacy of these manipulations to interfere with WBC function, as characterized by the expression of adhesion molecules, WBC/EC interaction, tissue infiltration, phagocytosis, Chemotaxis, oxygen free radical production, and release of cytotoxic metabolites, has hardly, if ever, been demonstrated. As was pointed out recently by Schmid-Schönbein (1993), documentation of systemic neutropenia after employing antileukocyte interventions is not sufficient, because it may merely represent a shift of circulating PMNLs into the marginated pool. Also, most studies focused only on the acute effects after therapy. Therefore, it was not possible to exclude that a beneficial treatment effect represented merely a transient improvement followed by delayed maturation of the ischemic lesion.
Mechanism of WBC-mediated secondary brain damage. While some studies have demonstrated a positive correlation between WBC accumulation and brain edema formation after cerebral ischemia (Matsuo et al., 1994) and traumatic brain injury (Schoettle et al., 1990; Biagas et al., 1992), clear-cut evidence as to the mechanism of WBC-mediated injury in the brain has not yet been provided. In this respect, intravital videomicroscopy could offer a powerful tool to shed light on WBC/EC interactions in the cerebral microcirculation. As has been demonstrated recently by Suematsu et al. (1993, 1994) using fluorescent dyes to visualize oxidative stress and irreversible nuclear damage in the rat mesentery and muscle, intravital videomicroscopy allows the complex interplay between WBCs and functional changes in endothelial and parenchymal cells to be shown in vivo. It also enables the investigator to control the efficacy and check for potential microcirculatory side effects of a particular antileukocyte intervention. The translation of similar experiments into the setting of cerebral ischemia and brain injury using closed cranial window preparations may become fruitful in the future. Uhl et al. (1995) have recently reported activation of WBCs in the early reperfusion period after forebrain ischemia in gerbils with intact dura mater and equipped with closed cranial windows. Preliminary results from our laboratory indicate that traumatic brain injury in a chronic cranial window model in rabbits leads to significant WBC activation in postcapillary pia venules as soon as 30 min after insult (Härtl et al., 1996).
Evidence against WBC-mediated brain damage. A role for PMNLs or WBCs in the development of brain damage in the acute phase of cerebral ischemia and brain injury has also been denied (Modha et al., 1988; Aspey et al., 1989; Schott et al., 1989; Anderson et al., 1990; Schürer et al., 1990b; Chang et al., 1992; Takeshima et al., 1992). Several investigators have reported that lymphocytes, but also PMNLs, might actually confer neuroprotection in early stages of ischemic stroke (Grau et al., 1994) and after cryogenic brain injury (Schürer et al., 1990b).
Summary
Taken together, these experimental results make it worthwhile to further pursue this avenue of research. Experimental findings strongly suggest that benefits would be more likely in transient focal cerebral ischemia. This supports the rational clinical testing of rapid reperfusion with effective antileukocyte strategies. In combination with recent advances leading toward a more “human” MoAb technology and granulocyte adhesion receptor biology with reduced side effects, a number of promising strategies may be appropriate (Riechmann et al., 1988; Clendennin et al., 1992). Pharmaceutical approaches for the future may also include the use of soluble forms of adhesion molecules (Dana et al., 1991; Watson et al., 1991) and (Fab)2 antibody fragments. Finally, other forms of acute cerebral disorders, such as traumatic brain injury, have only begin to be evaluated in terms of WBC involvement and represent a fertile field for future research (Means and Anderson, 1983; Schoettle et al., 1990; Biagas et al., 1992; Uhl et al., 1992; Zhuang et al., 1993; Härtl et al., 1996).