
Abstract
In neonates, asphyxia is a common cause of neuronal injury and often results in seizures. The authors evaluated whether blockade of a-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptors during asphyxia and early recovery with 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo-(F)-quinoxaline (NBQX) ameliorates neurologic deficit and histopathology in 1-week-old piglets. Anesthetized piglets were exposed to a sequence of 30 minutes of hypoxia, 5 minutes of room air ventilation, 7 minutes of airway occlusion, and cardiopulmonary resuscitation. Vehicle or NBQX was administered intravenously before asphyxia (30 mg/kg) and during the first 4 hours of recovery (15 mg/kg/h). Neuropathologic findings were evaluated at 96 hours of recovery by light microscopic and cytochrome oxidase histochemical study. Cardiac arrest occurred at 5 to 6 minutes of airway occlusion, and cardiopulmonary resuscitation restored spontaneous circulation independent of treatment modalities in about 2 to 3 minutes. Neurologic deficit over the 96-hour recovery period was not ameliorated by NBQX. Seizure activity began after 24 to 48 hours in 7 of 10 animals with vehicle and in 9 of 10 of animals with NBQX. In each group, four animals died in status epilepticus. Neuropathologic outcomes were not improved by NBQX. The density of remaining viable neurons was decreased in parietal cortex and putamen by NBQX treatment. Metabolic defects in cytochrome oxidase activity were worsened by NBQX treatment. Seizure activity during recovery was associated with reduced neuronal viability in neocortex and striatum in piglets from both groups that survived for 96 hours. This neonatal model of asphyxic cardiac arrest and resuscitation generates neurologic deficits, clinical seizure activity, and selective damage in regions of basal ganglia and sensorimotor cortex. In contrast to other studies in mature brain, AMPA receptor blockade with NBQX failed to protect against neurologic damage in the immature piglet and worsened postasphyxic histopathologic outcome in neocortex and putamen.
Partial or complete asphyxia in newborns or young children leads to transient oxygen deprivation or circulatory arrest. Even if children can be successfully resuscitated, encephalopathy and neurologic abnormalities such as movement disorders, epilepsy, and developmental delay may result. Neurologic abnormalities occur in 0.5% to 0.6% of near-term or term human births experiencing perinatal asphyxia (Sunshine, 1997). In those with moderate or severe grades of encephalopathy, seizures often develop within the first 48 hours of recovery (Sunshine, 1997). In nonsurvivors, neuropathologic study frequently shows cortical laminar necrosis and selective injury in basal ganglia, thalamus, brain stem, and cerebellar Purkinje neurons (Larroche and DeVries, 1996; Low et al., 1989). In survivors with handicaps, magnetic resonance imaging often indicates damage in basal ganglia, thalamus, and sometimes in cortex along the Rolandic fissure (Enzmann, 1997; Roland et al., 1998). Other than early work in monkeys (Myers, 1977) and more recent work in fetal sheep (Mallard et al., 1995), few models have been developed in newborn animals that mimic clinical outcome and regional brain damage found in asphyxic newborn humans near term. The commonly used model of hypoxia-ischemia in the postnatal rat produces lesions in striatum and cortex, but selective neuronal vulnerability has not been well delineated because of the focal nature of the insult and the formation of cavitary lesions (Hagberg et al., 1994; Towfighi et al., 1991).
We recently showed that postasphyxic brain damage in immature pigs does not occur in a random pattern but rather is system preferential (Martin et al., 1997a). Somatosensory and motor cortex, basal ganglia, thalamic sensory nuclei, and specific structures of brain stem are predominantly damaged, similar to postmortem neuropathologic outcome in term infants who died secondary to hypoxic–asphyxic episodes and similar to magnetic resonance imaging findings in survivors with severe handicaps. In contrast to mature brain, CA1 hippocampus is not injured. Energy metabolism increases at different stages during development in different regions. In the l-week-old piglet, vulnerable regions generally showed histochemical evidence of greater basal activity of cytochrome oxidase (Martin et al., 1997a). Thus, regional vulnerability corresponds to interconnected primary sensory and motor pathways with a high basal metabolic rate, consistent with early work of Myers (1977) in monkeys. Moreover, excitotoxicity may be important in putamen because we found early decreases in glial- and neuronal-based glutamate transporters in postasphyxic piglets (Martin et al., 1997b).
Studies in postnatal rats at 6 to 7 days have emphasized enhanced excitotoxicity to N-methyl-
Because of limited information on the use of an AMPA receptor antagonist in global brain asphyxia in immature brain, in the current study we used a model of hypoxic hypoxia followed by complete asphyxia and cardiac arrest to produce a global insult. We tested the hypothesis that administration of the AMPA receptor antagonist NBQX before and after complete asphyxia ameliorates neurologic deficit, incidence of seizures, and neuropathologic results and associated decrements in cytochrome oxidase histochemical activity in selectively vulnerable brain regions.
METHODS
Animal preparation
All procedures received approval from the Animal Care and Use Committee of the Johns Hopkins Medical Institutions. Thirty-eight piglets of mixed breed, 1 to 2 weeks old and weighing 3.75 ± 0.67 kg (mean ± SD), were studied. Anesthesia was induced with pentobarbital (65 mg/kg body weight intraperitoneally). The trachea was intubated using a 4.5-mm cuffed tube, and the lungs were mechanically ventilated with a fractional inspired oxygen (Fi
A sterile cut-down was performed in the left groin, and the femoral artery and vein were cannulated with catheters advanced into the thoracic aorta and inferior vena cava. Catheters were subcutaneously tunneled to the back up to about 2 cm left of the vertebral column. Catheter dead space was filled and flushed as needed with 0.1 mL of isotonic saline containing heparin (0.02 U/mL). All incisions were closed, and piglets received prophylactic intravenous antibiotics (cephalothin, 50 mg/kg intravenously). Additional cephalothin was given at the same dosage every 6 hours up to 24 hours. For fluid maintenance, piglets received a fluid infusion of 10 mL/kg/h of lactated Ringer's solution. To maintain anesthesia, fentanyl (10 µg/kg intravenously in a 50-µg/mL solution) was administered twice before the insult and twice during the first 4 hours of recovery. To achieve muscle paralysis, pancuronium bromide (0.3 mg/kg intravenously) was given every hour until 1 hour before extubation was anticipated.
Insult and resuscitation
After a postsurgical stabilization period of 120 minutes, baseline measurements were obtained. Hypoxia was induced by decreasing Fi
Heart rate, MABP, central venous pressure, temperature, ventilatory rate, Fi
Experimental design
Piglets were assigned randomly to four groups. In group I (vehicle-treated sham, n = 3), piglets were anesthetized but were not subjected to the hypoxia-asphyxia insult. In group II (NBQX-treated sham, n = 4), the procedures were the same as in group I, except that NBQX treatment (30-mg/kg bolus plus 15 mg/kg/h for 4 hours) was administered. In group III (n = 18), piglets were subjected to the hypoxic–asphyxic insult with vehicle treatment. In group IV (n = 13), piglets were subjected to the hypoxic–asphyxic insult with NBQX treatment during room air ventilation after the hypoxic period (30 mg/kg intravenous bolus) followed by a continuous infusion (15 mg/kg/h intravenously) for 4 hours starting after resuscitation. The NBQX was dissolved in 0.1 mol/L NaOH, which was diluted in 5% dextrose to a concentration of 2.5 mg/mL with pH adjusted to about 7.5 by HCl. The dose of NBQX is similar to that used by others to show ischemic neuroprotection (Hagberg et al., 1994; Li and Buchan, 1993; Nellgard and Wieloch, 1992).
Neurologic evaluation
A new neurologic deficit score (NDS; 0 = best outcome, 150 = worst outcome) was developed to quantify neurologic function in piglets based on six different components: I) level of consciousness (maximum score 15); 2) brain stem function (respiration, olfaction, vision, light reflex, corneal reflex, auditory response, swallow reflex; maximum score 34); 3) muscle tone in trunk and limbs (maximum score 8); 4) motor response to touch and momentary pain (maximum score 8); 5) excitability (startle myoclonus, seizures; maximum score 15); and 6) behavior (maximum score 70). The first four components were similar to those described by LeBlanc and associates (1991). The newly developed evaluation system for behavioral function was divided into motor behavior (0 to 38 points), orientation (0 to 16 points), and activity (0 to 16 points). Motor behavior was assessed using a six-step scale for gait previously described for use in cats (Fleischer et al., 1989). Additionally, we evaluated extensor postural thrust and wheelbarrowing (walking on forelimbs with hind limbs elevated). The behavioral subscore for orientation was based on motor activity in avoiding obstacles, in the use of sniffing to explore the laboratory room, and in the reaction to being lifted. To evaluate the activity subscore, the pig was checked for appetite, vocalization, psychomotor activity, and motivation to explore the environment. The pig was evaluated at 6, 12, 24, 36, 48, 72, and 96 hours of recovery. Each neuroscore was obtained by a neurologist (K.B.) who was blinded to the circumstances of treatment and resuscitation.
Neuropathology
At 96 hours of recovery, piglets were anesthetized with pentobarbital (10 mg/kg intravenously), the trachea was orally intubated, and the lungs were ventilated. After injection of fentanyl (10 µg/kg) and pancuronium bromide (0.3 mg/kg), a left-side thoracotomy was performed. Through the left ventricle, an intraaortic catheter was placed, and exsanguination was started immediately with phosphate-buffered saline for at least 5 minutes. When clear venous reflux from the punctured right atrium was observed, the descending aorta was clamped, and the upper part of the body was perfused for 20 minutes with cold (4°C) 4% paraformaldehyde prepared in 0.1 mol/L phosphate buffer (pH 7.4). The brains were bisected midsagittally, and each hemisphere was cut into l-cm slabs. From the left hemisphere, consistent samples were taken coronally at an anterior striatal level and a midhippocampal level containing the thalamus and the midbrain, as well as sagittally at the level of the cerebellar vermis. These samples were processed for paraffin histologic study, and sections (10 µm) were stained with hematoxylin and eosin and coded. Investigators (L.J.M., A.M.B., and D.F.H.) blinded to treatment assessed neuronal damage using light microscopic examination at 1000× magnification. In each region, the number of viable neurons was counted in six nonoverlapping, contiguous microscopic fields. Neuronal viability was assumed when no evidence was found for eosinophilic cytoplasm, cytoplasmic vacuolation, and nuclear pyknosis. Areas counted included medial and lateral portions of parietal cortex, dorsal hippocampus (CA1, CA2, CA3, CA4), basal ganglia (putamen, caudate, and substantia nigra), thalamus, and cerebellum (Purkinje cells).
Cytochrome oxidase histochemistry
The right hemisphere was cryoprotected in phosphate-buffered 20% glycerol for 24 hours, frozen in isopentane, and stored at −80°C until processing. These samples were cut serially at 40 µm on a sliding microtome. Every 20th section was used for enzyme histochemical assay of cytochrome oxidase (CO) activity, as described by Wong-Riley (1979). Samples from each pig were prepared concomitantly. Brain sections were rinsed (1 hour) with several changes of phosphate buffer. The freshly prepared reaction medium consisted of 100 mmol/L phosphate buffer (pH 7.4), 0.1% horse heart cytochrome c, 117 mmol/L sucrose, and 1.4 mmol/L diaminobenzidine tetrahydrochloride. During the reaction, brain sections were incubated for 2.5 hours at 37°C in a Dubnoff metabolic shaker incubator. Subsequently, sections were rinsed in phosphate buffer, mounted on glass slides, covered with coverslips, and analyzed densitometrically, as described by Martin and others (1997a). For negative controls, 10 mmol/L of KCN was added to the medium and was found to completely block formation of the reaction product.
Statistical analysis
All values are reported as mean ± SD. Physiologic variables and viable neuron counts in asphyxic piglets were compared between the vehicle and NBQX groups by t test. Densitometric measurements of CO activity in the vehicle-sham group, vehicle-asphyxia group, and NBQX-asphyxia group were compared by analysis of variance and the Duncan multiple range test. Indices of insult duration, NDS, and CPR parameters were compared between the vehicle and NBQX groups by the Mann-Whitney test. Survival, seizure incidence, and seizure prevalence were tested with χ2 tests. Probabilities of 5% (P < 0.05) were interpreted as statistically significant.
RESULTS
Insult and success of resuscitation
With asphyxia, 8 of 18 piglets treated with vehicle (group III) and 3 of 13 piglets treated with NBQX (group IV) before airway occlusion were excluded for a variety of reasons. Three piglets in group III and one piglet in group IV could not be resuscitated within 3 minutes. Seven animals were excluded because of various cardiopulmonary problems after successful resuscitation (i.e., repeated cardiac arrest [n = 1, group III], tension pneumothorax [n = 2, group III; n = 1, group IV], and heart failure [n = 2, group III]). One animal (group IV) developed acute renal failure on the second day of recovery and subsequently died. At autopsy, this animal had large pleural and peritoneal effusions and yellow crystals deposited within the kidneys. A total of 20 asphyxiated animals (group III: n = 10, group IV: n = 10) remained for evaluation of outcome.
Arterial O2 saturation decreased to approximately 30% during the 30-minute period of hypoxic hypoxia, partially recovered to about 60% during the 5-minute period of room air ventilation, and then decreased to approximately 5% by 5 minutes of airway occlusion (Fig. 1). For the first minute of asphyxia, progressive tachycardia and hypertension were observed followed by a sudden drop in heart rate to about 50% of baseline during the second minute. Within the second minute of asphyxia, blood pressure decreased to about 75 mm Hg and then progressively declined until circulation virtually ceased at 5 to 6 minutes and pulselessness (MABP less than 25 mm Hg without pressure fluctuation) was observed. Thus, with the termination of asphyxia (after 7 minutes), a period of 1 to 2 minutes of cardiac arrest had passed before CPR was initiated. No significant differences were noted between groups III and IV in arterial blood gases, pH, arterial O2 saturation, MABP, heart rate, or tympanic membrane temperature at any time point during hypoxia, asphyxia, resuscitation, and recovery (Table 1, Fig. 1). There were no differences in the magnitude of the insult based on the duration of MABP less than 60 mm Hg or less than 25 mm Hg during the 7-minute airway occlusion period, the duration of pulseless electrical activity, the duration of CPR until spontaneous circulation was restored, or the combined duration with MABP less than 25 mm Hg plus CPR time (Table 2).
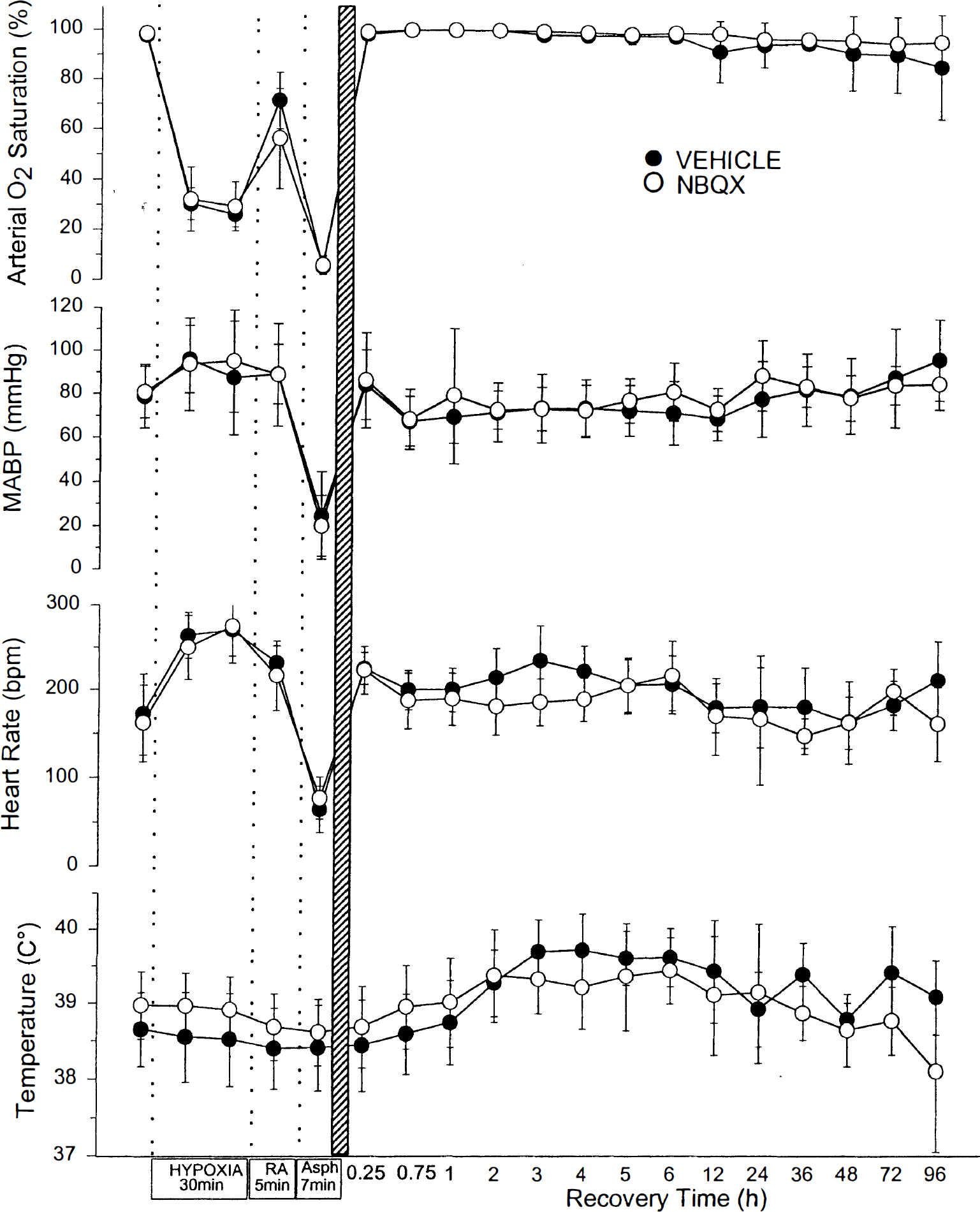
Arterial O2 saturation, MABP, heart rate, and temperature are plotted for piglets treated with vehicle or 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo-(F)-quinoxaline (NBQX). Values are means ± SD measured at baseline, at 10 and 27 minutes of hypoxia, at 5 minutes of room air (RA) ventilation, at 5 minutes of a 7-minute period of complete asphyxia (Asph), and during the 96-hour recovery period. There were no differences between groups. bpm, beats per minute.
Arterial blood analysis during hypoxia-asphyxia and recovery
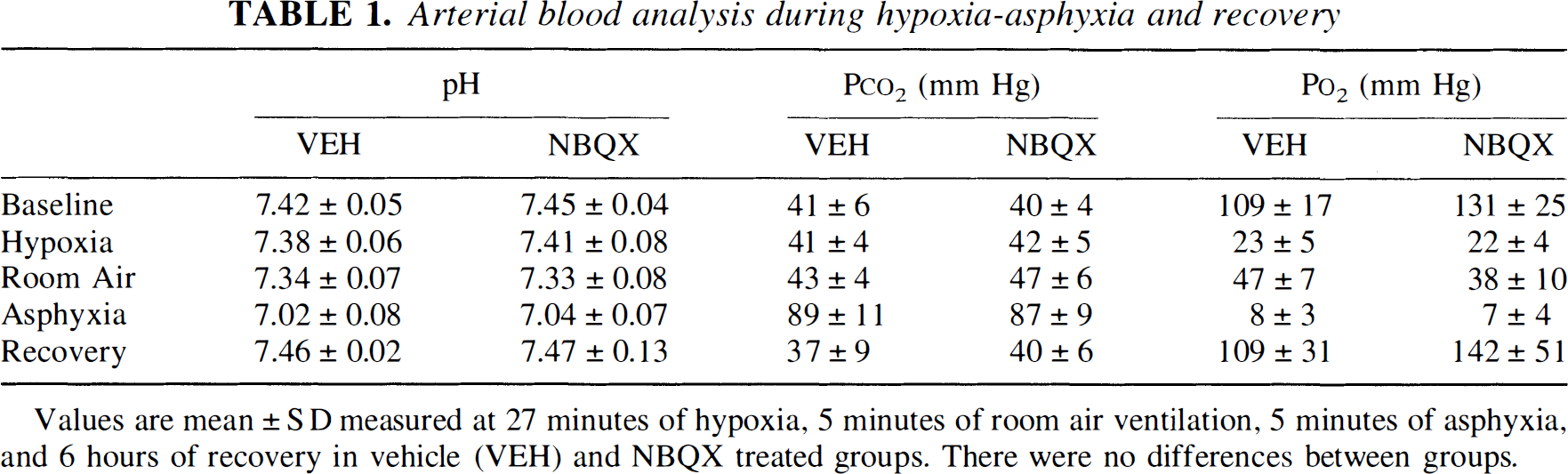
Values are mean ± SD measured at 27 minutes of hypoxia, 5 minutes of room air ventilation, 5 minutes of asphyxia, and 6 hours of recovery in vehicle (VEH) and NBQX treated groups. There were no differences between groups.
Survival and seizures
All sham animals (group I and II) survived for 96 hours, whereas 4 of 10 piglets in group III and 4 of 10 piglets in group IV did not survive the full 96-hour recovery period. No sham animal presented with seizure activity. In postasphyxic piglets, the prevalence of clinically apparent seizure activity at individual time points of recovery was similar in both groups III and IV (Fig. 2). The cumulative incidence of seizure activity indicated that clinical seizures started by 24 to 48 hours and that there was no significant difference in the cumulative incidence by 96 hours between the vehicle (70%) and NBQX (90%) groups. When animals showed clinical evidence of seizure activity, they were treated with repetitive doses of diazepam (0.1 mg/kg up to 0.3 mg/kg intravenously over 1 hour) until seizures stopped. Despite diazepam treatment, seizure episodes increased in frequency and duration to finally merge into continuous seizure activity, that is, status epilepticus, in 4 of 10 piglets in each group. All piglets in status epilepticus died from ventilatory and circulatory arrest. Thus, the mortality noted between 20 and 48 hours (Fig. 2) was related to status epilepticus.
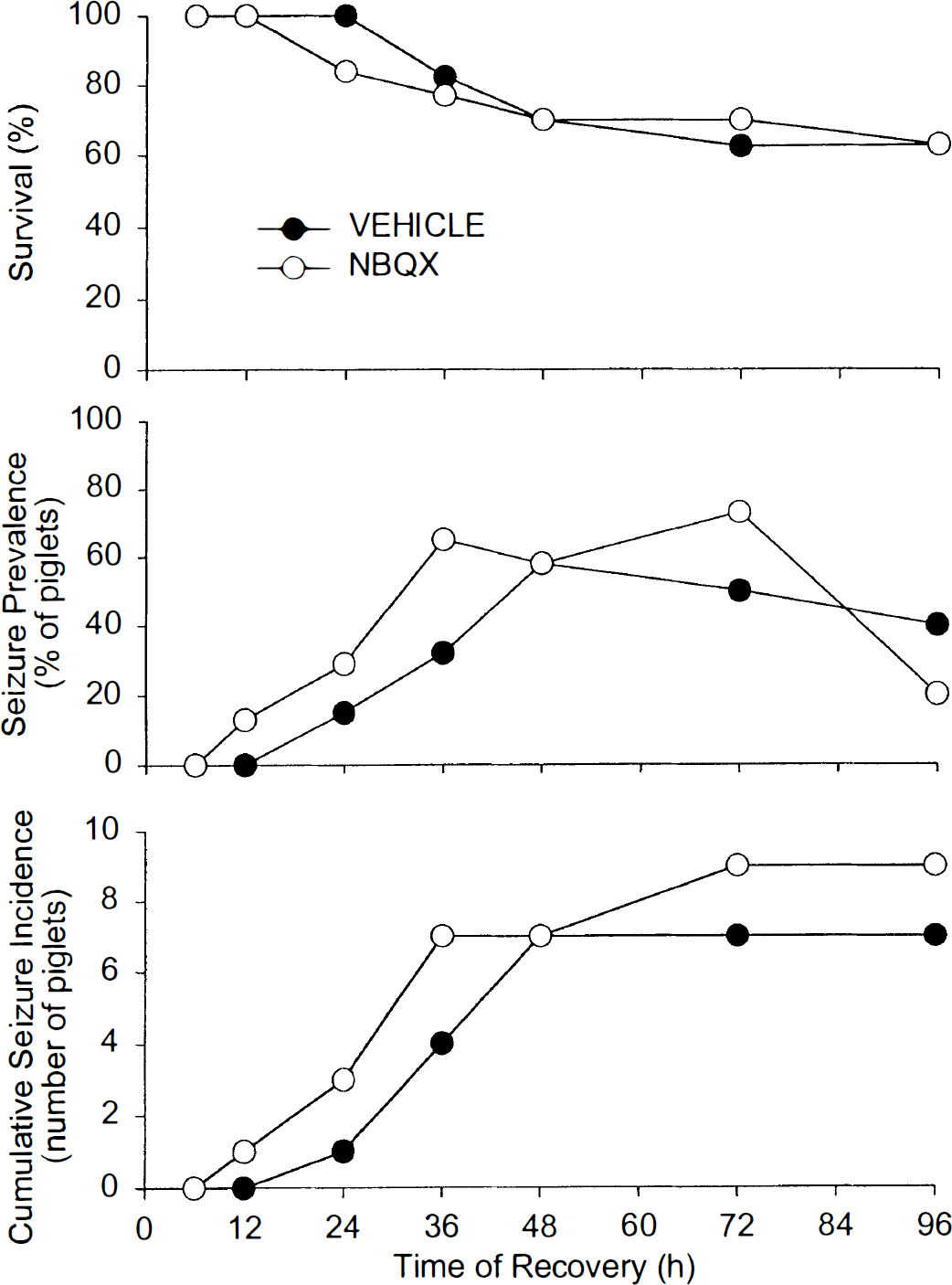
Percent survival, percent prevalence of seizures at a particular time, and cumulative incidence of seizures in the 10 piglets in the vehicle group and in the 10 piglets in the NBQX group are plotted over the 96-hour recovery period after asphyxia. There were no significant differences between groups.
Neurologic deficit
No neurologic deficit was noted in group I (sham + vehicle), and animals had fully recovered from anesthesia by 12 hours. However, sham animals with 4-hour NBQX infusion (group II) appeared sedated for many hours afterward, resulting in a NDS up to 7 points (hypoactive and depressed vocalization, psychomotoric activity or environmental motivation) at 12 hours before recovering to normal at 24 hours.
In postasphyxic animals, neurologic deficit was not improved with NBQX treatment. Moreover, animals of group IV (NBQX) required more time for recovery of brain stem reflexes and spontaneous ventilation and for extubation (Table 2). Group IV animals had a significantly higher NDS than group III animals at 6 hours. Thereafter, there was no significant difference in NDS between groups (Fig. 3A). Scores in Fig. 3A include all animals that were alive at each time point. When analyzed separately, the NDS of only those animals that survived 96 hours (n = 12) was not different at any time point from that of the entire cohort (n = 20). As with the entire cohort, there was no beneficial effect of NBQX on neurologic outcome in this subset of animals (Fig. 3B). Piglets that developed seizures had a greater NDS than those without clinically apparent seizures (Fig. 3C). This difference became significant by 24 hours, which preceded the onset of clinical seizures. NBQX treatment did not improve neurologic outcome in either of these subgroups.
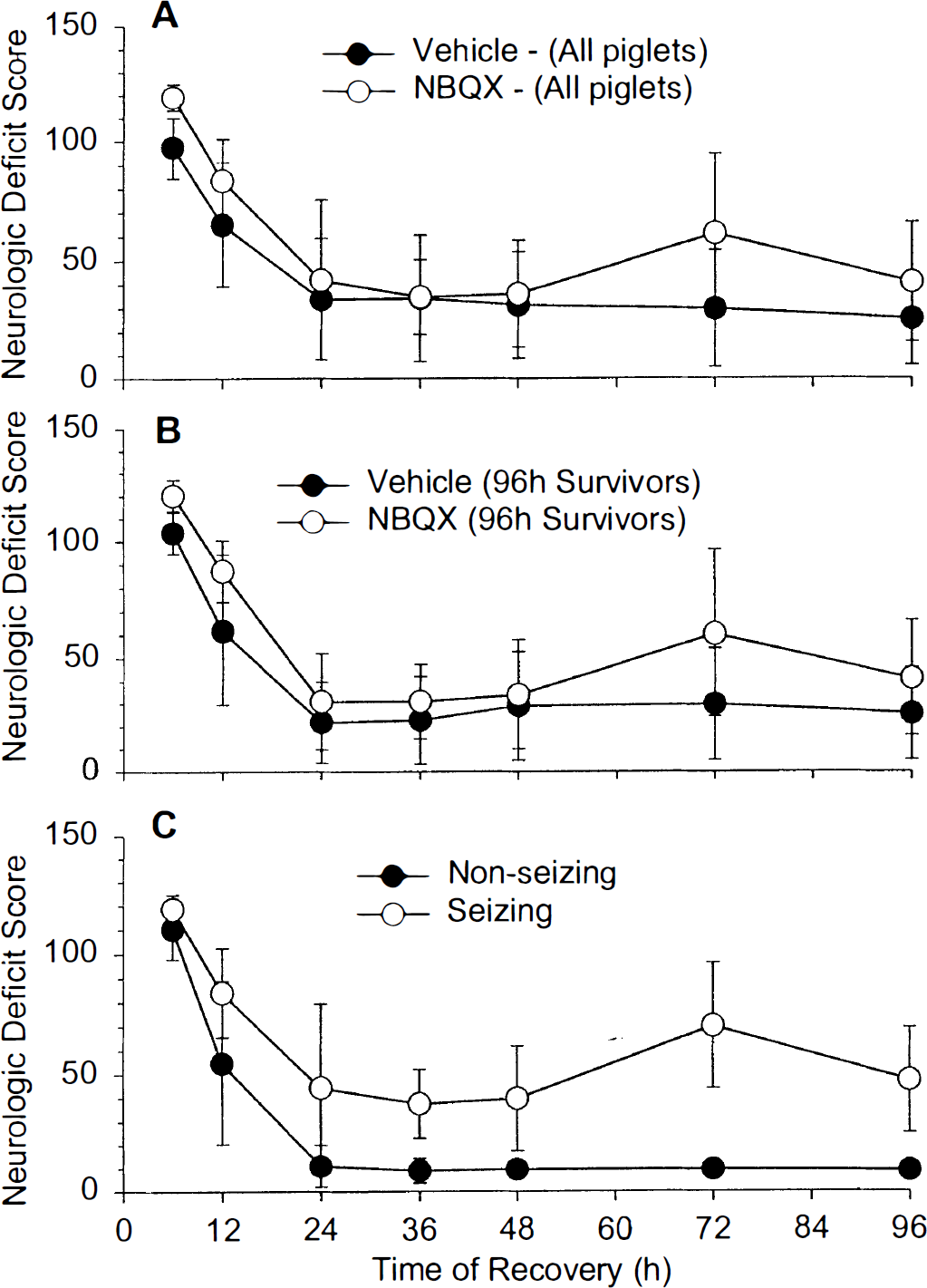
Values are means ± SD of the neurologic deficit score.
Indices of insult duration and cardiopulmonary resuscitation (CPR) measurements
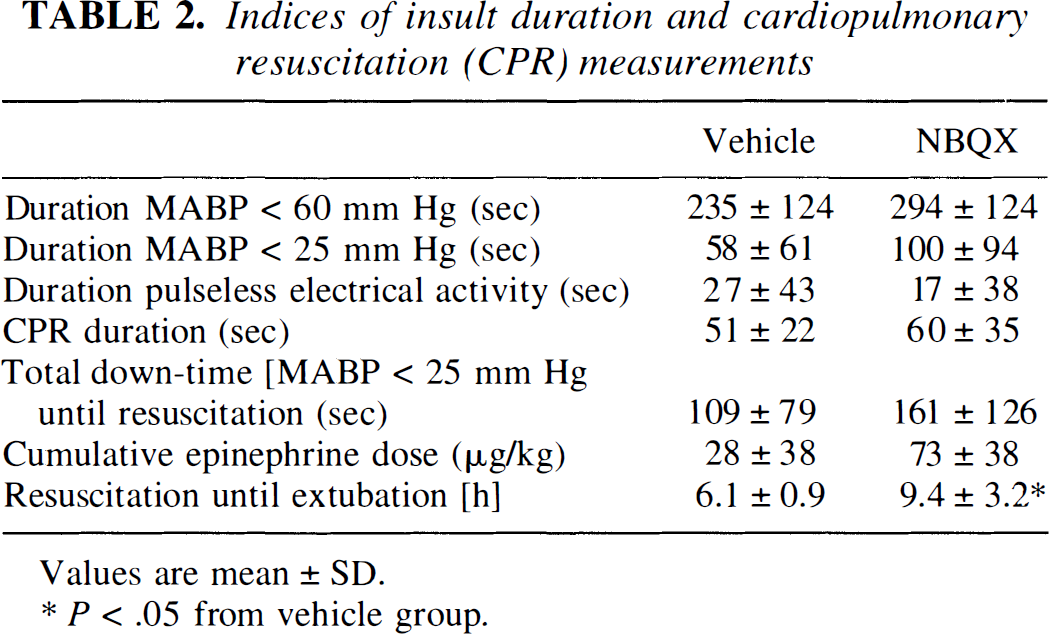
Values are mean ± SD.
P < .05 from vehicle group.
Neuropathology
At 96 hours after vehicle or NBQX treatment, sham-operated animals had less than 3% neuronal injury (expressed as percentage of the total number of cells per area counted) in neocortex, striatum, and hippocampus, possibly because of anesthesia, perfusion-fixation, brain compression during removal of the skull, or histologic processing. This background damage was assumed to be the same for all animals and thus was considered a systematic error not affecting the comparison of neuropathologic data between the asphyxic groups. Only postasphyxic animals that survived for 96 hours (n = 6, group III; n = 6, group IV) underwent neuropathologic evaluation. Gross examination revealed no evidence of intracerebral hemorrhage or cavitary or cystic lesions in any piglet. To quantify brain injury in postasphyxic animals, viable neurons were counted and expressed on a regional basis as a percentage of the mean number of cells counted in same areas of combined sham groups (n = 7).
Neuropathologic outcome at 96 hours after asphyxia was not improved with NBQX treatment. In parietal cortex of vehicle-treated postasphyxic piglets, the pattern of injury was primarily confined to selective laminar damage (Fig. 4B, b) compared with that of sham control piglets (Fig. 4A, a). In contrast, all NBQX-treated asphyxic animals showed global pan-laminar necrosis of superior parietal cortex with virtually complete necrosis of neurons in layers II, III, IV, and V (Fig. 4C, c). Neuronal viability in parietal cortex with NBQX treatment (28% ± 18%) was less than that with vehicle treatment (67% ± 20%). When analyzed on an individual gyral basis, viability was significantly reduced in the medial-most gyrus (superior parietal cortex), and in the laterally adjacent sulcus and laterally adjacent gyrus (midparietal cortex) (Fig. 5A).
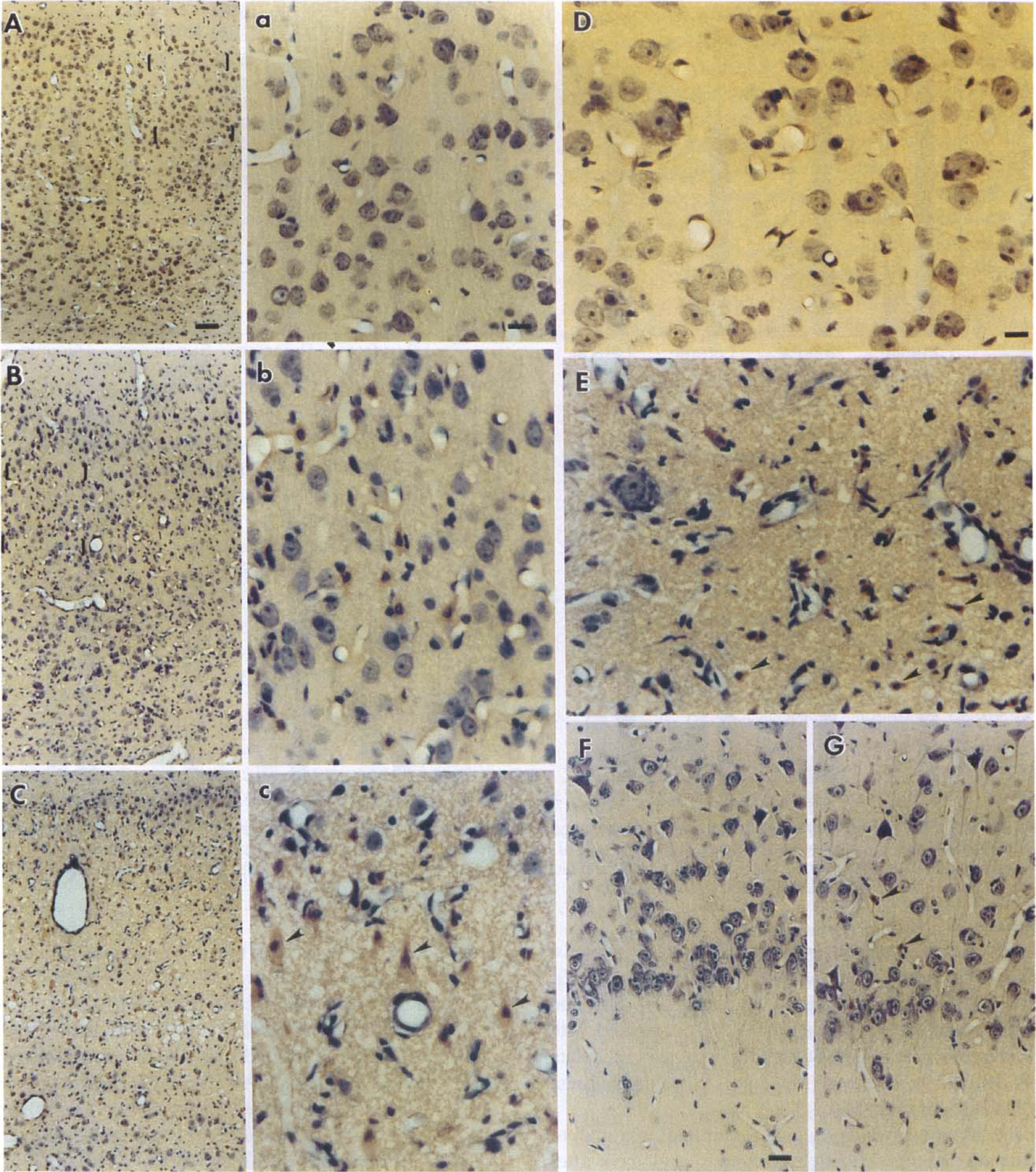
Neuropathologic features in hematoxylin and eosin–stained sections of piglet brain 96 hours after sham surgery or asphyxia.
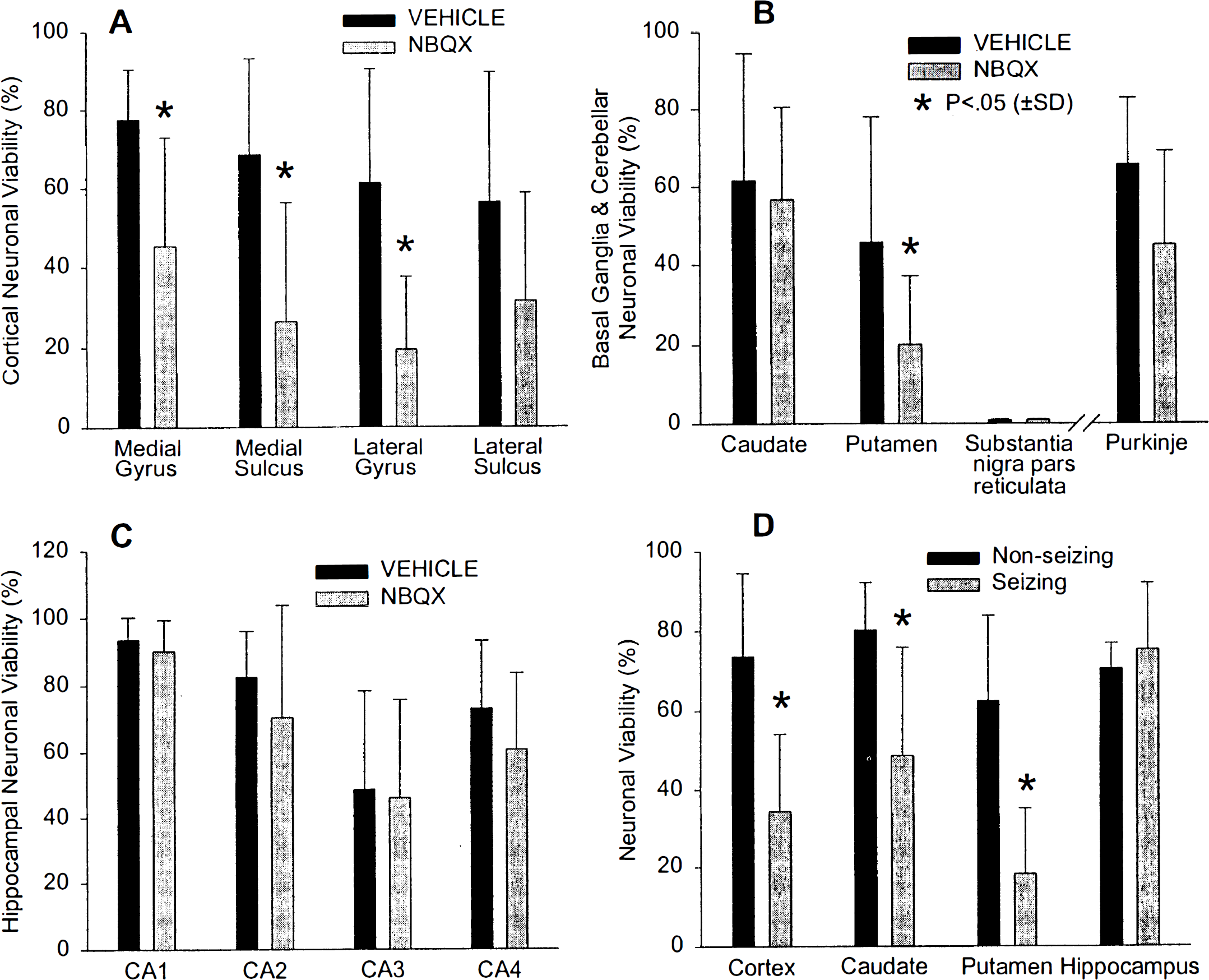
Neuronal viability in superior parietal cortex
Basal ganglia were damaged in all postasphyxic piglets (Fig. 4E). In the vehicle group, injury to the caudate nucleus was predominantly lateral, whereas injury in the NBQX group extended into medial caudate nucleus. Neuronal damage in the central putamen was significantly more severe with NBQX treatment (Fig. 5B), and most neurons were eliminated (Fig. 4E) compared with sham controls (Fig. 4D). Substantia nigra pars reticulata was completely infarcted (less than 1% viable neurons) in both groups (Fig. 5B).
The hippocampal formation exhibited minimal to moderate changes in different regions at 4 days after asphyxia in both asphyxic groups (Fig. 5C). In the 1- to 2-week-old piglets, CA1 neurons were not highly vulnerable to hypoxia–asphyxia (Fig. 4G) compared with sham controls (Fig. 4F). Treatment with NBQX did not alter the vulnerability. Postasphyxic loss of neuronal viability in CA3 and CA4 was significantly different from sham controls (P < 0.05). Neuronal survival did not differ significantly between NBQX- or vehicle-treated cohorts in any hippocampal region (Fig. 5C). In cerebellum, there was a significant loss of Purkinje cells in both groups compared with sham controls, and NBQX treatment did not provide protection (Fig. 5B).
In pooling piglets from groups III and IV that did not display clinical seizure activity, neuronal survival in neocortex and basal ganglia at 96 hours was greater than in pooled cohorts that exhibited seizures (Fig. 5D). There was no association of neuronal survival and seizure activity in hippocampus or cerebellum.
Cytochrome oxidase histochemical activity
Densitometric analysis of sham-operated controls revealed that baseline CO histochemical activity varied between individual brain regions (Table 3). Primary somatosensory neocortex exhibited high basal activity. In contrast, limbic cortex (i.e., cingulate cortex), as well as hippocampus and striatum, showed lower activity levels. In the postasphyxic vehicle group, CO histochemical activity in parietal cortex was reduced about 20%, compared with a 70% reduction with NBQX treatment (Table 3). In caudate nucleus, a decrease in CO histochemical activity was found in asphyxic piglets treated with NBQX compared with sham controls. In putamen, CO activity was decreased in both vehicle- and NBQX-treated piglets after asphyxia compared with sham controls. In contrast, no decrease in CO activity was detected after asphyxia in hippocampal CA1 region. In the CA3 region, postasphyxic CO activity increased in both the vehicle and NBQX groups.
Regional alterations in cytochrome oxidase activity
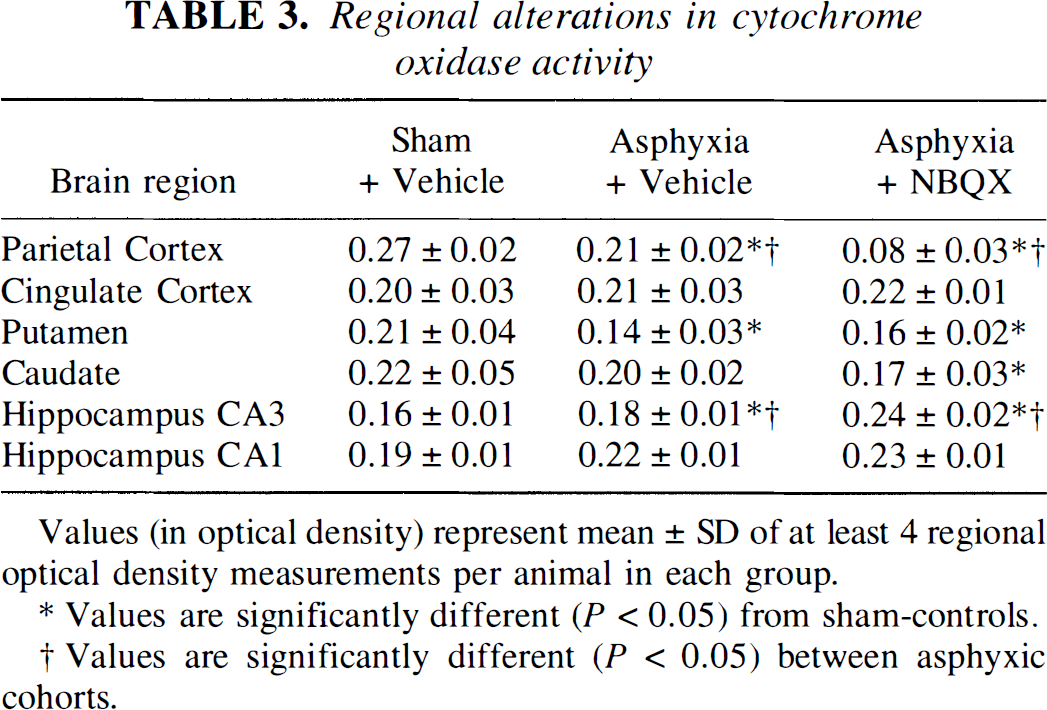
Values (in optical density) represent mean ± SD of at least 4 regional optical density measurements per animal in each group.
Values are significantly different (P < 0.05) from sham-controls.
Values are significantly different (P < 0.05) between asphyxic cohorts.
DISCUSSION
These results demonstrate that administration of the AMPA receptor antagonist NBQX before and after asphyxic cardiac arrest does not ameliorate outcome in 1- to 2-week old piglets. Survival and neurologic performance during the 96-hour observation period was not improved, and postasphyxic neuropathologic outcome and deficits in histochemically determined CO activity were worsened in selected regions with NBQX treatment.
Experimental model
Experimental models of hypoxia–ischemia in piglets with documented histopathologic necrosis in neocortex, basal ganglia, and hippocampus include severe systemic hypoxia titrated to produce depressed electroencephalographic activity (Thorensen et al., 1996), carotid occlusion plus hypoxia and hypotension (LeBlanc et al., 1991), and neck cuff inflation plus hypotension (Laptook et al., 1994). The global insult in our model is different from those described by others in the same species in that we combined a severe hypoxic challenge with a subsequent asphyxic episode. Our previous experiments using 7-minute asphyxia without hypoxia did not produce cerebral edema (Rose et a!., 1995) or adequate brain damage (unpublished observation) in piglets, whereas prolonged asphyxic periods (longer than S minutes) caused prolonged circulatory arrest that could not be routinely reversed by CPR. In pilot experiments, periods of hypoxia immediately before 7-minute asphyxia significantly impaired success of CPR. With a 5-minute period of room air ventilation intervening between hypoxia and asphyxia, we were able to worsen the insult and still successfully resuscitate most animals. Our insult paradigm appears to closely mimic the clinical situation of a severe perinatal hypoxia seen in the stressed fetus during difficult labor, followed by an asphyxic episode, which frequently occurs immediately after delivery of a severely depressed newborn. The observed outcome consisting of behavioral deficits, seizures, and selective neuropathologic findings in somatosensory cortical regions and striatum in piglets has a strong parallel to that seen in term infants with hypoxic–ischemic encephalopathy.
Neurologic outcome and seizure activity
After cerebral ischemia in large animals, neurologic deficit scoring often is heavily weighted by consciousness and brain stem function. Animals with primary cortical and subcortical lesions cannot be easily differentiated without better assessments of behavior. Our scoring system for neurologic deficit is adapted from those used by others (Fleischer et al., 1989; LeBlanc et al., 1991) but is expanded to place more emphasis on behavior. This NDS differentiates between different types of neurologic sequelae, that is, mild, moderate, and severe neurologic deficit. Piglets exhibiting mild neurologic damage appeared to be less active, more lethargic, and uninterested in the environment. Moderately injured piglets had alternating periods of hypoactivity and hyperactivity; they showed ataxic gait and circling movements and seem to be disoriented (e.g., they did not avoid obstacles). More severely injured animals were stuporous (not alert or reactive) and presented with hind limb paralysis or continuous paddling of legs (running movements). Many of these movement abnormalities are consistent with damage to basal ganglia. This variation in neurologic deficit after asphyxic cardiac arrest in piglets is analogous to the different grades of hypoxic–ischemic encephalopathy seen in term neonates (Sunshine, 1997).
All of the severely and most of the moderately damaged animals developed clinically apparent seizures between 24 and 48 hours of recovery. A poor NDS at 24 hours (i.e., before seizures became clinically apparent) correlated with seizure activity thereafter. Moreover, histologic damage in cortex and basal ganglia was more severe in animals that displayed seizures. It is not clear from this study whether seizures propagate neuronal injury and result in greater neurologic deficit or are a consequence of greater neuronal injury by 24 hours. In addition, a worse NDS at 24 hours in piglets that eventually develop clinically obvious seizures may result from sub-clinical seizure discharges at 24 hours, which would diminish behavioral performance.
Ordinarily, NBQX acts as an antiepileptic (Swedberg et al., 1995; Jensen et al., 1995). Clearly, neither the 4-hour NBQX treatment nor postinjury diazepam treatment arrested the seizures in our study. Perhaps continuous NBQX treatment for several days rather than for 4 hours would have suppressed seizures. Alternatively, Berg and coworkers (1993) report a lower seizure threshold to kainic acid in rats after NBQX pretreatment and speculated that NBQX blocks the activity of interneurons more effectively than pyramidal cell activity. Receptors for AMPA are expressed by neocortical interneurons (Martin et al., 1993). Thus, postasphyxic hyperexcitability in our animals may have been enhanced by NBQX treatment or its subsequent withdrawal.
Neuropathology
Cortical necrosis has been described in piglets using different models of hypoxia–ischemia (Laptook et al., 1994; LeBlanc et al., 1991; Thorensen et al., 1996). In our model, laminar necrosis occurs selectively in the region of primary somatosensory cortex (Martin et al., 1997a). Although some damage is present in layers II and III, as also occurs in mature brain (Lin et al., 1990), damage in piglets is most prominent in layers IV and V, where basal CO is greatest at this stage of development (Martin et al., 1997a). With NBQX treatment, the piglets exhibited pan-laminar necrosis, which resulted in lower numbers of viable neurons. The cortical injury also extended medially and laterally to cover a greater area. One explanation for augmented cortical injury is that the NBQX may reduce the activity of inhibitory neurons and result in disinhibition of excitatory neurons. An additional consideration is that NMDA antagonists such as MK-801 appear to provide protection in postnatal rats by reducing metabolic demand during a period of hypermetabolism after reoxygenation and by preserving mitochondrial function (Gilland et al., 1998). In contrast, NBQX has been found to impair metabolic recovery compared with control or MK-801 treatment in piglets during cardiopulmonary bypass after hypothermic cardiac arrest (Aoki et al., 1994). Thus, NBQX may adversely affect metabolic demand and mitochondrial function during reperfusion. Furthermore, we have observed that neuronal injury matures at a slower rate in cortex than in striatum. In immature brain, there may be an augmented component of target deprivation–induced cell death arising from injury in the primary sensory and motor pathways. Suppression of spontaneous synaptic activity during the early recovery period by NBQX could enhance a connectivity-based target deprivation component of delayed cortical neuronal death.
Striatum is considered to be selectively vulnerable in mature brain, although longer ischemic durations are required than that for producing hippocampal damage (Inoue et al., 1992). In fetal sheep, striatal damage is selectively enhanced by the use of short, repeated insults (Mallard et al., 1995). The prominent damage that we observed in putamen and lateral caudate nucleus may have been enhanced by the use of a brief period of reoxygenation interposed between hypoxemia and complete asphyxia. Repeated bouts of brief ischemia enhance a delayed increase in striatal microdialysis glutamate concentration during reperfusion (Lin et al., 1992). Thus, we postulated that administration of NBQX during this brief intervening period of reoxygenation and during the first 4 hours of reperfusion would provide striatal neuroprotection. However, we failed to see protection in caudate, and neuronal viability in putamen was worsened. Increases in striatal dialysate dopamine concentration can be detected with relatively moderate degrees of cerebral ischemia in rat (Kondoh et al., 1994) or tissue hypoxia in piglet (Pastuszko et al., 1993), and this increase in dopamine may amplify glutamate release (Globus et al., 1988; Yamamoto and Davy, 1992). Thus, dopaminergic activation may occur during the period of hypoxemia before NBQX administration and accelerate glutamate release during subsequent asphyxia. The lack of protection in striatum with NBQX suggests that AMPA receptor activation is not the primary mechanism involved in striatal injury. This lack of protection with NBQX is consistent with our recent observations indicating that striatal death in asphyxic piglets closely resembles NMDA receptor–mediated excitotoxicity rather than non–NMDA receptor–mediated excitotoxicity (Martin et al., 1998a). Substantia nigra pars reticulata, which undergoes selective neuronal injury in mature rat (Inoue et al., 1992; Kawai et al., 1992), became completely necrotic in the postasphyxic piglet. This region, which receives inhibitory input from putamen, may undergo excitotoxic injury secondary to the marked neuronal loss in putamen. If so, our results indicate that NBQX was not protective in this excitotoxic process.
Others report hippocampal damage in piglets 72 hours after a hypoxic–ischemic insult (Laptook et al., 1994; LeBlanc et al., 1991; Thorensen et al., 1996), but the locus of the injury was not well described. In our model, moderate hippocampal injury was confined to CA3 and CA4 regions, whereas CA1 was spared. Although we cannot exclude that there may be delayed maturation of injury beyond the 96-hour recovery period in our study, we believe that the sparing of CA1 neurons is developmentally based and possibly related to immaturity of entorhinal–hippocampal circuitry. Hippocampal CA1 neurons are spared in newborn dogs undergoing hypothermic cardiac arrest (Mujsce et al., 1993). Although some CA1 damage has been reported in vivo in postnatal rats (Beilharz et al., 1995; Towfighi et al., 1991), others report relative sparing during normothermic hypoxia-ischemia (Schwartz et al., 1992). Much of the work on neuroprotection by AMPA receptor antagonists in mature brain has focused on CA1 pyramidal neurons (Li and Buchan, 1993; Nellgard and Wieloch, 1992), where AMPA receptor subunit expression changes postnatally in rat (Martin et al., 1998b; Standley et al., 1995). Thus, one reason for the lack of protection by NBQX in our study may be the age dependency of CA1 neuronal vulnerability. However, even in CA3 and CA4, we could not detect neuroprotection with NBQX.
In parietal cortex and striatum, we observed a decrease in CO histochemical activity in postasphyxic piglets that corresponded to cell loss in these regions. However, in CA3 hippocampus, CO activity was greater in both postasphyxic groups than in the sham control group, despite some neuronal loss in the CA3 region. This increase in CO activity is presumed to reflect an adaptive mechanism to the asphyxic stress. This increase also could be related to the postasphyxic seizures, although we could not detect a significant difference between seizing and nonseizing cohorts.
It is possible that NBQX, at the dosage delivered intraveneously, has toxic effects on the CNS and peripheral organs in the piglets. For example, intraperitoneal administration of NBQX was neuroprotective in an adult rats, whereas intravenous delivery failed to protect against ischemic injury and was nephrotoxic (Li and Buchan, 1993). We did not observe any adverse effect of NBQX treatment on cardiopulmonary function, blood gases, or pH at baseline or during recovery. The solution of infused NBQX was yellow, and we noticed yellow staining of all organs and in CSF of animals that could not be resuscitated, indicating rapid distribution of the drug in solution before asphyxic cardiac arrest. All secretions (i.e., gastric content, urine) were stained yellow for 8 to 12 hours in surviving animals. All surviving NBQX-treated animals showed yellow crystals in the acidic environment of the pelvis of their kidneys even after 96 hours, but only one animal showed evidence of renal failure and was excluded from the protocol. Nevertheless, we cannot exclude that potential NBQX toxicity in peripheral organs added to the CNS insult and that other doses over different durations would have provided neuroprotection.
In conclusion, the applied model of hypoxemia followed by asphyxic cardiac arrest in neonatal piglets closely resembles the clinical and neuropathologic features of postasphyxic human newborns. However, AMPA receptor antagonism with NBQX treatment over the first 4 hours of reoxygenation failed to improve survival, neurologic performance, or neuropathologic outcome 96 hours after resuscitation. Damage in selectively vulnerable striatum and somatosensory cortex with relatively high CO activity was augmented with NBQX treatment, indicating that NBQX is unlikely to afford protection in immature brain by simply reducing metabolic demand. Immaturity of hippocampal excitatory circuitry is presumed to account for the sparing of CA1 pyramidal neuronal injury in this model and, consequently, for the inability of NBQX to provide hippocampal neuroprotection.
Footnotes
Acknowledgments
The authors thank Lydia Burnett for her help in preparing this manuscript.