
Abstract
This experiment examined the effects of nitric oxide (NO) synthase inhibition on brain intracellular pH, regional cortical blood flow, and NADH fluorescence before and during 3 h of focal cerebral ischemia using in vivo fluorescence imaging. Thirty fasted rabbits under 1% halothane were divided into four treatment groups receiving Nω-nitro-L-arginine methyl ester (L-NAME) intravenously at 20 min prior to ischemia (0.1, I, and 10 mg/kg and 1 mg/kg + 5 mg/kg L-arginine) and two control groups (nonischemic and ischemic). In ischemic controls, brain pHi declined to 6.73 ± 0.03 at 30 min and remained acidotic through the remainder of the ischemic period. In the 0.1 mg/kg group, brain pHi fell after 30 min of ischemia to 6.76 ± 0.05 (p < 0.05), but then improved progressively despite occlusion. In the 1 mg/kg group, brain pHi remained normal despite middle cerebral artery (MCA) occlusion. In the 10 mg/kg group and in the combined L-NAME + L-arginine group, pHi fell after 30 min of ischemia to 6.81 ± 0.03 (p < 0.05) and remained acidotic. During occlusion, regional cortical blood flow dropped in a dose-dependent manner. After 3 h of ischemia, regional cortical blood flow was 33.9 ± 10.9 and 25.1 ± 8.9 ml/100 g/min at doses of 0.1 and 10.0 mg/kg, respectively. L-NAME treatment did not significantly alter the increased NADH fluorescence that accompanied occlusion. This study shows that L-NAME can prevent intracellular brain acidosis during focal cerebral ischemia independent from regional cortical blood flow changes. This experiment suggests that NO is involved in pHi regulation during focal cerebral ischemia.
Emerging evidence suggests that nitric oxide (NO) may play a role in ischemic brain injury (Huang et al., 1994). Direct measurements have shown a large increase in NO during focal cerebral ischemia (Malinski et al., 1993). However, the role and mechanisms of NO-mediated neurotoxicity remain speculative. During ischemia, release of excitatory amino aicds activates N-methyl-D-aspartate (NMDA) receptors producing a calcium transient.
This enhances NO production by activating nitric oxide synthase (NOS), which is calcium-dependent (Dugan and Choi, 1994). Dawson et al. (1991) observed that NMDA-mediated neuronal injury could be blocked by inhibition of NO production. The effects of NOS inhibition during brain ischemia in vivo remain controversial, with some groups reporting protective (Nowicki et al., 1991; Ashwal et al., 1994; Nishikawa et al., 1994) and some reporting toxic (Zhang and Iadecola, 1994; Yamamoto et al., 1992; Kuluz et al., 1993) effects. This controversy may reflect the fact that in vivo nonselective inhibition of endothelial and neuronal NOS may produce opposite effects on neuronal injury.
The purpose of this experiment was to examine the effects of NOS inhibition by Nω-nitro-L-arginine methyl ester (L-NAME), administered prior to focal cerebral ischemia, on serial changes in intracellular brain pH (pHi), regional cortical blood flow, and the nicotinamide adenine dinucleotide (NADH) redox state as measured by in vivo fluorescence imaging (Anderson et al., 1992). We also tested if coadministration of L-NAME and L-arginine could reverse any effect produced by L-NAME.
MATERIALS AND METHODS
Animal preparation
Upon approval from the Animal Care and Use Committee, 30 overnight-fasted New Zealand white rabbits, weighing 3.5–4.5 kg, were induced with thiamytal sodium 40 mg/kg (Surital), and operated on at 2.0% and studied under 1.0% halothane anesthesia, respectively. A tracheotomy was performed, and animals were placed on a Harvard respirator (Harvard Apparatus, Millis, MA, U.S.A.). Animals were given 0.15 mg/kg pancuronium bromide (Pavulon) (Organon Inc, West Orange, NJ, U.S.A.) to abolish respiratory efforts. Animals were kept normoxic and normocarbic during the surgical procedure and throughout the experimental procedure with supplemental CO2 and O2.
Catheters were inserted into the right femoral artery and vein for monitoring blood pressure, sampling arterial blood gases, and administration of drugs. A PE-50 catheter was inserted into the right lingual artery so that its tip was located at the origin of the external carotid artery for retrograde delivery of the indicator umbelliferone into the internal carotid artery.
Skin, subcutaneous tissue, and muscle were excised over the right supraorbital ridge and parietal area. A craniectomy was performed utilizing a high speed air drill (Hall Surgical, Division of Zimmer, Santa Barbara, CA, U.S.A.) with the aid of an Olympus operating microscope (Olympus, Tokyo, Japan). The majority of the frontal and parietal cortex was exposed for imaging. The dura was removed and carefully cauterized at the margins of the craniectomy, then covered with Saran Wrap to prevent surface oxygenation and to keep the brain moist. Blood loss for surgical preparation was not >5 ml.
Following surgical preparation, the animal was moved from the operating table and placed on an intravital-type microscope stand. The microscope was focused on an area centered about the suprasylvian gyrus, with 1.5 cm2 of cortex imaged for pHi, regional cortical blood flow and NADH fluorescence measurements. Arterial blood pressure was measured by a Statham strain gauge (Statham, Oxnard, CA, U.S.A.) attached to the femoral artery catheter and recorded on a Grass model 78 polygraph. Animals were kept normothermic by the use of a heating blanket (K-Pad, Gorman-Rupp, Bellville, OH, U.S.A.) and core body temperature was monitored with a rectal digital thermometer. Arterial Paco2, Pao2, and pHa measurements were performed on a London Radiometer blood gas analyzer (PHM-73) (Copenhagen, Denmark).
Brain temperature was not measured in this study since earlier infrared microscopy studies in this laboratory had shown only small decreases (<1.5°C) in brain temperature during focal cerebral ischemia in the squirrel monkey (Sundt et al., 1976). It is known that brain tissue is a poor conductor of thermal activity. Therefore, in a model of focal cerebral ischemia, in which small volumes of tissue are affected, there would be small variations in brain temperature compared with that of global ischemia.
Focal cerebral ischemia was induced by occlusion of the middle (MCA) and anterior (ACA) cerebral arteries using Mayfield miniature aneurysm clips. The contralateral common carotid artery was also ligated. Animals were studied over a period of 180 min of focal ischemia. Thirty rabbits were divided into six groups of five each. There was a nonischemic control group and an ischemic control group, and four drug groups that received 0.1, 1, 10 mg/kg L-NAME, or 1 mg/kg L-NAME with simultaneous administration of 5 mg/kg L-arginine. Drugs were administered intravenously at a rate of 1 ml/min for 5 min at 20 min prior to the onset of ischemia.
In vivo video fluorescent instrumentation
Instrumentation was designed to perform serial panoramic video imaging of cortical brain pHi and regional cortical blood flow with umbelliferone fluorescence (Anderson et al., 1992). The optical characteristics were such that the majority of the entire hemisphere could be studied simultaneously through a large craniectomy. The use of umbelliferone as a noninvasive in vivo technique for measuring brain pHi and cortical blood flow has been previously described (Anderson et al., 1992; Anderson et al., 1987). Umbelliferone is nontoxic, fat-soluble, and freely diffusible across the blood-brain barrier (BBB), it is equilibrated rapidly across cell membranes and is distributed through the cytoplasm as an uncharged molecule (Anderson et al., 1992). Umbelliferone was prepared for injection by dissolving 0.2 g of indicator in 200 ml of 5% glucose-saline solution at 90°C for 30 min. The solution was then filtered through a 0.22 mesh filter before injection. The volume of injectate was 1.5 ml in this study.
The pH-sensitive indicator umbelliferone has two fluorophors, anionic and isobestic, that are excited at 370 and 340 nm, respectively, and have a common emission at 450 nm. Fluorescence of the anionic form varies directly with pH, whereas fluorescence of the isobestic form varies directly only with indicator concentration. Therefore, it is possible to create a nomogram from the ratio of 340-nm: 370-nm excitations to determine brain pHi. Acquired images were corrected for background NADH fluorescence before processing. NADH fluorescence images were stored for later analysis of mitochondrial function. Images from the 340–nm excitation were processed to compute regional cortical blood flow using the 1-min initial slope index. The regional cortical blood flow image was then displayed and stored on tape for final analysis. For processing of the pHi image, ratios of the paired images from the 340-nm:370-nm excitations were made, and the resultant pHi image was then displayed and stored on tape for final analysis. Umbelliferone has been shown to be a reliable indicator of cortical blood flow. Anderson et al., (1987) compared the different techniques of measuring cerebral blood flow (CBF) to that of umbelliferone. They found that a distinction can be made between cerebral blood flow (CBF), as measured by radiolabeled compounds, and cortical blood flow, as measured by umbelliferone. CBF, by definition, indicates areas of flow that contain major vessels as well as capillaries and arterioles, as measured by radiolabeled compounds. Cortical blood flow, as measured by umbelliferone, indicates areas that are relatively avascular and contain primarily arterioles and capillary beds. The imaging system allows the measurement of regional cortical blood flow by allowing the investigator to outline cortical areas of interest that are devoid of major surface conducting vessels.
Statistical analysis
Because of anatomical variation of the microvasculature from animal to animal, single points along an x,y-coordinates cannot be averaged, frame by frame, from different animals for the same time period. Therefore, measurements of regional pHi, regional cortical blood flow-umbelliferone, and NADH fluorescence were made in areas devoid of major vessels. Measurements of these parameters were made over these relatively avascular areas by averaging 10,000–15,000 pixels and the mean and standard deviation tabulated. Analysis of variance (ANOVA), followed by Newman-Keul's test for multiple comparisons were used to test the statistical significance of differences between groups. A p value of <0.05 was considered significant.
RESULTS
There were no significant differences over time between animals studied in each of the six groups in the measurements of Paco2, Pao2 body temperature, glucose, lactate, and hematocrit (Table 1). Mean arterial blood pressure (MABP) increased after L-NAME administration and this was dose-dependent (p < 0.05 at 10 mg/kg, Table 1).
Systemic parameters a
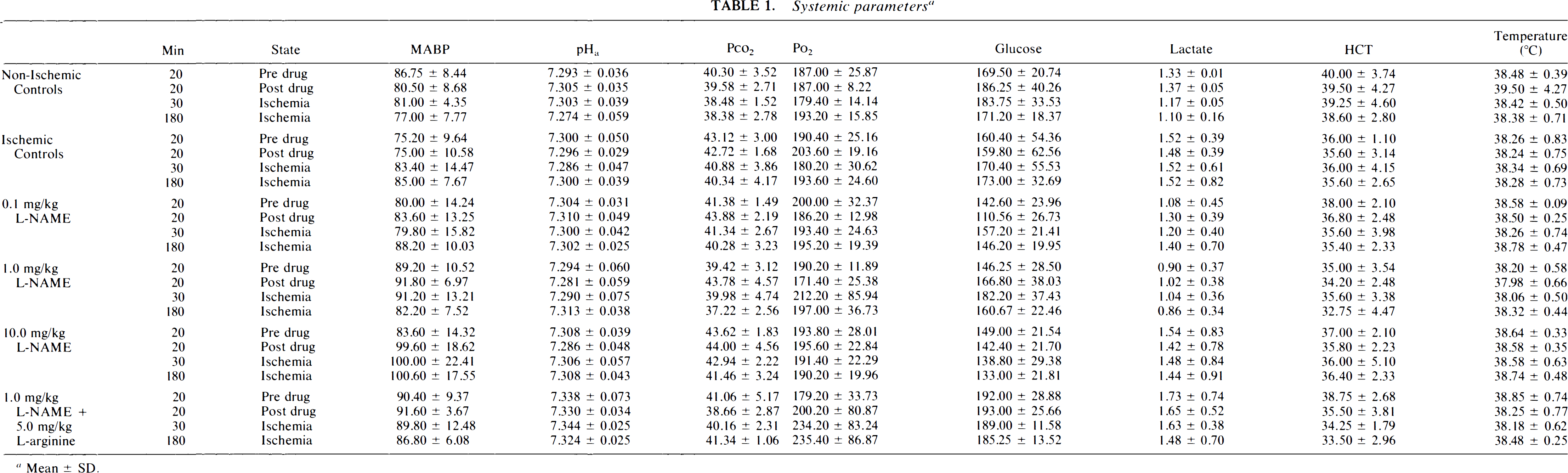
Mean ± SD.
Intracellular brain pH
Baseline brain pHi (Fig. 1) was uniform over the entire exposed cortex in all groups, measuring 6.99 ± 0.02. In the nonischemic control group, brain pHi remained stable throughout the experimental protocol and was 7.00 ± 0.03 after 240 min. During occlusion, pHi fell significantly (p < 0.05) in the ischemic-control group and was 6.73 ± 0.03 after 30 min of focal ischemia and did not recover after 180 min (6.77 ± 0.04). Drug treatment with either L-NAME or L-NAME + L-arginine did not affect preocclusion brain pHi. In the 0.1 mg/kg L-NAME group, pHi fell significantly from a baseline of 7.01 ± 0.02 to 6.76 ± 0.05 (p < 0.05) at 30 min. At 60 min, pHi was still acidic (6.79 ± 0.05, p < 0.05), but then steadily improved towards normalization, measuring 6.87 ± 0.04 at 120 min and 6.93 ± 0.07 at 180 min. In the 1 mg/kg L-NAME group, brain pHi remained normal throughout the whole experimental protocol including the period of focal ischemia (6.99 ± 0.03 at 30 min; 6.97 ± 0.04 at 60 min, and 6.98 ± 0.04 at 180 min). These improvements in brain pHi were not associated with regional cortical blood flow changes, as discussed below. In the 10 mg/kg L-NAME group, pHi decreased significantly to 6.81 ± 0.03 after 30 min of focal ischemia. Similarly, pHi fell to 6.81 ± 0.03 (p < 0.05) after 30 min of ischemia in the combined L-NAME + L-arginine group. Both of these groups showed persistent and significant intracellular acidosis for the remainder of the ischemic period (6.75 ± 0.05 at 60 min, 6.75 ± 0.05 at 120 min, and 6.77 ± 0.06 at 180 min for the 10 mg/kg L-NAME group, and 6.79 ± 0.03 at 60 min, 6.78 ± 0.03 at 120 min, and 6.84 ± 0.04 at 180 min for the mixed L-NAME + L-arginine group).
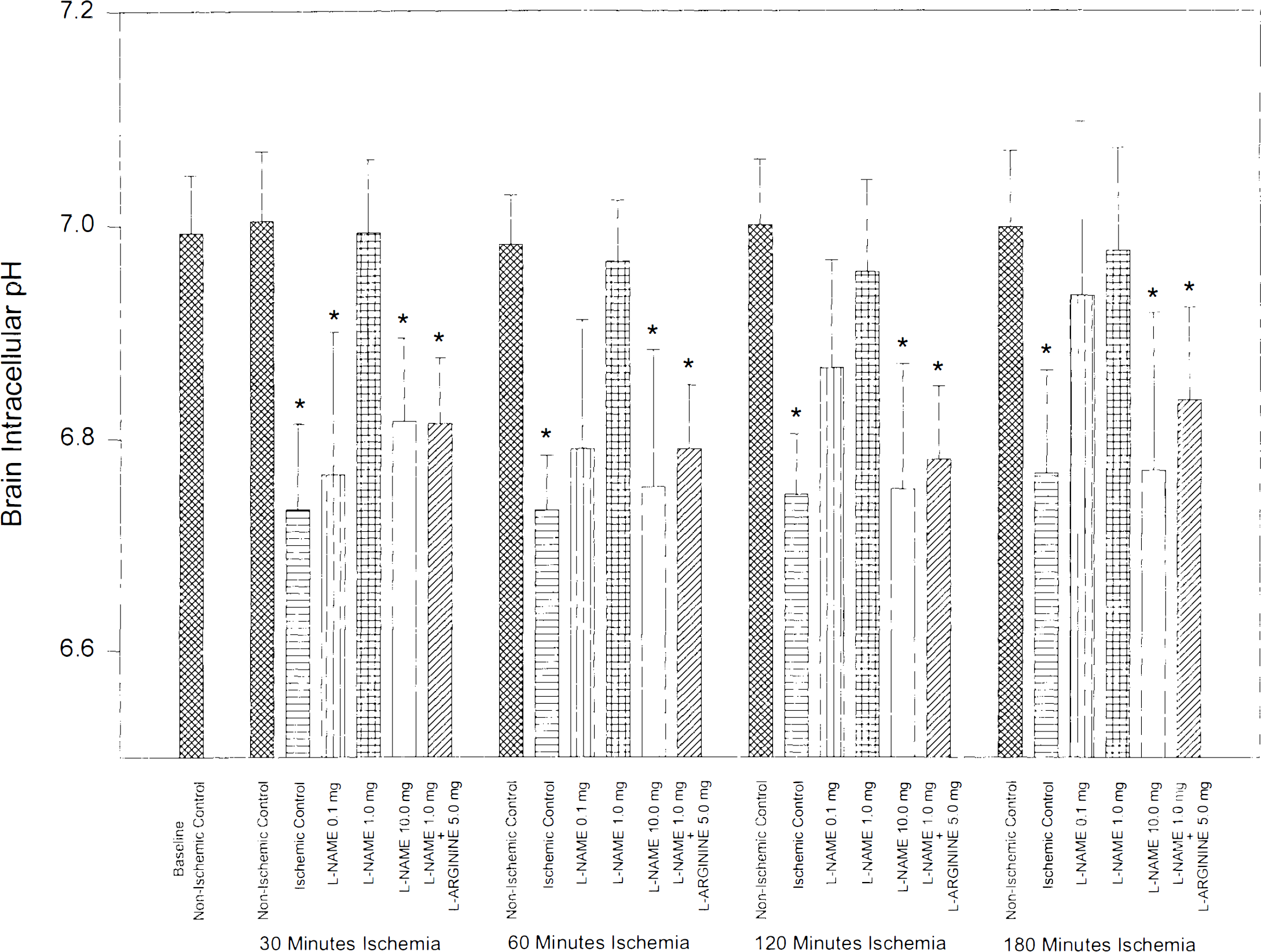
Bar graph showing intracellular brain pH (pHi) of the six study groups (n = 5 in each group) throughout the experimental protocol. Focal cerebral ischemia was produced by MCA occlusion at time 0 and maintained for 180 min. Drug administration did not affect pHi. The 1 mg/kg L-NAME ischemic group did not develop acidosis during focal cerebral ischemia, and pHi was not different from that of nonischemic controls throughout the experimental protocol. The 0.1 mg/kg L-NAME ischemic group showed early significant acidosis that recovered steadily after the 60 min time frame. Ischemic controls, the 10 mg/kg L-NAME, and the 1 mg/kg L-NAME + 5 mg/kg L-arginine ischemic groups all showed early significant acidosis during focal ischemia that did not recover during the 180 min of observation. Values are mean ± SD, * indicates p < 0.05.
Regional cortical blood flow
Regional cortical blood flow (Fig. 2) was stable (49.6 ± 2.8 ml/100 g/min) throughout the experimental protocol in the nonischemic control group. In the ischemic control group, baseline regional cortical blood flow measured 49.9 ± 1.5 ml/100 g/min. Regional cortical blood flow fell significantly (p < 0.05) to 28.7 ± 3.7 ml/100 g/min after 30 min of occlusion and remained near this level for the remainder of the ischemic event, being significantly different from nonischemic control values. Baseline regional cortical blood flow was 52.2 ± 5 ml/100 g/min in the four drug treatment groups. At 5 and 20 min after drug infusion, regional cortical blood flow was not different from control values in the 0.1 and 1 mg/kg L-NAME and the L-NAME + L-arginine groups. In the 10 mg/kg L-NAME group, there was a 30% decrease in regional cortical blood flow after drug infusion that was significant at 20 min (38.2 ± 5.8 ml/100 g/min, p < 0.05). During occlusion, regional cortical blood flow decreased in a dose-dependent manner. After 3 h of ischemia, regional cortical blood flow was 33.9 ± 10.9 and 25.1 ± 8.9 ml/100 g/min at doses of 0.1 and 10.0 mg/kg, respectively.
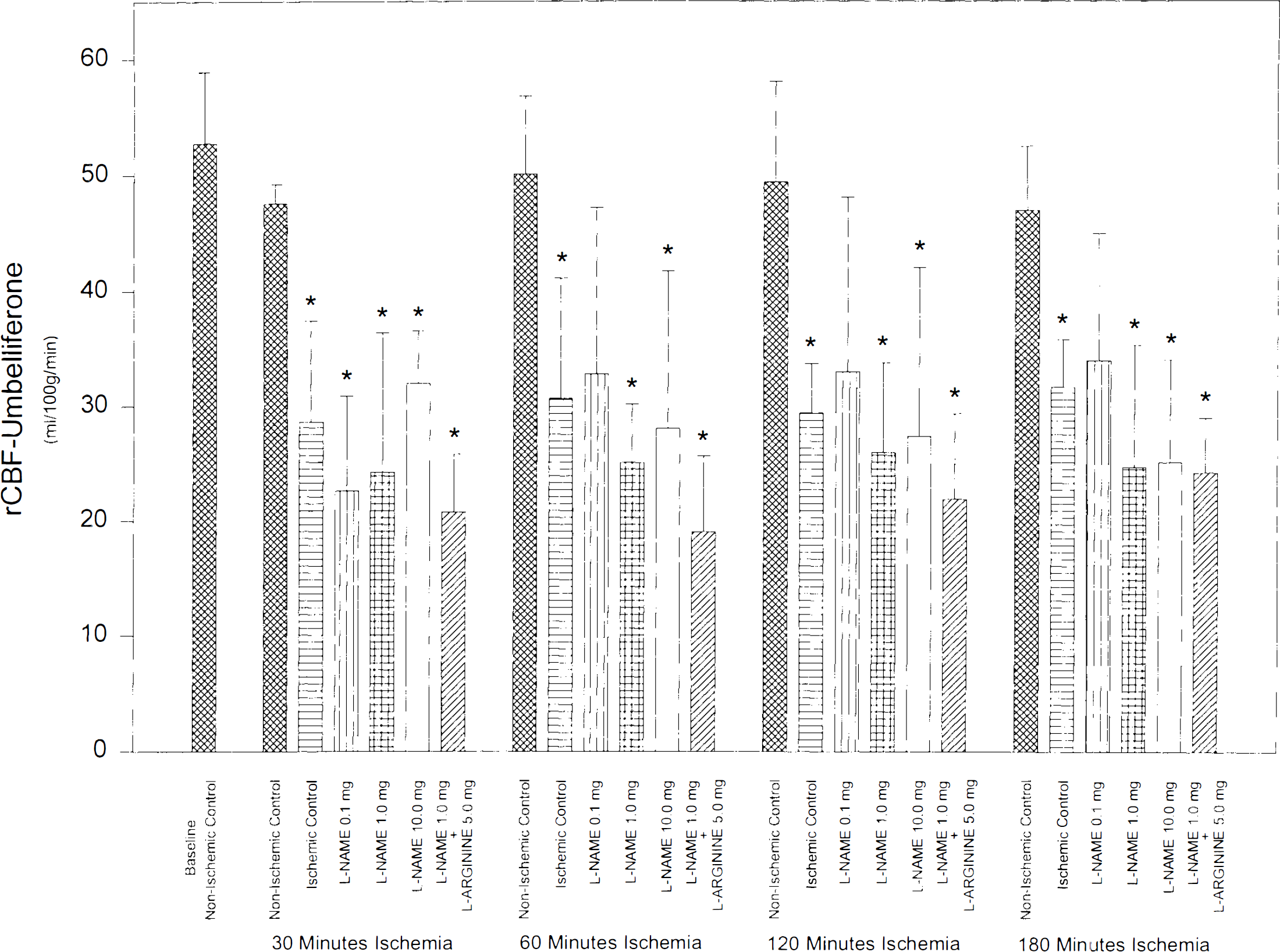
Bar graph showing regional cortical blood flow regional cortical blood flow of the six study groups (n = 5 in each group) throughout the experimental protocol. Focal cerebral ischemia was produced by MCA occlusion at time 0 and maintained for 180 min. Drug infusion decreased regional cortical blood flow by 30% in the 10 mg/kg L-NAME group prior to MCA occlusion. In all other groups, rCBF did not change after drug infusion. MCA occlusion decreased regional cortical blood flow in all ischemic groups by the same extent, and was significantly different from baseline. Values are mean ± SD, * indicates p < 0.05.
NADH fluorescence
NADH fluorescence measured over the exposed cortex remained stable throughout the experimental protocol in nonischemic control animals. NADH fluorescence was not affected by drug treatment, and values after infusion did not differ from control values. Thirty minutes after ischemia, NADH fluorescence increased in the ischemic control group by 155 ± 12% (p < 0.05) and still measured 143 ± 9% (p < 0.05) after 180 min of occlusion. A similar massive increase in ischemic NADH fluorescence was recorded in the L-NAME + L-arginine group: 149 ± 5% at 30 min and 146 ± 10% at 180 min of focal ischemia. All three L-NAME treatment groups showed an increase in NADH fluorescence (∼141 ± 7% at 30 min and 122 ± 8% at 180 min). The percent increase was smaller than in ischemic control and L-NAME + L-arginine groups, but was not statistically different.
DISCUSSION
This study examined the effects of NOS inhibition by L-NAME on brain pHi, regional cortical blood flow, and NADH fluorescence during focal cerebral ischemia in vivo. The most consistent observation was that intravenous administration of low dose L-NAME prior to occlusion prevented intracellular brain acidosis during focal cerebral ischemia. This effect was not related to regional cortical blood flow changes since blood flow decreased to the same extent during ischemia in all groups. Coadministration of L-NAME and L-arginine abolished the protective effect on brain acidosis, which suggests that the mediator was NOS. Administration of high dose L-NAME (10 mg/kg) decreased regional cortical blood flow prior to occlusion and ischemic brain acidosis was not prevented in this group.
Nitric oxide is increasingly being recognized as a major regulator of central nervous system (CNS) function. In the CNS, NO can originate from the endothelial cells of the cerebral vessels and from NOS containing neurons (Bredt and Snyder, 1994; Bruhwyler et al., 1993) and plays a role in CBF regulation (Iadecola et al., 1994). Neuronal NOS is a constitutive enzyme that is activated by Ca2+ (Bredt and Snyder, 1990), and has been shown to be coupled primarily to activation of the NMDA-receptor (Dawson et al., 1993). NO is synthesized from L-arginine by NOS, which is competitively inhibited by L-NAME (Rees et al., 1990). Dawson et al., (1991) showed that NMDA toxicity was blocked by NOS inhibitors in vitro. NO measurements in vivo have demonstrated that NO increases significantly during focal cerebral ischemia (Malinski et al., 1993). Reports on the effects of NO in ischemic neuronal injury in vivo have been contradictory. Inhibition of NO synthesis has been shown, in some laboratories, to enhance injury (Zhang and Iadecola, 1994; Yamamoto et al., 1992; Kuluz et al., 1993) or to provide substantial protection (Nowicki et al., 1991; Ashwal et al., 1994; Nishikawa et al., 1994). Nonselective inhibition of vascular and neuronal isoforms could mask a neuroprotective effect evoked by selective neuronal NOS inhibition. A recent study by Huang et al., (1994), using mice deficient in neuronal NOS (thus avoiding this dual action of NO), provides substantial evidence that lack of neuronal NOS activity is associated with reduced ischemic damage. In our study, L-NAME prevented brain acidosis only at lower doses, doses at which no regional cortical blood flow changes could be detected. Alternatively, prevention of acidosis in the high-dose L-NAME group could have been obscured by a hemodynamic effect due to endothelial NOS inhibition that resulted in a 30% decrease in regional cortical blood flow which, in turn, would exacerbate ischemic damage. In addition, L-NAME at higher dosages may directly affect mitochondrial respiration (via interfering with cytochrome c reduction by Fe2+) and other iron-chelated reactions (Peterson et al., 1992).
Intracellular acidosis during focal cerebral ischemia has long been regarded as a mediator of cell death provided that the acidosis is severe (Siesjö et al., 1993; Kraig et al., 1987). Heinzel et al., (1992) showed that as the pH of the environment becomes more acidotic, NO production increases. In the present study, we showed that inhibition of NOS could be neuroprotective. However, we did not perform histology in these animals and, therefore, could not determine if treatment with L-NAME was neuroprotective. The cellular and molecular mechanisms by which intracellular acidosis is postulated to be detrimental include disruption of cell volume regulation with edema formation, accentuation of the rise in intracellular Ca2+, inhibition of mitochondrial respiration, inhibition of lactate and NADH regeneration, and promotion of free radical generation by facilitating the Haber-Weiss reaction (Siesjö et al., 1993; Rehncrona, 1985). In spite of the evidence in favor of acidosis being injurious, discrepant results have been published suggesting that if not too pronounced, acidosis may, in fact, be protective (Traynelis and Cull-Candy, 1990; Tang et al., 1990).
Several possible mechanisms by which NOS inhibition might decrease intracellular pH during focal cerebral ischemia include DNA damage by deamination (Nguygen et al., 1992), free radical-mediated production of peroxynitrite anions (ONOO−) (Beckman and Crow, 1993; Lipton et al., 1993), 5-nitrosylation of proteins, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) inhibition (Zhang and Snyder, 1992; McDonald and Moss, 1993), and formation of NO-iron complexes with several enzymes including complexes I and II of the mitochondrial electron transport (Weiss et al., 1993).
NO, like other free radicals, can damage DNA by base deamination (Nguyen et al., 1992; Wink et al., 1991). Zhang et al., (1994) demonstrated that NO activates poly(adenosine 5′-diphosphoribose) synthetase (PARS) in association with damage to DNA, and that PARS inhibitors prevent NMDA neurotoxicity with relative potencies paralleling their inhibition of the enzyme. This enzyme activation can lead to cell death associated with depletion of ATP and a change in the NAD+/NADH redox state (Gaal et al., 1987). Inhibition of NOS during ischemia should retard this pathway and decrease consumption of both ATP and oxidized NAD+. An increase in ATP availability during ischemia certainly favors cell function, including pHi regulation involving the Na+/H+ exchanger and the HCO−3/Cl− transporter, which are energy-dependent. In addition, lack of PARS activation should decrease cellular consumption of NAD+ and reduce the drive for NADH regeneration coupled to lactic acid synthesis.
Free radical-mediated damage is another key common pathway of injury (Siesjö et al., 1989). When NO reacts with superoxide anions (O−2) it forms peroxynitrite anions (ONOO−) (Beckman and Crow, 1993; Lipton et al., 1993). These decompose to yield highly damaging hydroxyl free radicals, which have been suggested to initiate lipid peroxidation (Watson, 1993). In addition to producing active metabolites, peroxidation may alter cell membrane fluidity (Thaw et al., 1983), with potential detrimental effects on membrane associated H+ transporter and antiporter involved in brain pHi regulation. Lei et al. (1992), showed that NO may be neuroprotective against NMDA toxicity, while others have shown no neuroprotective effects (Regan et al., 1993; Pauwels and Leysen, 1992). NO may exert both neurodestructive and neuroprotective effects, depending on its oxidation-reduction status, with NO− being neurodestructive and NO+ being neuroprotective (Lipton et al., 1993).
Another possible mechanism is the stimulation by NO of the 5-nitrosylation of various proteins (Lipton et al., 1993). NO can also modify GADPH, an important enzyme of glycolysis (McDonald and Moss, 1993). Inhibition of GAPDH will also suppress lactate formation and could reduce brain tissue acidosis (Hillered et al., 1985). NO has also been reported to inhibit iron-sulfur enzymes of the citric acid cycle and respiratory chain (Weiss et al., 1993). Improved respiratory metabolism and NOS inhibition should decrease intracellular acidosis during focal ischemia.
In conclusion, this study demonstrates that NOS inhibition by L-NAME prevents intracellular acidosis during focal cerebral ischemia independent from regional cortical blood flow changes. This observation has not been reported before, but based on this study, we postulate that brain acidosis may reflect NO-mediated neurotoxicity during focal ischemia. The multiplicity of actions attributed to NO in the CNS complicate studies in in vivo ischemia models. The molecular mechanisms by which L-NAME prevents acidosis during focal ischemia remain uncertain, but further exploration of pHi modulation by NO during focal cerebral ischemia may help better understand the complex pathways of ischemic neuronal injury.
Footnotes
Acknowledgment:
The authors are indebted to Ms. Heidi Martin and Mr. Robert Carlson for their technical assistance, and Ms. Mary Soper for preparation of the manuscript. We are most appreciative of the valuable suggestions of W. K. Tan, Ph.D. This work was supported by NIH grant RO1 25374 and grants from the Foundation SICPA and the Foundation Decker of Lausanne, Switzerland (L.R.).