
Abstract
We have previously shown that treatment with selective kappa-opioid receptor agonist BRL 52537 hydrochloride [(±)-1-(3,4-dichlorophenyl) acetyl-2-(1-pyrrolidinyl) methylpiperidine] (1) has a long therapeutic window for providing ischemic neuroprotection and (2) attenuates ischemia-evoked nitric oxide (NO) production in vivo in rats. Neuronally derived NO has been shown to be deleterious in the male, but not in the female, rodent model of focal ischemic stroke. We sought to determine if the agent fails to protect ischemic brain when neuronal NO synthase (nNOS) is genetically deleted in male, but not female, mice. Halothane-anesthetized adult male and female nNOS null mutants (nNOS−/-) and the genetically matched wildtype (WT) strain were subjected to transient (2 h) middle cerebral artery occlusion by the intraluminal filament technique. Vehicle or BRL 52537 treatment with continuous intravenous infusion was instituted at the onset of reperfusion and continued for 22 h. In WT male mice, infarct volumes measured at 72 h of reperfusion were robustly decreased with BRL 52537 treatment. In contrast, BRL 52537 did not decrease infarct volume in male nNOS−/- mice. BRL 52537 had no effect in the WT or nNOS−/- female mice. These data support that BRL 52537% mechanism of neuroprotection in vivo is through attenuation of nNOS activity and ischemia-evoked NO production. Neuroprotective effects of BRL 52537 are lost in the male when nNOS is not present; therefore, BRL 52537 likely acts upstream from NO generation and its subsequent neurotoxicity.
Introduction
Kappa (κ)-opioid receptors (KOR) have been shown to play an important modulatory role in ischemic brain injury. Several studies have shown KOR agonists to attenuate histologic brain injury (Mackay et al, 1993; Birch et al, 1991; Baskin et al, 1994) as well as improve functional recovery in animal models of global and focal cerebral ischemia (Itoh et al, 1993; Genovese et al, 1994; Goyagi et al, 2003; Zhang et al, 2003; Chen et al, 2004, 2005). We have previously shown that a highly selective KOR agonist has a long therapeutic window for providing ischemic neuroprotection after transient focal ischemia in a rat model of transient focal ischemia with middle cerebral artery occlusion (MCAO) (Chen et al, 2004), and this neuroprotective action is dose dependent (Goyagi et al, 2003), receptor selective (Zhang et al, 2003), and observed only in male animals (Chen et al, 2005).
Abundant literature shows the effects of KOR agonists on a variety of neurotransmitter systems (You et al, 1999; Schoffelmeer et al, 1997; Shippenberg et al, 2001). While some in vivo studies have shown that KOR agonists modulate dopaminergic neurotransmission in the substantia nigra, neostriatum, and the mesolimbic system (You et al, 1999; Schoffelmeer et al, 1997; Shippenberg et al, 2001), we did not observe modulation of ischemia evoked acute release of dopamine or its metabolites (Chen et al, 2004). Nitric oxide (NO) derived from constitutively expressed nitric oxide synthase (NOS) in neurons (nNOS) and the inducible isoform expressed by other cellular elements (iNOS) are important mediators in the excitotoxic cascade of ischemic brain injury (Iadecola, 1997; Samdani et al, 1997). We have previously shown that selective KOR agonist attenuates ischemia-evoked NO production in the striatum in vivo, and have postulated that this may account for its neuroprotective action (Goyagi et al, 2003), although the precise signaling of this interaction remains unclear.
It is well known from epidemiologic studies that biologic sex plays a major role in the incidence and prevalence of ischemic stroke (Hurn and Brass, 2003). In addition, mounting evidence demonstrates gender-specific responses to brain injury after experimental cerebral ischemia (Alkayed et al, 1998; Hurn and Macrae, 2000; McCullough and Hurn, 2000). While animal studies have shown these observations across different animal models and genetic strains and implied that endogenous sex steroids are a major influence in outcome after ischemic stroke (Alkayed et al, 1998, 2000; Hurn and Macrae, 2000; McCullough and Hurn, 2003), emerging data suggest that excitotoxic pathways might have a differential effect in males versus females. For example, neuronally derived NO has been shown to be deleterious in the male, but not in the female, rodent model of focal ischemic stroke (Sampei et al, 2000; McCullough et al, 2005). We have previously shown that the selective KOR agonist provides ischemic neuroprotection in male, but not in female, rats, and that the lack of protection by BRL 52537 is not due to female sex steroids (Chen et al, 2005).
In this study, we tested the hypothesis that the highly selective KOR agonist BRL 52537 hydrochloride [(±)-1-(3, 4-dichlorophenyl) acetyl-2-(1-pyrrolidinyl)methylpiperidine)] (Vecchietti et al, 1991, 1992) acts in a sex-specific manner to provide ischemic neuroprotection in a well-characterized murine model of transient focal ischemia. We further sought to determine if BRL 52537 fails to protect ischemic brain when nNOS is genetically deficient (nNOS−/-). If so, then these results would strongly implicate neuronal NO as an important target in BRL 52537's neuroprotective mechanism(s).
Materials and methods
Experimental protocols were approved by the Institutional Animal Care and Use Committee, and conform to the National Institutes of Health guidelines for the care and use of animals in research. All methods have been previously described in mice (Sampei et al, 2000, Sawada et al, 2000).
Middle Cerebral Artery Occlusion in Mice
The nNOS null mutant (nNOS−/-) mice were produced on a purebred C57BL/6 background as described previously (Huang et al, 1993). Adult male and female nNOS−/- and wildtype (WT) C57BL/6 14 mice (21 to 28 g) were anesthetized with 1% to 1.2% halothane in oxygenenriched air. With aseptic surgical techniques, the right jugular vein was cannulated, tunneled subcutaneously, exteriorized, and tethered to the skin for vascular access and drug administration. Rectal temperature was maintained throughout surgical procedures, during ischemia, and until emergence from anesthesia.
Transient focal ischemia (2 h) was produced by MCAO, using an intraluminal suture technique in combination with laser-Doppler flowmetry (LDF) (Moor Instruments Ltd, Model MBF3D, England) over the ipsilateral parietal cortex, as described previously (Sampei et al, 2000; Sawada et al, 2000). Animals were then allowed to waken in separate cages and housed at ambient room temperature. Daily neurologic deficit scoring (NDS) was performed as follows: 0 = normal motor function, 1 = flexion of torso and of contralateral forelimb on tail lift, 2 = circling to the contralateral side but normal posture at rest, 3 = leaning to contralateral side at rest, 4 = no spontaneous motor activity. Animals that did not show reduction in LDF signal or that had an NDS ≤1 on emergence from anesthesia were excluded from the study. After completion of treatments at 22 h of reperfusion, the jugular catheter was carefully removed. At 72 h of reperfusion, brains were harvested for assessment of injury volume. The forebrain was sliced into five 2-mm thick coronal sections, which were stained with 1% triphenyltetrazolium chloride (TTC), as described previously (Eliasson et al, 1997; Sampei et al, 2000, Sawada et al, 2000). Infarct volume was measured with digital imaging. The infarcted area was numerically integrated across each section and over the entire ipsilateral hemisphere. Infarct volumes were measured separately in the cerebral cortex and caudoputamen (CP) complex, and expressed as percentage volumes of the contralateral structure (correction for swelling), as described previously in the rat model of MCAO (Zhang et al, 2003; Chen et al, 2004).
Experimental Groups
All experiments were performed in a randomized fashion with the investigator masked to treatment. In the first series of experiments, male WT and nNOS−/- mice were treated with continuous intravenous infusion of vehicle (saline) or BRL 52537 (1 mg/kg h). In the second series of experiments, female WT and nNOS−/- mice were treated with continuous intravenous infusion of vehicle or BRL 52537 (1 mg/kg h). Treatments were started at the onset of reperfusion after 2 h of MCAO and continued for 22 h. All intravenous infusion rates were 0.05 mL/h.
Statistical Analysis
Laser-Doppler flowmetry and rectal temperature measurements among groups were subjected to repeated-measures analysis of variance (ANOVA). Differences in infarct volume were determined by one-way ANOVA. Post hoc analysis comparisons were made with the Newman–Keuls test. Data are presented as means ± s.d. Neurologic deficit scoring score is presented as median (with 25% and 75% quartiles) and analyzed by the nonparametric Mann–Whitney U-test. A value of P < 0.05 was considered significant.
Results
In the first set of experiments, premature mortality (prior to 72 h) was 2/12 in WT-vehicle-, 1/12 in WT-BRL-, 2/11 in nNOS−/--vehicle-,and 4/18 in nNOS−/--BRL-treated male mice. Two mice in the WT-BRL group sustained a small hemorrhage in the infarct and were excluded from the final analysis. Thus, the animals that were included in the final analysis were as follows: male WT-vehicle, n = 10; male WT-BRL, n = 9; male nNOS−/--vehicle, n = 9; male nNOS−/--BRL, n = 14.
Rectal temperatures were similar among different experimental groups. Percent body weight loss was significantly less in WT-BRL 52537-treated mice as compared with WT-vehicle-treated counterparts (Table 1). Infarct volume was significantly attenuated in BRL 52537-treated male WT (cortex: 29% ± 22%; CP complex: 55% ± 22%) as compared with vehicle-treated WT male mice (cortex: 53% ± 21%; CP complex: 74% ± 6%). Neurologic deficit scoring was also attenuated in BRL 52537-treated male WT mice. BRL 52537 did not confer any additional protection to vehicle-treated male nNOS−/- (Figure 1). In the second set of experiments, premature mortality rates were as follows: 4/12 in WT-vehicle, 2/10 in WT-BRL, 3/12 in nNOS−/- vehicle, and 15/26 in nNOS−/--BRL in female mice. The effect of BRL on mortality rate was determined by the Mantel–Haenszel statistic for testing conditional independence on layered sets of 2 × 2 frequency tables (SPSS, Inc., Chicago IL, USA). There was no significant effect of BRL on mortality rate conditioned on sex and nNOS gene deletion.
Summary of physiological variables and NDS in male mice
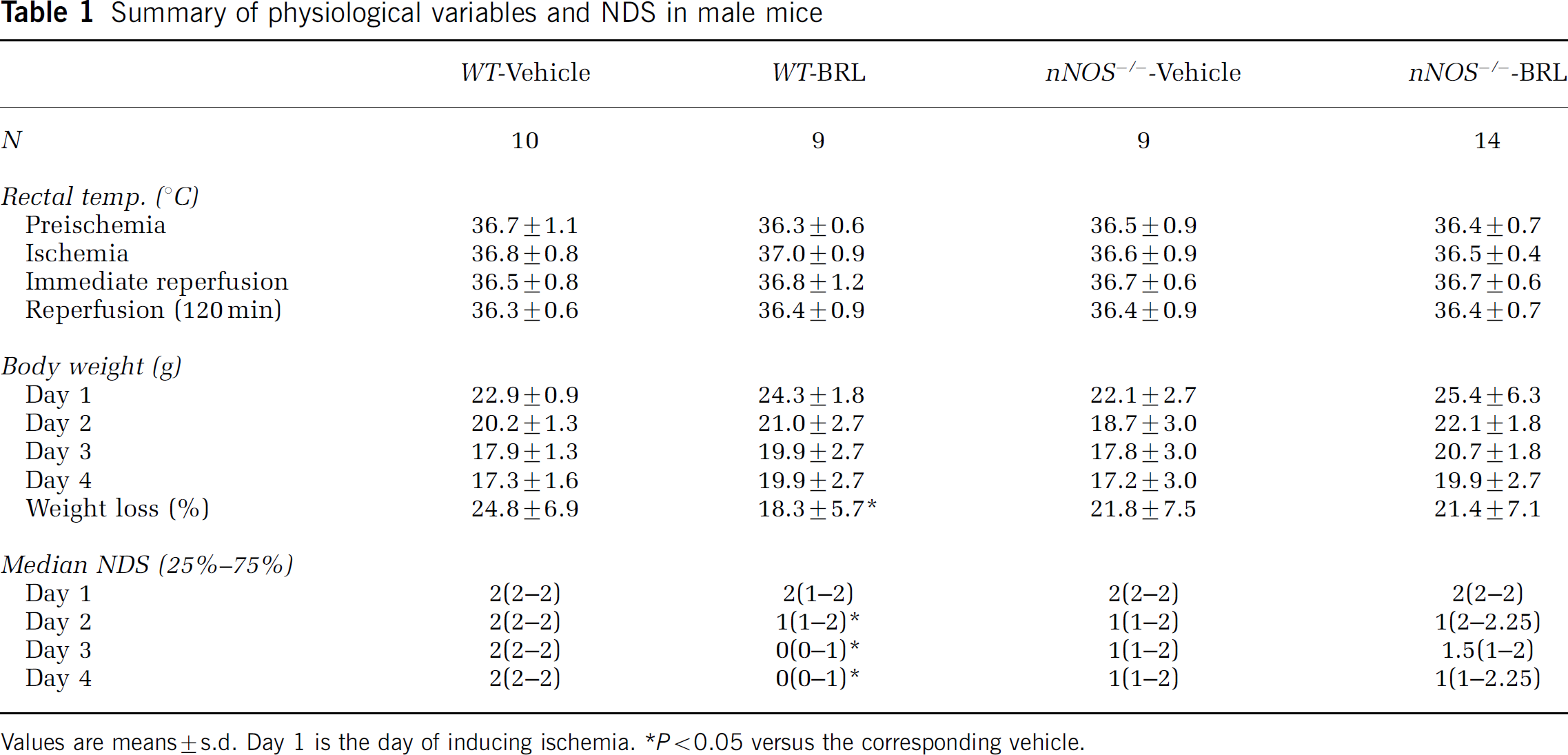
Values are means ± s.d. Day 1 is the day of inducing ischemia.
P < 0.05;versus the corresponding vehicle.
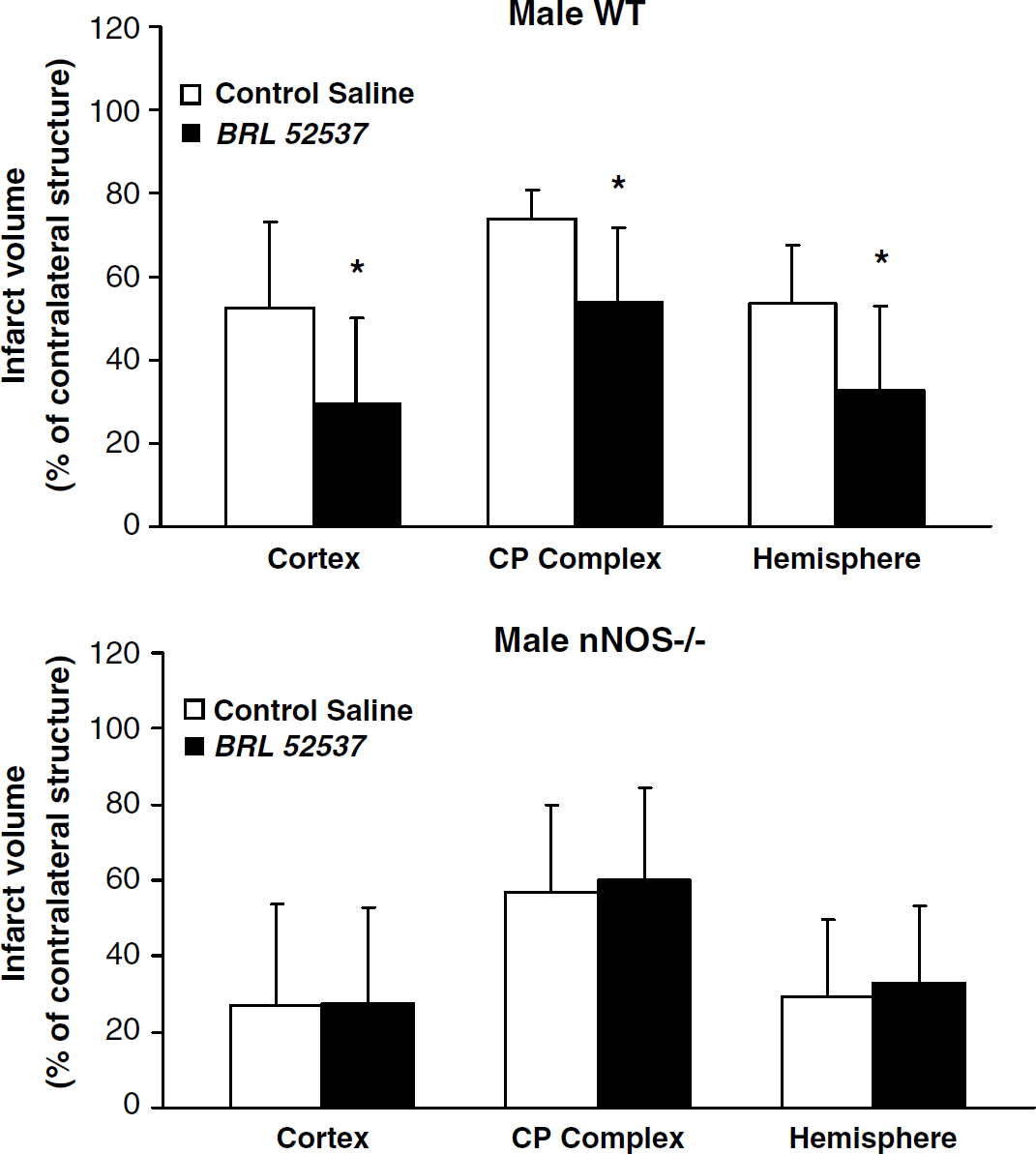
Triphenyltetrazolium chloride-determined infarct volume, corrected for swelling (% of contralateral structure) at 72 h of reperfusion (mean ± s.d.) in vehicle-treated (n = 10) or BRL 52537-treated (n = 9) male WT mice (top panel) and vehicle (n = 9) or BRL 52537-treated (n = 14) male nNOS−/- mice (lower panel). Treatments were begun at reperfusion after 2 h of MCAO and continued for 22 h. *P < 0.05 versus vehicle treatment.
The animals that successfully completed the experimental protocol were as follows: female WT-vehicle, n = 8; female WT-BRL, n = 8; female nNOS−/--vehicle, n = 9; female nNOS−/--BRL, n = 11. Reduction in LDF signal (from preischemic baseline) during MCAO was similar among treatment groups (WT-vehicle: 89.4% ±3.7%; WT-BRL: 90.4% ±3.7%; nNOS−/--vehicle: 87.3% ± 6.9%; nNOS−/--BBL: 86.7% ± 5.4%) in female mice. Rectal temperature was similar in all treatment groups at baseline, during MCAO, and at immediate and 2-h reperfusion. Percent body weight loss was similar across all treatment groups. BRL 52537 treatment did not confer improvement in NDS compared with vehicle-treated WT or nNOS−/- counterparts (Table 2). BRL 52537 also did not reduce infarct volume in female WT or nNOS−/- mice compared with those treated with vehicle (Figure 2).
Summary of physiologic variables and NDS in female mice
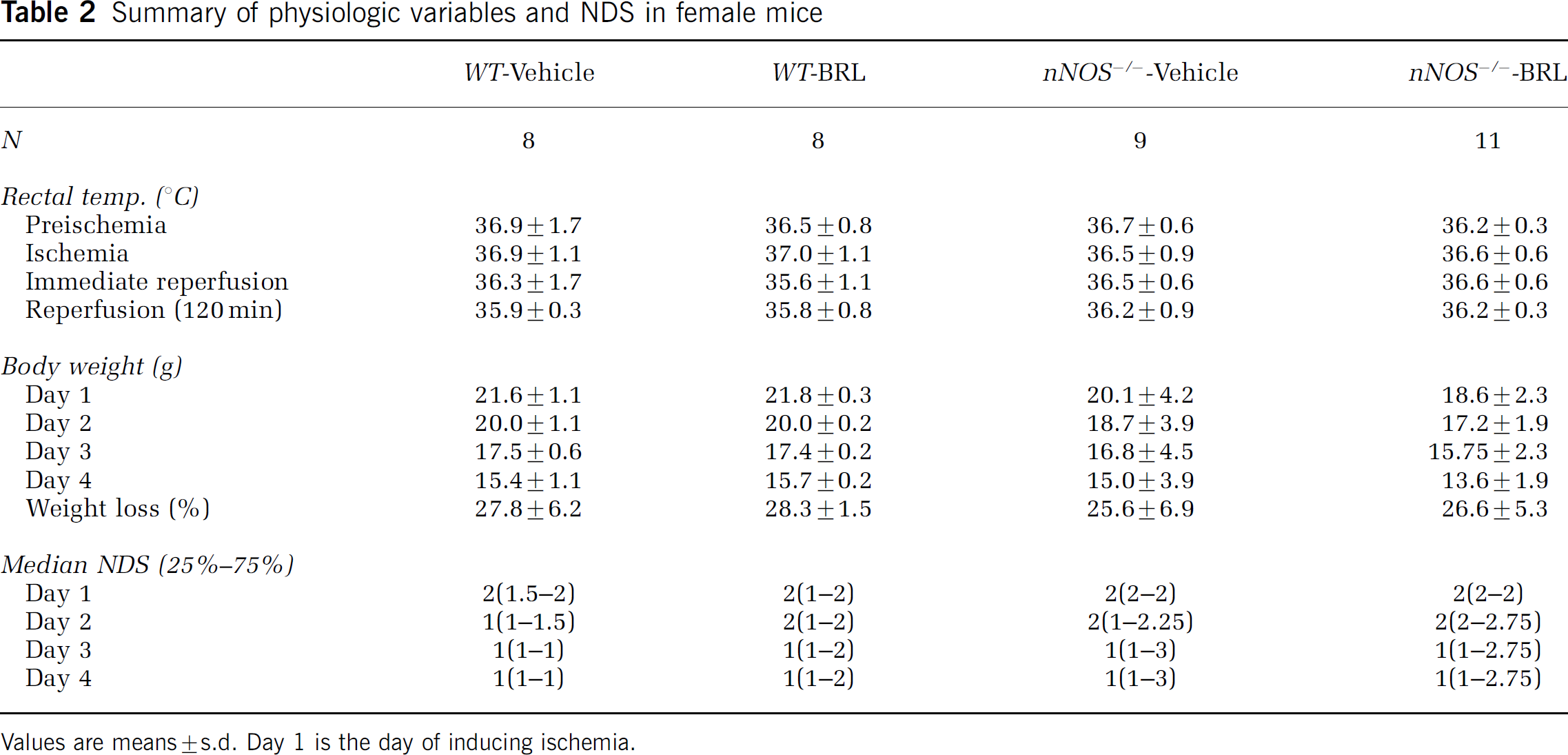
Values are means ± s.d. Day 1 is the day of inducing ischemia.
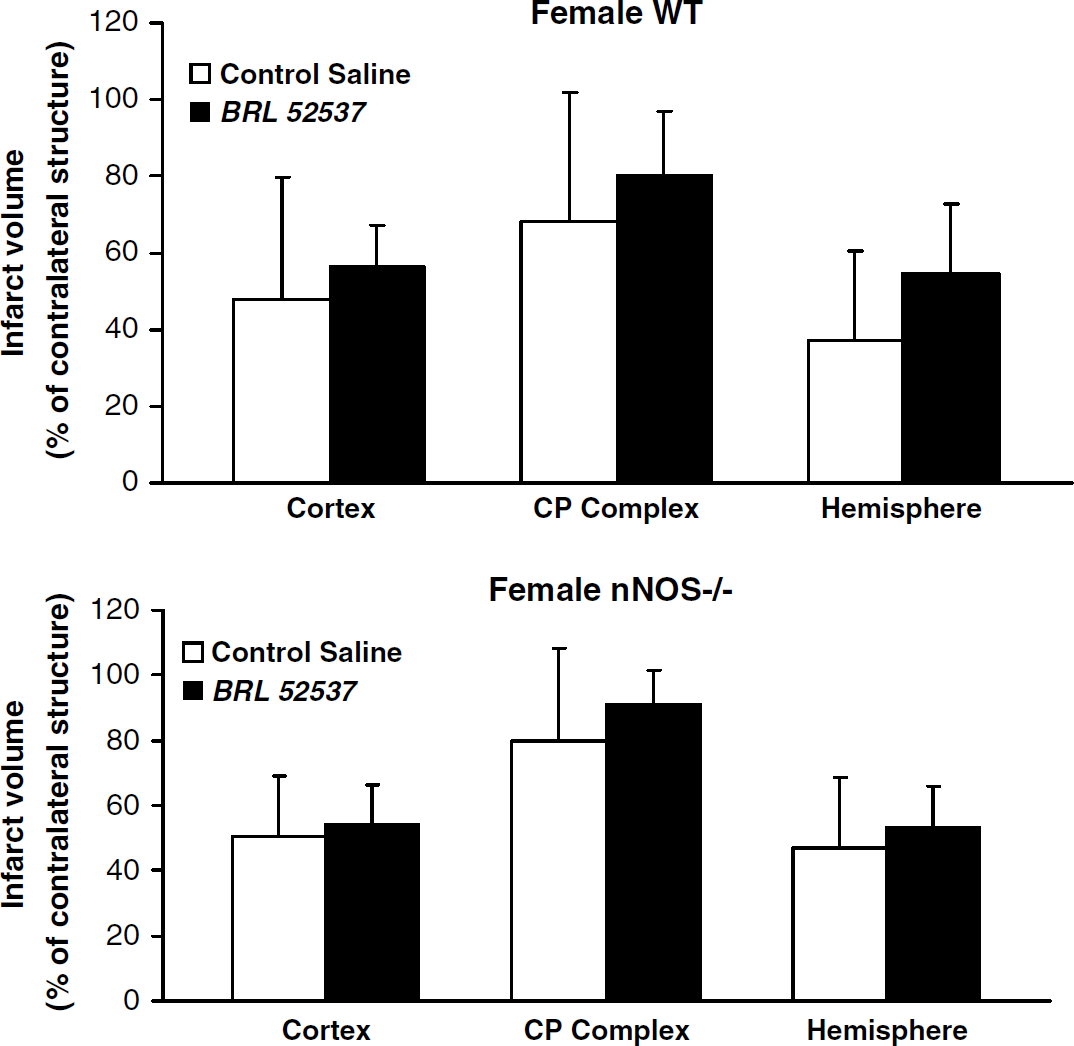
Triphenyltetrazolium chloride-determined infarct volume, corrected for swelling (% of contralateral structure) at 72 h of reperfusion (mean ± s.d.) in vehicle-treated (n = 8) or BRL 52537-treated (n = 8) female WT mice (top panel) and vehicle-treated (n = 9) or BRL 52537-treated (n = 11) female nNOS−/- mice (lower panel). Treatments were begun at reperfusion after 2 h of MCAO and continued for 22 h.
Discussion
This study demonstrates three important findings. First, intravenous administration of a selective KOR agonist, BRL 52537, confers ischemic neuroprotection that is specific to the male animal. Second, ischemic neuroprotection seen in male mice is due to an interaction between the KOR agonist and nNOS, and supports the results of our previous study (Goyagi et al, 2003), which suggests that one mechanism by which BRL 52537 reduces ischemic damage in male animals is through reduction of early NO toxicity. Third, the present study confirms our previous findings in the rat that ablation of nNOS activity does not alter ischemic outcome in female brain, and supports the finding that BRL52537's sex-specific efficacy is related to its potential to reduce NO toxicity. Based on these data, we speculate that the compound would be best employed as an antiischemic therapy for the male.
Ischemic Neuroprotection with BRL 52537
κ-Opioid receptors have been of interest as potential therapeutic targets for ischemic neuroprotection for several years. As in our previous studies (Goyagi et al, 2003; Zhang et al, 2003; Chen et al, 2004, 2005), we used BRL 52537 hydrochloride (Vecchietti et al, 1991, 1992), a water-soluble agent that is a highly specific KOR agonist (e.g., Ki κ—0.24 nmol/L; Kiμ—1560 nmol/L). We have previously shown that BRL 52537 provides significant ischemic neuroprotection when the onset of treatment is delayed for up to 6 h of reperfusion after 2 h of MCAO in the rat in a dose-dependent fashion (Chen et al, 2004). This observed neuroprotection with BRL 52537 is without alteration in core body temperature (Zhang et al, 2003), without any significant effects on physiologic parameters evaluated within our studies (Goyagi et al, 2003; Zhang et al, 2003; Chen et al, 2004), and is gender specific (Chen et al, 2005). Furthermore, prolonged treatment (4 days) at these neuroprotective doses does not result in gross neuropathology or myelin injury in naive nonischemic rats (Goyagi et al, 2003). In the present study in mice, we utilized the same dose (1 mg/kg h) of BRL 52537 that conferred neuroprotection in our rat studies previously (Goyagi et al, 2003; Zhang et al, 2003; Chen et al, 2004, 2005). Data from the present study confirm our previous findings in the rat model of focal ischemia that intravenous administration of BRL 52537 provides significant ischemic neuroprotecton in the murine model of MCAO in male animals only (Figure 1).
BRL 52537 and Neurotransmitter System Interactions
Data from several previous studies support the premise that antiexcitotoxic mechanisms are important in ischemic neuroprotection provided by KOR agonists. In vitro studies have demonstrated that KOR agonists modulate glutamate toxicity via inhibition of presynaptic glutamate release, possibly by closing N-type Ca2+ channels and inhibiting excitatory postsynaptic potentials by attenuating presynaptic Ca2+ influx (Gross and Macdonald, 1987; Xiang et al, 1990). Others have shown attenuation of glutamate release (Mackay et al, 1993) with graded ischemia in experimental stroke with a KOR agonist, as well as modulation of the inhibitory neurotransmitter γ-aminobutyric acid (Hjelmstad and Fields, 2003). In contrast to previous in vitro studies, neuroprotective doses of BRL 52537 did not alter the acute release of dopamine or its metabolites in the ischemic striatum in vivo in our study (Chen et al, 2004). However, in keeping with prior in vitro studies, we have shown that neuroprotective doses of BRL 52537 attenuate ischemia-evoked striatal NO production, and have postulated that the consequent reduction in early NO toxicity may represent one mechanism for this KOR agonist's neuroprotective action in ischemic stroke (Goyagi et al, 2003).
In the present study, we reasoned that if BRL 52537 acted via nNOS-linked mechanisms, the agent would have no effect on stroke outcome in nNOS−/- mice. As in previous reports (Huang et al, 1994), nNOS−/- mice in this study sustained reduced infarct volumes as compared with WT controls (Figure 1). BRL 52537 had no efficacy in nNOS−/- mice. These findings implicate nNOS activity as a key link to BRL 52537's mechanism of action in ischemic brain. Other isoforms of NOS, for example, the Ca2+ -independent inducible isoform of NOS, are expressed by neurons, endothelial cells, and microglia (Iadecola et al, 1995, 1997), and clearly play a role in stroke outcome. Treatment with selective inhibitors or genetic deletion of iNOS improves the infarct volume. Effects of BRL 52537 on iNOS activity are unknown at present, and experiments in the present study were not designed to specifically exclude interactions between BRL 52537 and iNOS. However, based on the data from our previous study (Goyagi et al, 2003), in which we measured NOS activity in situ via recovery of labeled citrulline after local labeled arginine infusion utilizing in vivo microdialysis, it seems unlikely that iNOS activity contributed to large increases in citrulline recovery by 1 h of MCAO with steady recovery throughout the 5-h monitoring period. Such an acute timeframe of NO production would be too early to reflect iNOS activity. However, we cannot exclude the possibility that BRL 52537 influences iNOS as well as nNOS-linked mechanisms, because our treatment period was 22 h.
Gender-Selective Ischemic Neuroprotection with BRL 52537
Epidemiologic studies have highlighted gender differences in the incidence and prevalence of human ischemic stroke (Hurn and Brass, 2003). Mounting evidence from laboratory-based studies indicate gender-specific responses to many forms of brain injury, and endogenous sex steroids (estrogen and progesterone) have been implicated in this differential outcome after experimental ischemic stroke (Alkayed et al, 1998, Hurn and Macrae, 2000; Murphy et al, 2002). Previously, we showed in rats that BRL 52537 protects only the male brain and performs no better in females with or without native ovarian sex steroids (Chen et al, 2005). There we concluded that the sex specificity of BRL 52537 could not be attributed to the ability of protective estradiol or progesterone to obscure its actions. We did not show worsened ischemic injury in female nNOS−/- mice as compared with their WT counterparts. In the present study, the reperfusion time was 72 h versus the 22 h of our earlier study (McCullough et al, 2005), so infarct volume was evaluated at 2 different time points in injury maturation (early versus mature). The mortality rate was 58% in the nNOS−/--BRL-treated group as compared with 25% in nNOS−/--vehicle-treated (25%) female mice in our study. Although not significant, the higher mortality rate on average with BRL might have biased the infarct volume data by excluding those with the largest infarct volume. Lastly, the results of the present study are consistent with a previous study in which infarct volume was similar in female WT and nNOS−/- mice after permanent MCAO (Sampei et al, 2000). The findings of our study are also consistent with an emerging concept that excitotoxic signaling pathways may differ in females, independent of hormone status. Such differential response has also been reported with other brain functions; for example, brainstem painmodulating circuitry has been shown to be sexually dimorphic with respect to KOR receptor function (Tershner et al, 2000). Furthermore, in vitro studies in cultured neurons from males and females are differentially susceptible to nitrosative stress and excitotoxicity, and respond to therapies that are sexually dimorphic (Du et al, 2004).
Neuronally derived NO has been shown to be deleterious in the male, but not in the female rodent model of focal ischemic stroke (Sampei et al, 2000). The present study confirms previous reports (Sampei et al, 2000; Du et al, 2004) that male mice lacking nNOS are significantly protected from brain injury after focal ischemia, while female knockout mice do not sustain reduction in infarct volume as compared with their WT counterparts (Figure 2). Other studies have reported that loss of nNOS or poly (ADP-ribose) polymerase-1 (PARP-1) by genetic knockout or pharmacologic inhibition increases ischemic damage in the female (McCullough et al, 2005), and that loss of PARP-1 does not protect female newborn mice from hypoxic–ischemic injury (Hagberg et al, 2004).
Our study has several limitations. We acknowledge the possibility of unique compensatory responses in life-long, genetically deficient nNOS−/- mice that might have obscured BRL 52537's benefit. For example, our experiments were not designed to specifically exclude interactions between BRL 52537 and iNOS, and it is plausible that this interaction exists over the period of our observation of 72 h. However, iNOS does not contribute to ischemic injury after focal ischemia in the first 24 h, and does not contribute to long-term outcome in female mice (Loihl et al, 1999). It is also plausible that ischemic neuroprotection observed in male nNOS−/- mice is maximal and that additional neuroprotective benefit is therefore masked. We utilized a simple neurologic functional outcome score to discern sensorimotor deficits in our study over a period of 4 days. A more elaborate neurobehavioral battery may show the benefits and harmful effects in long-term functional outcome studies in this treatment paradigm.
In conclusion, these data in mice show and confirm our previous findings in rats that a continuous intravenous infusion of the potent and selective KOR agonist BRL 52537 attenuates stroke damage in male, but not female, rodents subjected to transient focal ischemia. BRL 52537 did not amplify ischemic neuroprotection observed with chronic nNOS inhibition. Therefore, we conclude that BRL 52537 acts by a mechanism upstream from neuronally generated NO toxicity. Notably, thus far, few preclinical studies of ischemic neuroprotection have been stratified by sex in examining experimental stroke outcome. Our study further highlights the importance of utilizing animal models of both sexes in experimental studies of ischemic neuroprotection.
Footnotes
Acknowledgements
The authors thank Tzipora Sofare, MA, for her editorial assistance in preparing this manuscript.