
Abstract
Ischemia induces immediate-early genes (IEGs) in brain. Since prolonged expression of some IEGs may precede neuronal death, some researchers have suggested that these IEGs mediate neuronal death. We therefore examined the effect of 5 and 10 min of global ischemia on the expression of the IEGs NGFI-A, NGFI-B, NGFI-C, egr-2, egr-3, and Nurr1 in gerbil brain. All of the IEGs were induced after 30 min of reperfusion in the hippocampus. Most of them were induced in several other regions as well, including cortex, hypothalamus, thalamus, and amygdala. The acute IEG induction decreased in most brain areas by 2–6 h. However, at 24 h following 5 min of ischemia NGFI-A continued to be expressed in the CA1 region and dentate gyrus. In the dentate gyrus, NGFI-C continued to be expressed for 24 h and egr-3 for as long as 72 h. In other brain areas, all of the IEGs returned to control levels by 72 h except in CA1, where most messenger RNA (mRNA) levels were decreased; this decrease correlated with marked neuronal loss. The persistent expression of NGFI-A in CA1 neurons destined to die and the persistent expression of NGFI-A, NGFI-C, and egr-3 genes in dentate granule cell neurons that survive may indicate that some transcription factors modulate cell death whereas others support cell survival when expressed for prolonged periods. The protein products of several transcription factors, including c-fos, are known to downregulate their own expression. The persistent expression of NGFI-A in the CA1 neurons destined to die could therefore be due to ischemia-induced transcriptional activation caused by, e.g., increased intracellular calcium levels plus a lack of negative feedback caused by the blockade of the translation of NGFI-A mRNA into protein.
The responses of the fos and jun proto-oncogene families to cerebral ischemia have been extensively studied in the brain (An et al., 1993; Gass et al., 1993; Kogure and Kato, 1993). The protein products of fos and jun are transcription factors, which regulate the expression of so-called late-response genes. In the brain, the expression of fos and jun immediate-early genes (IEGs) has been used as a marker of activated cells (Sagar et al., 1988). Fos and jun belong to a larger group of IEGs that respond rapidly and transiently to diverse types of stimuli without requiring prior protein synthesis. Like the leucine zipper Fos and Jun proteins, the zinc finger proteins can bind to DNA and act as transcription factors, thereby leading to long-term alterations in cellular gene expression. However, the DNA sequences recognized by the zinc finger transcription factors differ from the AP-1 site recognized by Fos and Jun. Therefore, the protein products of zinc finger genes may have separate target genes in the brain. Some of the zinc finger genes are expressed in brain, where they can be induced by several stimuli, including stress (Honkaniemi et al., 1994), seizures (Crosby et al., 1991; Mack et al., 1995), ischemia (An et al., 1992; Hsu et al., 1993; Kiessling et al., 1993; Collaco-Moraes et al., 1994; Dragunow et al., 1994; Kinouchi et al., 1994; Neumann-Haefelin et al., 1994; Wang et al., 1995), and focal brain injury (Honkaniemi et al., 1995).
During development of the CNS, neurons that do not establish appropriate connections with target cells die (Oppenheim, 1991). This process, termed programmed cell death or apoptosis, may be regulated by growth factors provided to the neurons by the target cells. Apoptosis is characterized by fragmentation of genomic DNA into internucleosomal-sized fragments and chromatin condensation. Eventually, cells are digested without inflammation. In general, this is caused by a specific genetic program and the synthesis of novel proteins, although in some models the death machinery is already present in the cell in an inactive state, waiting for a suitable signal (Jacobson et al., 1994; Schulze-Osthoff et al., 1994; Steller, 1995). In addition to occurring during development, apoptosis may also occur in mature brain. Neurons exhibiting DNA fragmentation and therefore probably undergoing apoptotic death have been demonstrated after cerebral ischemia (Linnik et al., 1993; MacManus et al., 1993, 1994; Ferrer et al., 1994; Kihara et al., 1994; Sei et al., 1994; Charriaut-Marlangue et al., 1995; Nitatori et al., 1995), kainic acid–induced seizures (Filipkowski et al., 1994; Pollard et al., 1994), and adrenalectomy (Schreiber et al., 1994). It has also been suggested that apoptosis plays a role in some neurodegenerative processes, such as Alzheimer's disease, amyotrophic lateral sclerosis, and Parkinson's disease (Loo et al., 1993; Johnson, 1994; Yoshiyama et al., 1994; Thompson, 1995).
A link between the expression of the c-fos IEG and apoptotic neuronal death has been suggested (Smeyne et al., 1993). Kainic acid–induced seizures produce prolonged expression of c-fos, which precedes apoptotic neuronal death especially in the hippocampus. A similar association between prolonged expression of c-jun and neuronal death following cerebral ischemia and seizures has also been suggested (Popovici et al., 1990; Schreiber et al., 1993; Dragunow et al., 1993; Taniguchi et al., 1994). Global ischemia causes selective neuronal death, especially in the CA1 region, that occurs several days after the ischemic insult. This delayed neuronal death involves fragmentation of genomic DNA, indicative of apoptosis (Nitatori et al., 1995). To determine whether the zinc finger genes would also show this kind of prolonged induction, we examined the effect of global ischemia on the expression of NGFI-A (also known as krox-24, zif268, TIS8, and egr-1), NGFI-B (also known as Nur77 and TIS1), NGFI-C (also known as egr-4 and pAT133), egr-2 (also known as krox-20), egr-3, and Nurr1 (also known as RNR-1, NOT, and TINUR) in gerbil brain using in situ hybridization.
MATERIALS AND METHODS
Animal treatments
Adult male Mongolian gerbils were subjected to global ischemia. Four intact animals served as controls. The gerbils subjected to ischemia were deeply anesthesized with methoxyflurane via inhalation (Pitman-Moore, Mundelein, IL, U.S.A.). The common carotid arteries were exposed and occluded with aneurysm clips for 5 or 10 min. The rectal temperature was maintained at 37.5 ± 0.5°C with a heating lamp until the gerbils recovered. After recovering, the subjects became hyperthermic, which was an invariant response of the animals to the ischemic injury. The animals were killed 30 min (n = 2 for 5-min ischemia and n = 2 for 10-min ischemia), 2 h (n = 4 and 4), 6 h (n = 4 and 4), 24 h (n = 2 and 2), or 72 h (n = 2 and 2) after ischemia.
In situ hybridization
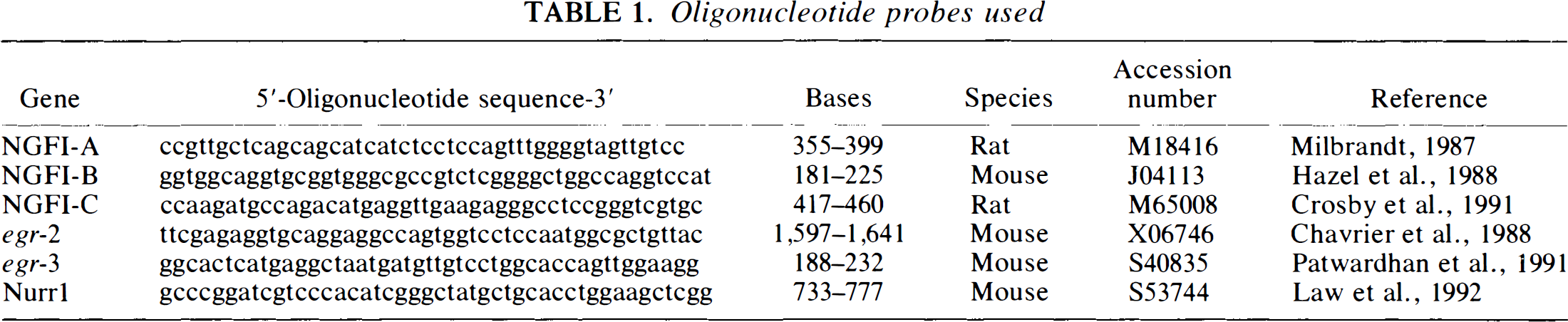
The brains were removed and frozen on dry ice. Coronal sections (14 μm) were cut through the hippocampus with a cryostat and mounted onto Fisherbrand Superfrost Plus slides (Fisher Scientific, Pittsburgh, PA, U.S.A.). At least four sections were hybridized with each oligonucleotide probe for each subject. The oligonucleotide probes used are listed in Table 1. Searches of the Genbank database using Blast and Fasta software revealed no significant homology of these oligonucleotide sequences with any previously characterized gene. We have previously shown that these oligonucleotides detect single bands of appropriate size on Northern blots (Honkaniemi et al., 1995).
Oligonucleotide probes used
The oligonucleotides were labeled with 35S-dATP (DuPont–NEN Research Products, Boston, MA, U.S.A.) using terminal deoxynucleotidyltransferase (TdT; DuPont–NEN or Gibgo–BRL, Gaithesburg, MD, U.S.A.). The sections were air-dried at room temperature and hybridized at 42°C for 18 h with a mixture of 4 × SSC, 50% formamide, 1 × Denhardt's solution, 1% sarcosyl, 0.02 M phosphate buffer (pH 7.0), 10% dextran sulphate, 500 μg/ml heat-denaturated salmon sperm DNA, 200 mM dithiothreitol, and 1–10 × 107 cpm/ml of the labeled probe. After hybridization, the sections were washed four times for 15 min each in 1 × SSC at 55°C and thereafter left to cool for 1–3 h at room temperature. The sections were then dipped in distilled water and subsequently in 75 and 90% ethanol and air-dried at room temperature. Sections were covered with Kodak SB5 film (Rochester, NY, U.S.A.) and exposed for 1–2 weeks. Films were developed with Kodak GBX developer, fixed, washed, and dried. The MCID M4 image analysis system was used to analyze the optical densities. The relative optical densities of CA1 and dentate gyrus after ischemia were normalized by comparing them with the background value obtained from the adjacent white matter. Statistical analysis were performed using the BMDP statistical software (BMDP Statistical Software Inc., Los Angeles, CA, U.S.A.).
RESULTS
The expression of zinc finger IEGs in the normal gerbil brain was comparable with that previously described in the normal rat brain (Honkaniemi et al., 1994, 1995). Relatively high basal expression of NGFI-A and egr-3 was observed in the cerebral cortex, layers CA1–CA3 of the hippocampus, and the caudate–putamen (Fig. 1). Though the baseline expression of NGFI-B and NGFI-C was much lower than that of NGFI-A and egr-3, the patterns of expression for all four genes in normal brain were similar (Fig. 1). Nurr1 was expressed in medial habenula, deep layers of the cortex, and layers CA1–CA3 of the hippocampus. Egr-2 appeared to be expressed slightly above background levels in cortex.
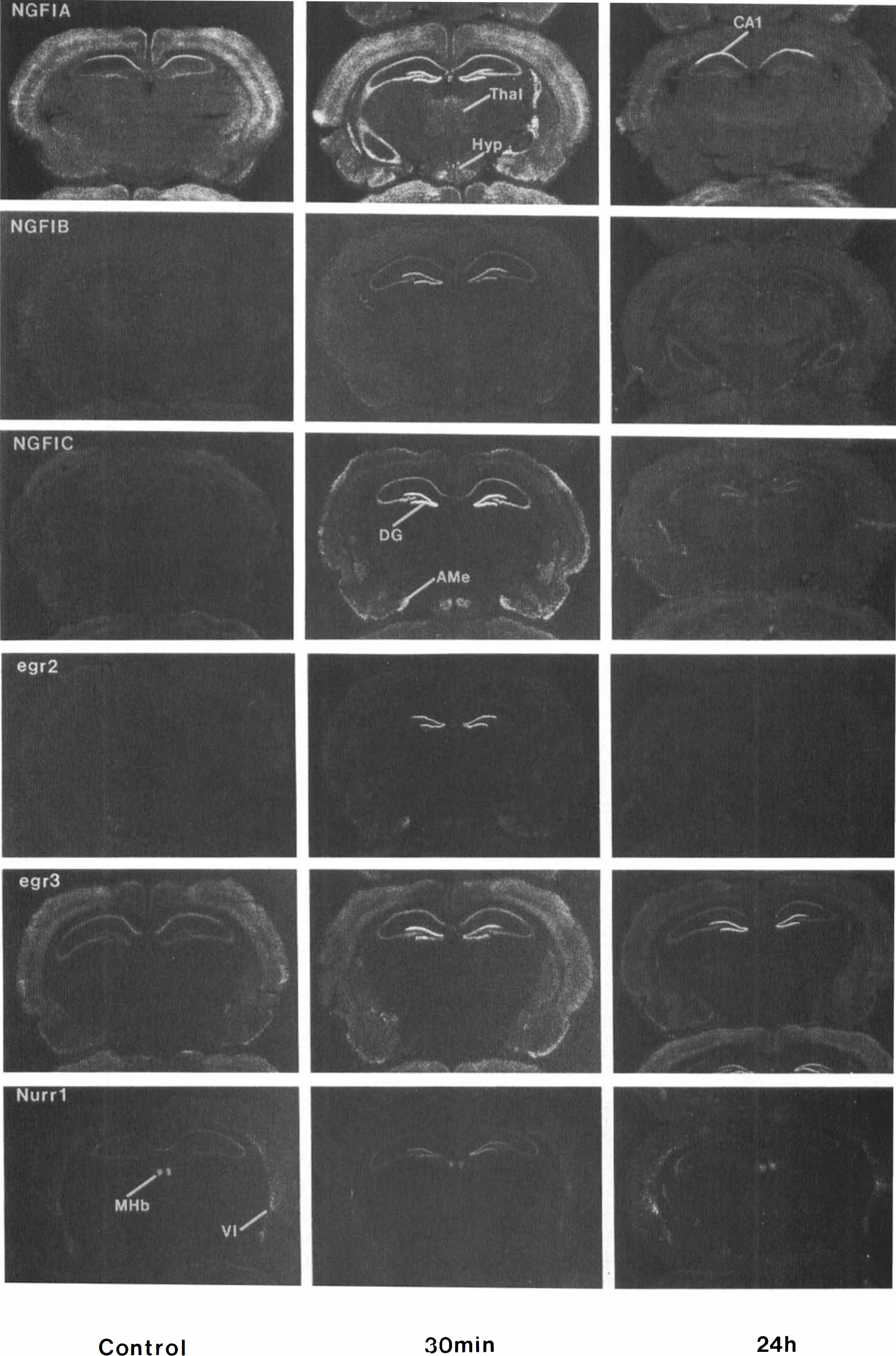
In situ hybridization autoradiographs of control gerbil brains and of gerbil brains at 30 min and 24 h after 5 min of global ischemia. The expression of mRNAs for the zinc finger immediate-early genes NGFI-A, NGFI-B, and NGFI-C and egr-2, egr-3, and Nurr1 increased in dentate gyrus of the hippocampus and in other selected brain regions 30 min after the ischemia. By 24 h after the ischemia, the gene expression had decreased except for egr-3 and NGFI-C, which continued to be expressed in the dentate gyrus, and NGFI-A, which continued to be expressed in the CA1 region of hippocampus and dentate gyrus. (DG, dentate gyrus of hippocampus; CA1, CA1 subfield of hippocampus; Thai, thalamus; Hyp, hypothalamus; AMe, medial amygdala; MHb, medial habenula; VI, layer VI of cortex.)
Each of the genes was induced in specific brain regions by 30 min after the ischemia, with no differences in the patterns of induction being observed for the animals subjected to 5 and 10 min of ischemia. Not surprisingly, ischemia induced many of the genes in hippocampus. Although most of these zinc finger genes were not expressed in normal dentate cells, all of the genes examined were markedly induced in dentate gyrus granule cell neurons (Figs. 1 and 2). NGFI-A, NGFI-B, NGFI-C, and egr-3 were also induced in the CA1 and CA3 pyramidal cell layers (Figs. 1 and 2). The data shown in Fig. 2 demonstrate small standard deviations for the optical densities for the combined group of animals for each gene. This finding, along with the consistent bilateral CA1 neuronal cell death observed in the 72-h group, reflects the consistency of the global ischemic injury obtained using the present model.
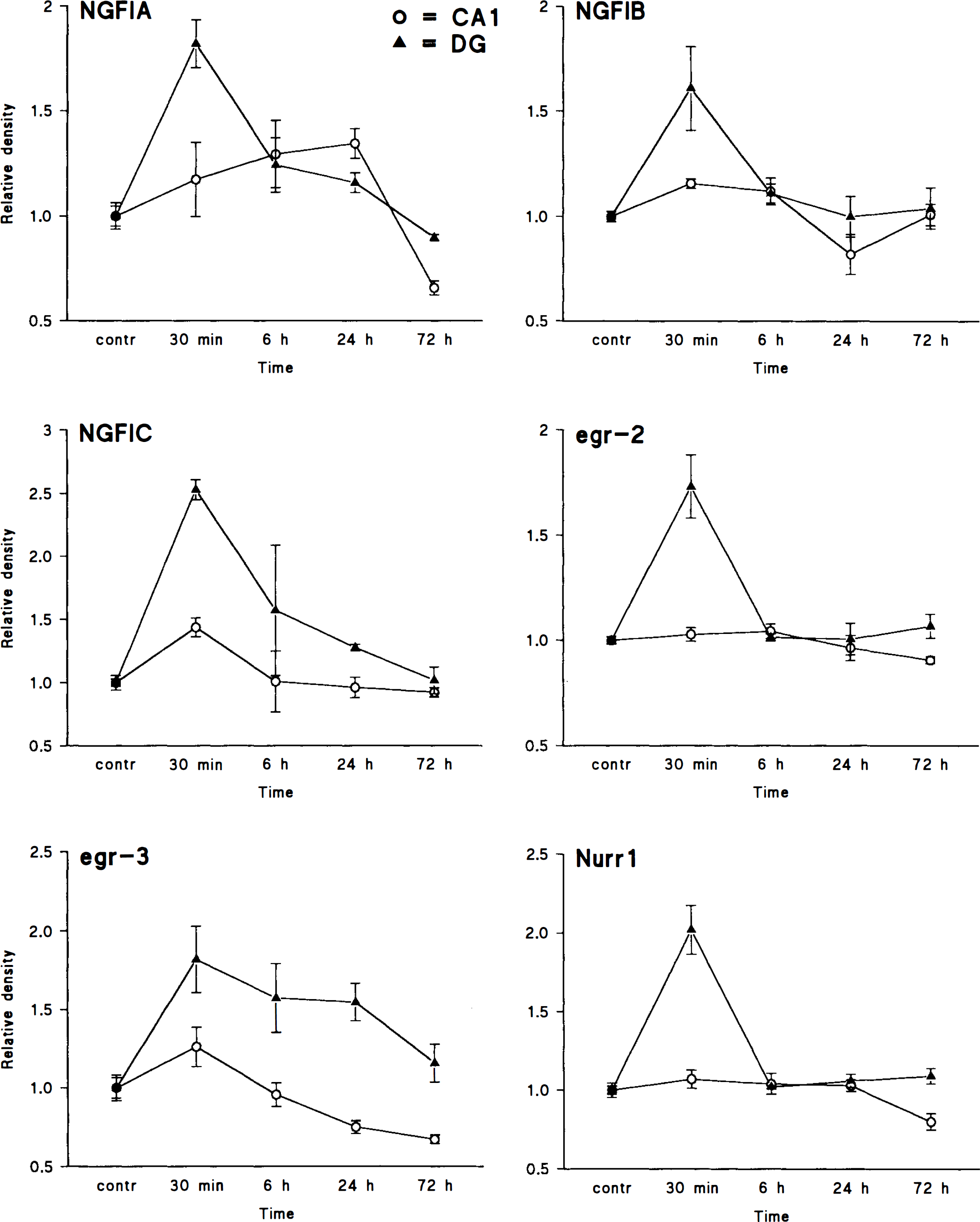
The relative optical densities of NGFI-A, NGFI-B, NGFI-C, egr-2, egr-3, and Nurr1 from in situ hybridization autoradiographs are shown at the indicated time points after 5 min of ischemia in adult male gerbils (control = 1). Optical densities were measured either over the CA1 pyramidal cell layer (circles) or over the granule cell layer of the dentate gyrus (triangles) and normalized to white matter adjacent to the hippocampus. The values are expressed as means ± SD. Using the Student t-test with Bonferroni's correction, the significance levels were p ≤ 0.05 when comparing the highest value to control levels for all the genes studied except for egr-2 and Nurr1 in the CA1 subfield.
Outside of the hippocampus, NGFI-C and to lesser extent NGFI-A, NGFI-B, and egr-2 were induced in cortex, mainly in superficial layers (Fig. 1). NGFI-A messenger RNA (mRNA) was also induced in thalamus, hypothalamus, and amygdala. NGFI-C was highly induced in caudate–putamen, ventral hypothalamus, and ventral medial amygdala. Although egr-2 was mostly induced in dentate granule cells, there was slight induction in the amygdala and ventral hypothalamus in addition to the cortical induction. The relatively high cortical egr-3 expression did not appear to change 30 min after ischemia.
The mRNAs induced at 30 min following global ischemia decreased between 2 and 6 h and returned to baseline levels by 24 h for most of the genes in most of the brain regions (Figs. 1 and 2). There were notable exceptions, however. NGFI-A mRNA continued to be expressed in the CA1 region of the hippocampus up to 24 h following 5 but not 10 min of ischemia. Elevated NGFI-A and NGFI-C mRNA levels were also observed in the dentate gyrus at 24 h following 5 and 10 min of ischemia. NGFI-A mRNA levels decreased below control levels in cortex by 24 h. Egr-3 mRNA also continued to be expressed in dentate gyrus neurons at 24 h following 5 and 10 min of global ischemia, but it decreased below control levels in cortex. NGFI-C mRNA also continued to be expressed in dentate gyrus neurons at 24 h following global ischemia.
By 72 h following the global ischemia, egr-3 mRNA continued to be overexpressed in dentate granule cell neurons (Figs. 1 and 2) but had returned to control levels in the cortex. The expression of all of the other IEGs had returned to basal levels, except in the CA1 region where the expression of all of the IEGs was decreased, probably due to the marked loss of CA1 neurons (data not shown).
DISCUSSION
In our study, global ischemia was shown to induce several family members of the zinc finger immediate-early genes in the brain. These results are in agreement with those from previous studies (An et al., 1992; Hsu et al., 1993; Kiessling et al., 1993; Collaco-Moraes et al., 1994; Dragunow et al., 1994; Kinouchi et al., 1994; Neumann-Haefelin et al., 1994; Wang et al., 1995) and expand the spectrum of transcription factors induced by ischemia from the widely studied fos and jun family members (An et al., 1993; Gass et al., 1993; Kogure and Kato, 1993) to the less well-characterized zinc finger transcription factors. All of the IEGs studied, with the exception of egr-2 and Nurr1, were induced in regions where neurons are known to die. However, the present results do not address the question of whether the observed gene induction provides protection from or contributes to neuronal injury following the ischemic insult. The induction of these genes was especially prominent in the hippocampus. As previously demonstrated for c-fos and c-jun (Dragunow et al., 1993; Taniguchi et al., 1994), NGFI-A is persistently expressed in CA1 neurons destined to die. A unique finding in this study, however, was the prolonged expression of NGFI-C and egr-3 in dentate granule cell neurons that survive the global ischemia. Therefore, our study showed that the persistent expression of IEGs does not necessarily correlate with whether cells are destined to die. However, the persistent expression of different IEGs in different cell groups could suggest different target genes and different functions for each transcription factor; some zinc finger genes possibly mediate cell death and others possibly support cell survival when expressed for prolonged periods.
Each of the zinc finger genes studied was induced in dentate gyrus neurons of the hippocampus by the global ischemic stimulus. We observed a similar induction of these IEGs in the dentate after focal, penetrating brain injury (Honkaniemi et al., 1995) and kainic acid–induced seizures (Honkaniemi et al., unpublished data). The explanation for this marked induction in dentate gyrus neurons is unclear. A wide variety of stimuli may activate dentate granule cells, resulting in the induction of many transcription factors in dentate gyrus. This explanation is consistent with the induction of most fos and jun family members and the zinc finger genes (Dragunow et al., 1993, 1994; Kiessling et al., 1993; Schreiber et al., 1993; Kinouchi et al., 1994; Honkaniemi et al., 1995) in dentate granule cell neurons following seizures, global ischemia, and trauma. Alternatively, the continuous birth of dentate granule cell neurons (Cameron et al., 1995) may make them particularly resistant to injury produced by global ischemia and seizures, and this resistance might be conferred in part by the target genes of various transcription factors, including the zinc finger genes (Honkaniemi et al., 1995). It is also possible that all regions of the hippocampus are activated by the ischemic stimulus, but that the threshold for ischemic induction of zinc finger IEGs is lower in the dentate granule cells than in the CA1 and CA3 pyramidal cell neurons.
The patterns of gene induction correlate in part with the patterns of injury expected following the global ischemia. Following 5 min of global ischemia, neurons only in the CA1 region of hippocampus are known to die, whereas following 10 min of global ischemia neurons in the CA1, CA3, and dentate hilar regions of hippocampus die in addition to neurons scattered throughout cortex (Linnik et al., 1993; MacManus et al., 1993, 1994; Ferrer et al., 1994; Kihara et al., 1994; Sei et al., 1994; Charriaut-Marlangue et al., 1995; Nitatori et al., 1995; Honkaniemi et al., unpublished observations). The fact that we did not observe any clear difference between animals subjected to 5 and those subjected to 10 min of ischemia may indicate that the acute IEG induction does not correlate with cell death but rather is due to other factors, such as ischemia-induced spreading depression.
The induction of NGFI-A, NGFI-B, NGFI-C, and egr-3 throughout CA1 and CA3 fields of hippocampus and the induction of the same genes in cortex appear to occur in the brain regions that sustain the greatest injury from global ischemia. The major exception to these findings is the induction of all of these genes, along with Nurr1 and egr-2, in dentate gyrus, where cells are not believed to die. It is possible that the dentate cells are similarly affected by the ischemic insult but are very resistant to the injury.
The type of stimulus greatly affects the anatomical pattern of the IEG induction. For example, although ischemia was not sufficient to induce Nurr1 in the cortex, kainic acid–induced seizures and penetrating brain injury produce an intense cortical expression of Nurr1 (Honkaniemi et al., 1995; and unpublished observations). It is possible that whether a gene is induced or not depends on the stimulus itself and whether it activates particular sets of receptors, such as the N-methyl-D-aspartate (NMDA) or kainate-α-amino-3-hydroxy-5-methyl-isoxazole-4-propionic acid (AMPA) glutamate receptors.
The interpretation of the persistent expression at 24 h of NGFI-A in CA1 neurons and NGFI-A, NGFI-C, and egr-3 in dentate gyrus neurons is of uncertain significance, since the CA1 neurons are destined to die and the dentate gyrus neurons are destined to survive. Whether the persistent mRNAs observed are due to persistent or increased mRNA synthesis or decreased mRNA degradation cannot be answered from the present results. The prolonged induction of egr-3 in the dentate granule cells resembles that observed after penetrating brain injury (Honkaniemi et al., 1995). NGFI-A, NGFI-C, egr-2, and egr-3 all bind to a similar consensus sequence in target genes (Christy and Nathans, 1989; Crosby et al., 1991; Patwardhan et al., 1991; Swirnoff and Milbrandt, 1995). The prolonged expression of egr-3, NGFI-A, and NGFI-C in the dentate gyrus may therefore continue to regulate the expression of the same target genes as egr-2, once the expression of egr-2 has returned to control levels by 6 h.
Several reports have suggested a connection between prolonged IEG expression and cell death. C-jun mRNA and protein remain at elevated levels for 24 h in hippocampus after hypoxic-ischemic injury and kainic acid–induced status epilepticus (Dragunow et al., 1993; Wessel et al., 1991). Similarly, hypoxic injury induces persistent cortical c-fos expression 1–7 days after the hypoxia (Taniguchi et al., 1994; Wessel et al., 1991). However, not all studies have demonstrated a prolonged IEG induction following ischemia (Kiessling et al., 1993). Kainic acid–induced seizures, on the other hand, elevate c-fos mRNA levels for up to 16 h (Schreiber et al., 1993) and c-fos protein for up to 12 h (Popovici et al., 1990) after the onset of seizures.
Using a transgenic mouse line containing the gene encoding β-galactosidase (LacZ) and driven by the c-fos promoter, Smeyne et al. (1993) demonstrated that after kainic acid–induced seizures, fos-LacZ is expressed for up to 16 h and reappears 4 days after the seizures in the hippocampus and amygdala, where cells are known to die. The connection between IEGs and neuronal death is also supported by in vitro studies. The majority of fibroblasts transformed with c-fos undergo apoptotic cell death after 2 days of serum deprivation, whereas most normal fibroblasts survive (Smeyne et al., 1993). Overex-pression of c-jun induces apoptosis in sympathetic neurons. Intracellular microinjections of c-jun-dominant negative mutant or neutralizing antibodies against c-Jun and the Fos family protect sympathetic neurons from apoptosis caused by NGF deprivation (Estus et al., 1994; Ham et al., 1995). NGF deprivation leads to increased c-fos, c-jun, and NGFI-A mRNA levels in cultured sympathetic neurons (Estus et al., 1994; Ham et al., 1995). NGFI-B is induced in apoptotic thymocytes and NGFI-B-dominant negative and antisense protect T cells from apoptosis (Liu et al., 1994; Woronicz et al., 1994). NGFI-B mRNA levels remain high for 14 h in T cell hybridomas undergoing apoptosis (Woronicz et al., 1994). The expression of the human homologue of Nurr1, TINUR, remains elevated in dying cells for up to 24 h (Okabe et al., 1995). Taken together, these findings support the idea that these IEGs may play a role in apoptotic cell death, especially when continuously expressed. The transcription factors may affect cell death programs by directly regulating the transcription of other genes, e.g., c-myc, p53, and members of the bcl-2 family, which directly induce or inhibit apoptosis. It has also been suggested that programmed cell death is due to aborted mitosis (Steller, 1995). Since many of these genes were originally characterized from growth factor- or serum-stimulated cell lines, the prolonged induction of these genes in neurons undergoing apoptosis may be a sign that these neurons are trying to enter the cell cycle, which in terminally differentiated neurons may lead to apoptotic death.
NGFI-A was the only zinc finger gene expressed for prolonged periods in the CA1 neurons destined to die. However, this was only true for 5-min ischemia. The lack of prolonged NGFI-A induction following 10-min ischemia in the CA1 and CA3 regions, where neurons are known to die following 10-min global ischemia, argues against the hypothesis that NGFI-A contributes to neuronal death. Although NGFI-A is induced in apoptotic cell lines (Estus et al., 1994), it is not known whether NGFI-A participates in the apoptotic death of CA1 neurons. Interestingly, basal NGFI-A expression is dependent on NMDA receptor activity, which may be an important contributing factor to ischemic neuronal death. Therefore, the prolonged NGFI-A expression may be due to persistent NMDA receptor activation caused by ischemia. Why this might occur following 5 but not 10 min of ischemia is uncertain, unless CA3 cell death after 10 min of ischemia leads to less CA1 activation.
The presence of mRNA is not necessarily an indication that a particular mRNA is translated to a functional protein. Global ischemia decreases protein synthesis in many brain areas. At 12 h after recirculation, protein synthesis returns to basal levels in most brain areas except in the CA1 neurons, in which protein synthesis is permanently disrupted (Thilmann et al., 1986). Therefore, the ischemia-induced increase in NGFI-A mRNA does not necessarily lead to increased protein levels in the CA1 subfield. Kiessling et al. (1993) demonstrated that global ischemia increases NGFI-A mRNA levels in the hippocampus; this increase is followed by an increase in immunoreactive protein only in the dentate granule cells and CA3 pyramidal cells and not in the CA1 region. In fact, the number of NGFI-A–immunoreactive neurons in the CA1 subfield decreased below control levels after 48 h of reperfusion (Kiessling et al., 1993). Although Kiessling et al. (1993) did not observe any persistent increased expression of NGFI-A in the CA1 subfield, it is possible that the prolonged NGFI-A expression demonstrated in the present study may not lead to increased protein levels. In fact, persistent failure of translation in the CA1 region may be a major contributing factor to the CA1 ischemic cell death, whereas restoration of protein synthesis may explain, the relative tolerance of dentate granule cells to ischemia. Since some transcription factor proteins autoregulate their own transcription, the absence of the appropriate protein could increase mRNA levels. For example, Fos protein suppresses its own transcription (Sassone-Corsi et al., 1988). The absence of the negative feedback provided by the Fos protein because of the failure of protein synthesis may lead to persistent transcription of c-fos mRNA. Furthermore, the c-fos promoter contains a calcium-dependent cyclic AMP-responsive element, through which increased intracellular calcium levels stimulate c-fos transcription (Vendrell et al., 1993). The combination of transcriptional activation caused by ischemia (Kiessling and Gass, 1994) and lack of negative feedback because of disrupted protein synthesis may explain the prolonged expression of c-fos and zinc finger mRNAs after ischemia. The ischemia-induced decrease in total protein synthesis must be considered when drawing conclusions from studies only involving analysis of mRNA expression.
Footnotes
Acknowledgment:
This study was supported by grants from the Finnish Academy of Sciences and the Finnish Neurology Association (J.H.), by NIH grants NS14543 and NS 28167, and by the Merit Review Program of the Department of Veterans Affairs (F.R.S.). The skillful technical assistance of Mrs. Leena Honkaniemi and Mr. Jose Espinoza is greatly appreciated.