
Abstract
Treatments that postpone hypoxic spreading depression (SD)-like depolarization (also called anoxic depolarization) facilitate recovery of function after transient cerebral hypoxia. Hypertonia reduces cerebral excitability, and we tested whether it also offers protection against SD-like depolarization and hypoxia. Oxygen was withdrawn from hippocampal slices bathed in normal artificial cerebrospinal fluid (ACSF) and, simultaneously, from slices cut from the same hippocampus but bathed in strongly hypertonic ACSF. Extracellular osmolarity (πo) was increased by adding 100 mM mannitol or fructose to ACSF. Slices in normal πo underwent SD-like negative extracellular voltage shift (ΔVo). The hypertonic slices usually showed no SD-like ΔVo but only a small, gradual negative voltage shift. Hypertonia also prevented the precipitate drop of interstitial calcium level ([Ca2+]o). When oxygenation and normal osmolarity were restored, synaptic transmission in the previously hypertonic slices recovered completely, but 3 h after reoxygenation orthodromically transmitted population spikes of the control slices recovered only 25.1% of the initial control amplitude. We conclude that hypertonic treatment during hypoxia improves subsequent recovery of synaptic function. The protection is probably due to the prevention of calcium uptake by blocking the SD-like depolarization, with the prevention of hypoxic cell swelling playing a lesser role.
The vulnerability of mammalian central neurons to acute hypoxic injury has been attributed to excessive uptake of Ca2+ and the consequent activation of autolytic processes (Siesjö, 1986; Siesjö and Bengtsson, 1989) or to the uptake of Na− and Cl− and the resulting osmotic cell swelling (Rothman, 1985; Rothman and Olney, 1987). We (Balestrino and Somjen, 1986; Young et al., 1991) and others (Roberts and Sick, 1988) have shown that withdrawing calcium from hippocampal tissue slices offers protection against the acute loss of neuron function caused by withdrawal of oxygen. The uptake of a great excess of Ca2+ during hypoxia appears to be mediated by the sudden accelerating depolarization that resembles spreading depression (SD) (Leão, 1947; Somjen et al., 1990) and that is associated with a nonspecific increase of membrane permeability to ions (Phillips and Nicholson, 1979; Czéh et al., 1992; Czéh et al., 1993). In the presence of normal [Ca2+]o, any treatment that reduces excitability and thereby postpones and curtails the time spent in hypoxic SD-like depolarization offers some protection against hypoxic neuron failure (Balestrino and Somjen, 1986; Somjen et al., 1990).
Reducing extracellular osmotic pressure (π0) enhances synaptic transmission in hippocampal tissue slices, and exposure to very strongly hypotonic solutions induces recurrent waves of SD (Andrew, 1991; Chebabo et al., 1995a, b; Bossut and Somjen, 1995). After hypotonic treatment, neuronal function recovers, except if very strongly hypotonic treatment lasts a long time (Huang et al., 1995a). By contrast, raising πo by adding mannitol to the bathing solution powerfully but reversibly depresses synaptic transmission as well as voltage-dependent membrane ion currents (Huang and Somjen, 1995a, b; Vreugdenhil et al., 1995).
In this study, we asked whether hypertonic treatment might also protect against irreversible hypoxic synaptic failure, because hypertonia both reduces synaptic excitation and counteracts cell swelling. Some of the results are published in an abstract (Huang et al., 1995b).
METHODS
Brain slices were prepared from male Sprague–Dawley rats of 75–180 g body weight. Under ether anesthesia, a hippocampus was removed and 400-μm transverse slices were cut with a modified Mcilwain tissue chopper, as previously described (Dingledine, 1984; Somjen et al., 1986). Alternate slices were transferred into two beakers serving as holding chambers and subsequently incubated in the two wells of a dual-interface recording chamber for 90 min in artificial cerebrospinal fluid (ACSF), which contained (in millimoles per liter) NaCl, 130; NaH2PO4, 1.25; NaHCO3, 24; CaCl2, 1.2; MgSO4, 1.2; and glucose 10, saturated with CO2/O2 (5%:95%) at 37°C. The dual wells shared the same temperature control and gas supply, with independent ACSF supplies. The perfusion rate of ACSF was 1.5 ml/min−1. The hypertonic solution was prepared by adding 100 mM of either mannitol or fructose and leaving other ingredients unchanged. The osmolarity of the hypertonic solution was 404 ± 3 mOsm/kg and that of the normal ACSF solution was 304 ± 2 mOsm/kg, measured by freezing-point osmometer (Advanced DigiMatic Osmometer Model 3DII).
After the 90-min incubation, control recordings were obtained from one slice in each well of the dual chamber. The perfusion of one well was then switched from normal ACSF to hypertonic solution, and after we allowed mannitol or fructose to penetrate the slice, oxygen was withdrawn from both wells. Two slightly different protocols were used. When mannitol was tested, hypoxia was induced in two steps; after 18 min of mannitol treatment, both wells were switched to solutions saturated by 95% N2 with 5% CO2, maintaining normal πo in the one and hypertonic condition in the other, and 12 min later, the gas supply of both wells was changed to N2 95% with CO2 5%, and this condition of severe hypoxia was maintained for 6.5 min. During this period, the N2 gas was flowing at 1,700 ml/min to ensure complete exchange of the gas phase of the chamber. When fructose was used as hypertonic agent, it was allowed to diffuse for 30 min before switching the gas phase to 95% N2, 5% CO2, without changing the gas saturating the bathing fluid, and hypoxia was maintained for 9 min.
After the hypoxic period, the gas was switched to 95% O2, 5% CO2 at the rate of 1,700 ml/min, and at the same time, oxygenated ACSF and normal πo were restored. Then after 5 to 10 min, gas flow was reduced to the normal rate of 1,000 ml/min. Recovery was observed for 3 h.
Extracellular micropipette recording electrodes filled with NaCl, 150 mM, with resistance of 7 to 15 mΩ were inserted in stratum (st.) pyramidale of the CA1 region. Monopolar microwires, insulated except at the tip, were positioned in the Schaffer collateral bundle in st. radiatum and used to evoke orthodromic responses. Five stimuli of 80 μA and 150 μA each, 0.1 ms, were delivered every 15 min. Extracellular voltage (Vo) signals were coupled to DC amplifiers, monitored on an oscilloscope and recorded on a pen-writing chart recorder. Evoked potentials were also saved on computer diskettes for later analysis with Pclamp software (Axon Instruments).
The amplitude of population spike was measured as the difference between the negative peak and the average of the two positive points at the base of the spike. Five responses evoked at each stimulation intensity were averaged.
Interstitial calcium level ([Ca2+]o) was recorded with double-barreled ion-selective microelectrodes from CA1 st. radiatum, as previously described (Somjen, 1981), with the reference barrel serving for Vo recording. The Fluka “cocktail” A: 21048 served as ion exchanger. [Ca2+]o and the associated Vo were recorded either on chart paper or with the Axotape program (Axon Instruments).
Statistical analysis was performed with the computer software package “StatView,” Abacus Concepts, Inc. One-factor analysis of variance (ANOVA) followed by the Bonferroni–Dunn test was used for ANOVA parameters at multicomparison significance level of at least p < 0.05 (in most cases of interest, however, p < 0.0002). The type of experiment was factorial.
RESULTS
Exposure to hypertonic solution in normal oxygen depressed the orthodromic responses (Figs. 1 and 2). After 30 min hypertonia, the orthodromic responses at 80 μA stimulation were completely suppressed. Population spikes evoked by 150 μA stimulation were reduced to 4.8 ± 10.3% (mean ± SD) of the initial control amplitude by mannitol and to 34.7 ± 17.2% by fructose. Withdrawal of oxygen from the gas phase completely suppressed the orthodromic responses in slices in both normal and hypertonic πo.
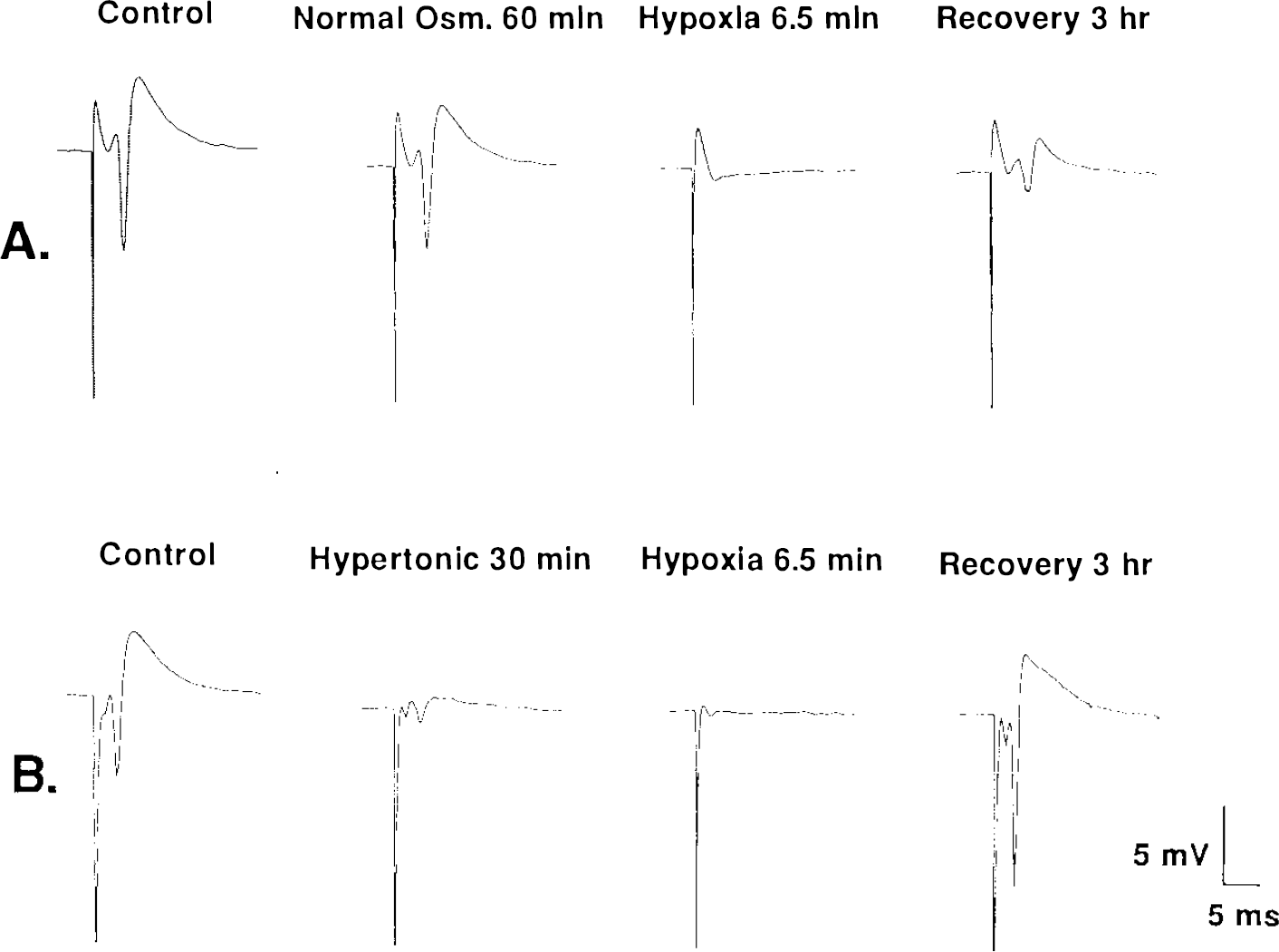
Sample orthodromic responses in CA1 st. pyramidale evoked by 0.1 ms, 150 μA pulses, recorded from two hippocampal tissue slices prepared from the same hippocampus, before and after 6.5 min of hypoxia.
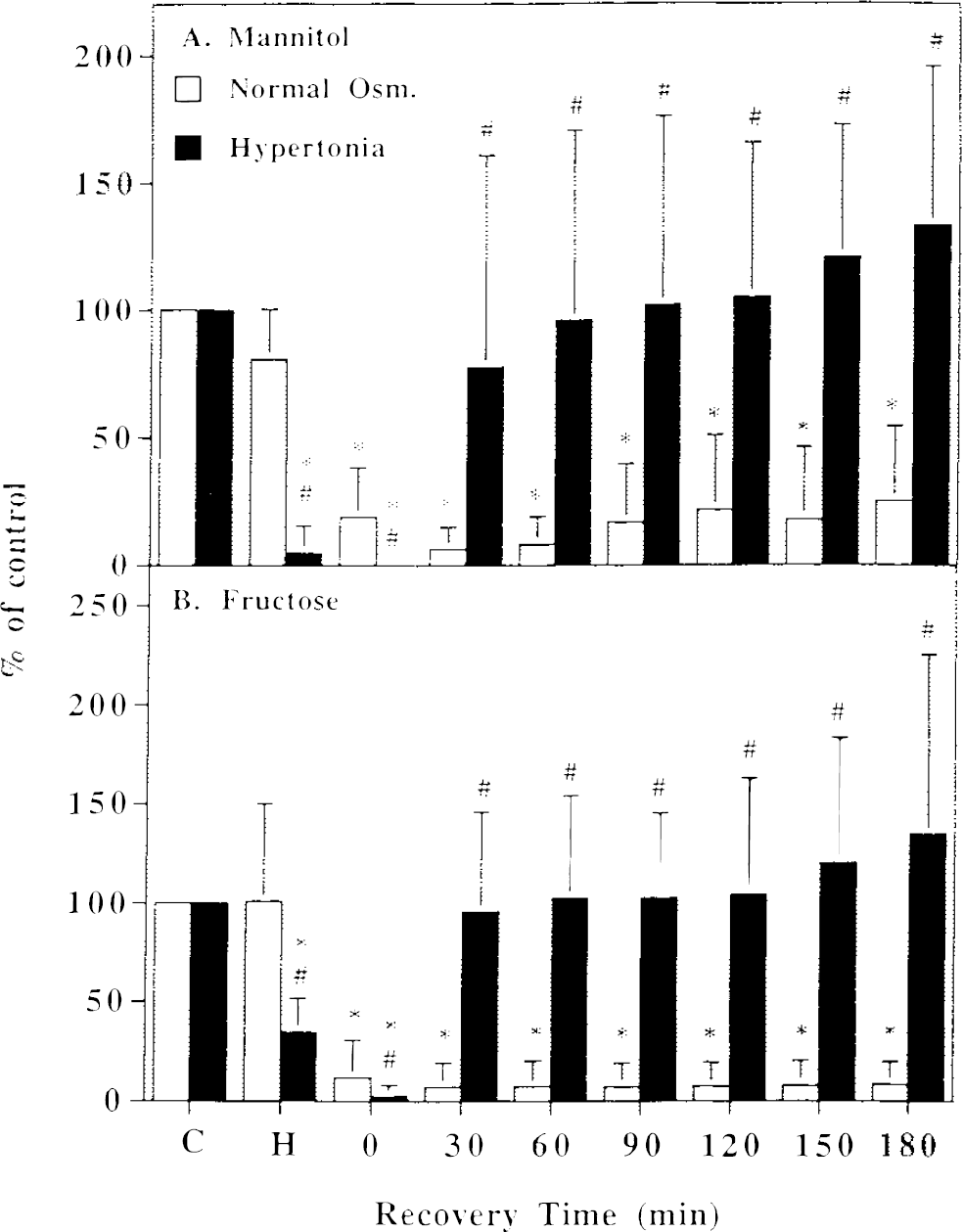
The average normalized orthodromic population spike amplitude (mean ± SD; 0.1 ms, 150 μA stimulation), before and after hypoxia, for paired slices in normal πo and in hypertonic solution.
In all the slices in normal πo, spreading depression (SD)-like depolarization occurred 1.5–2.5 min after changing to 95% N2, 5% CO2 in the gas phase, and these slices remained in an SD-like state until reoxygenation. SD-like depolarization was recognized by the accelerating negative shift of extracellular potential (ΔVo; Figs. 3 and 4; Leão, 1947; Czéh et al., 1993). No SD-like ΔVo occurred in seven of nine slices in mannitol-containing hypertonic solution (recorded in st. pyramidale) and in none of the 12 slices in fructose solution (six in st. pyramidale and six in st. radiatum). Instead, the DC potential trace shifted slowly in the negative direction, reaching −1.5 to −4.0 mV at the end of 6.5 or 9–10 min of hypoxia (Figs. 3 and 4). In two of the mannitol-hypertonic slices a ΔVo similar in wave form to SD was recorded, but it started 2.0 and 2.5 min later than in the paired slice in isotonic ACSF, and the maximal negative shift was only −4.5 and −5.8 mV, compared with the ΔVo of −8 to −17.5 mV recorded in the slices in hypoxic, isosmotic condition.
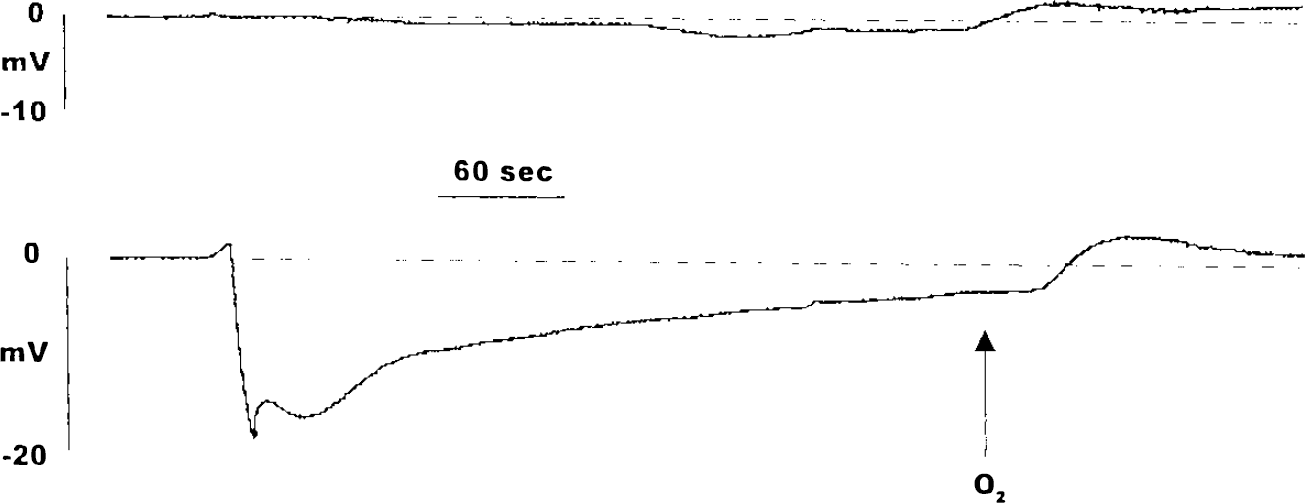
Extracellular potentials in CA1 st. pyramidale of two slices prepared from the same hippocampusand recorded simultaneously during exposure to hypoxia. Top: in hypertonic solution (100 mM mannitol); bottom: in normal πo. Recording by chart recorder, scanned by computer.
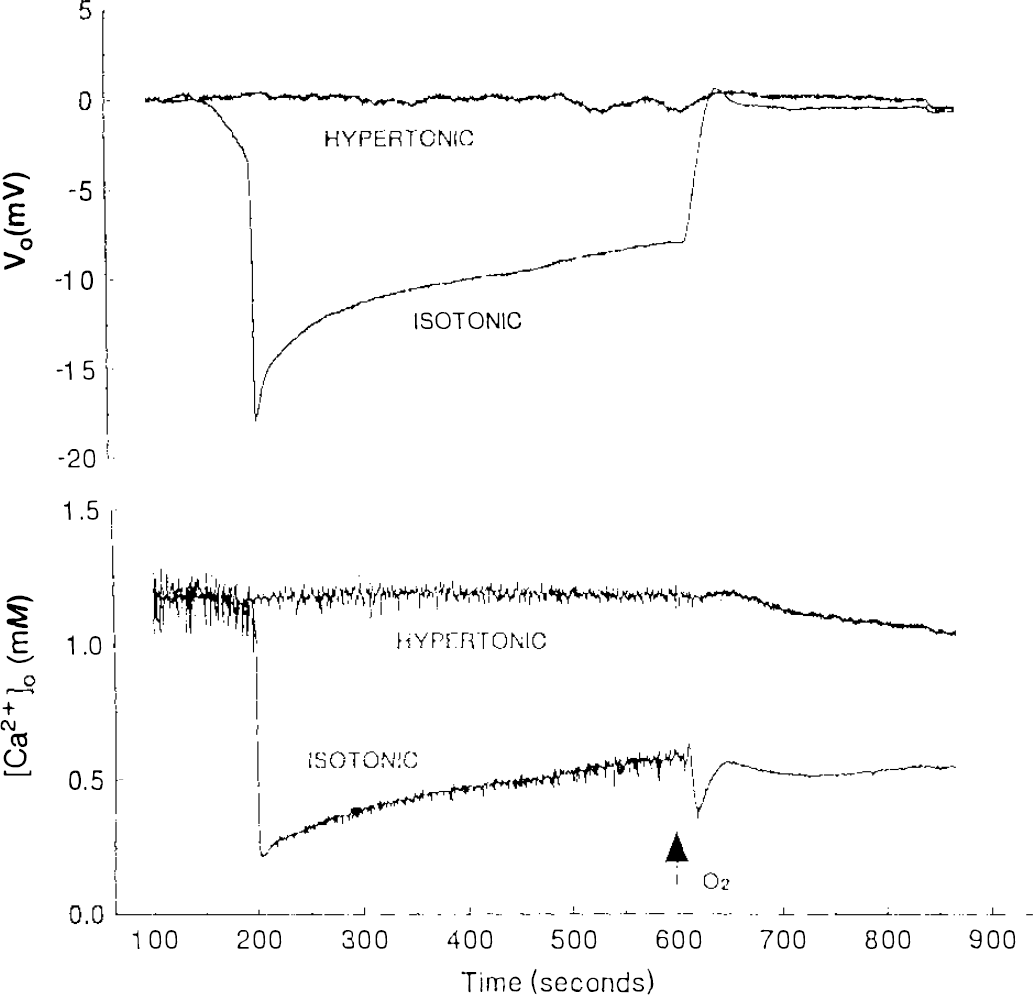
Extracellular voltage (Vo) and interstitial calcium ([Ca2+]o) during hypoxia in CA1 st. radiatum of two slices recorded simultaneously, one in normal πo, the other made hypertonic by the addition of 100 mM fructose. Oxygen was withdrawn at zero time (outside the graph) and restored at the arrow. Recording by computer; sampling frequency, 200 Hz.
Restoring normal oxygenation in both wells and normal πo in the hypertonic well started the recovery period. In the slices that had been in hypertonic solution during hypoxia, orthodromic transmission recovered completely. In the isosmotic slices, orthodromic responses remained depressed. Sample recordings are shown in Fig. 1 and averages and standard deviations of the response amplitudes in Fig. 2.
In six additional paired experiments, [Ca2+]o was recorded. In normal πo, withdrawal of O2 induced the expected precipitate decrease of [Ca2+]o associated with SD-like depolarization (Hansen, 1985).
After reoxygenation, a second, smaller, transient decrease of [Ca2+]o coincided with the recovery and overshoot of Vo in five of the six slices in normal πo. In slices made hypertonic by the addition of fructose, [Ca2+]o either did not change or it very briefly and slightly decreased and then increased very slightly and slowly, followed by a slow, slight decrease during reoxygenation (Fig. 4). The small changes of [Ca2+]o in the hypertonic slices during and after hypoxia could be due to changes of interstitial volume. In the six slices in normal πo during hypoxia, [Ca2+]o decreased from the control level of 1.20 mM to a mean minimal level of 0.21 ± 0.12 mM; in the hypertonic slices, the average maximal level reached during 9 to 10 min of hypoxia was 1.23 ± 0.18 mM. For the difference in hypoxic [Ca2+]o between hypertonic and isotonic slices p < 0.0001. After reoxygenation, [Ca2+]o recovered to near its control level in ∼20 min in both slices (not shown).
In the six experiments in which [Ca2+]o was recorded, the recovery of the evoked potentials was observed for only ∼30 min, and these data are not included in Fig. 2B.
DISCUSSION
The two main findings of this investigation are, first, that slices that have been maintained in a hypertonic environment recovered after oxygen deprivation to a greater extent than did slices in an isosmotic condition, by a wide margin. Second, that hypertonicity completely prevented hypoxic SD-like depolarization in most slices and postponed and mitigated it in the two cases in which it did occur. Hypertonia also prevented the decrease of [Ca2+]o associated with the SD-like ΔVo and, by inference, uptake of Ca2+ from interstitial fluid into cells. Earlier, Balestrino et al. (1992) reported delay of hypoxic SD by mannitol.
It is important to note that in each experiment, the two slices that were being compared were cut from the same hippocampus and were treated exactly alike, except for the difference in πo. Variations of age or genetic makeup of the animal or in experimental conditions, such as the duration or degree of hypoxia or temperature, could play no part in the difference of the outcome.
The fact that mannitol and fructose achieved the same degree of protection suggests that the effect was due to the elevated πo and not to a specific action of either compound. In the experiments with fructose, the hypoxia lasted 9 min compared with 6.5 min in the experiments with mannitol. As may be expected, with the longer hypoxia, the failure of synaptic function in the isotonic slices was more profound, yet the hypertonic treatment provided virtually complete protection (Fig. 2).
The question is, are the two effects, prevention of SD-like depolarization and improved posthypoxic recovery, causally related. We think there is good reason to believe that they are. In an earlier study, the effects of hypoxia on CA1 region and dentate gyrus were compared. Synaptic transmission was blocked earlier, SD occurred earlier, and recovery from hypoxic depression was less complete in CA1 than in DG (Balestrino et al., 1989). As we mentioned in the Introduction, all drugs and other treatments (for example, acidification of the bath) that curtailed the duration of hypoxic SD favored recovery from hypoxia (Balestrino and Somjen, 1986; Somjen et al., 1990; Tombaugh, 1994; Balestrino, 1995). We also showed that SD-like depolarization in the presence of normal oxygen can cause long-lasting loss of neuronal function, if the depolarization lasts long enough, and if calcium is present in the extracellular medium (Kawasaki et al., 1988; Jing et al., 1991a). These studies have also indicated that it is not the depolarization per se but the uptake of Ca2+ by cells that initiates the process that ultimately leads to loss of neuron function (Balestrino and Somjen, 1986; Jing et al., 1991a; Young et al., 1991). Hypertonicity has now joined the list of treatments that prevent both SD-like depolarization and posthypoxic neuron failure.
It is remarkable that hypertonicity was more effective than any of the other treatments so far tested in suppressing hypoxic SD-like ΔVo and the associated A [Ca2–]o, at least in hippocampal slices maintained at close to physiologic temperature in an interface chamber. Moreover, posthypoxic recovery of function was as complete in the hypertonically treated slices in the presence of normal bath [Ca2+] as it has been in slices that have been depleted of Ca2+ for the duration of hypoxia (Young et al., 1991). This suggests that hypertonia has virtually completely blocked the pathologic uptake of calcium.
The question then remains, was the hypertonic shrinkage of cell volume and the resulting prevention of hypoxic cell swelling also a factor in protecting neurons? Here our conclusion is less certain. We have tested whether exposure to very hypotonic solution causes long-lasting neuron failure in well-oxygenated hippocampal slices. It turned out that 60 min of severe hypotonic swelling is followed by depression of synaptic transmission without loss of antidromic conduction, in other words, without neuron death (Huang et al., in press, 1995a). We concluded that swelling may aggravate hypoxic neuron injury but cannot be its sole cause. As a corollary, cell shrinkage might aid the protection, but the prevention of cell swelling is not the main protective factor.
We should also ask in what way hypertonia prevents SD-like depolarization. Earlier we hypothetically proposed that SD-related depolarization is initiated by cell swelling, which would open stretch-gated membrane ion channels (Somjen et al., 1992). It would be tempting to conclude that the prevention of SD by hypertonic cell shrinkage supports this hypothesis. Unfortunately for the hypothesis, unlike glial cells (Kimelberg and Kettenmann, 1990), neurons show neither depolarization nor increased ion conductance when exposed to hypotonic media (Somjen et al., 1993; Bossut and Somjen, 1995; Vreugdenhil et al., 1995). Moreover, strongly hypotonic bathing solutions do induce waves of SD episodes in hippocampal slices but not an enduring state of depolarization (Chebabo et al., 1995b). The hypothesis of swelling-induced ion conductance increase predicts a sustained increase of membrane permeability during cell swelling. For these reasons, we had to abandon the idea that SD-like depolarization is due to stretch-gated membrane ion conductance increase.
Although for now we have no good explanation of the mechanism by which hypertonia suppresses SD, it may be pointed out that virtually any treatment that reduces neuron excitability or excitatory transmission or both is also capable of postponing both normoxic and hypoxic SD, even though none of these other interventions completely prevents SD (Balestrino and Somjen, 1986; Aitken et al., 1991; Jing et al., 1991b; Balestrino, 1995). It is clear that hypertonia is among the treatments that depress synaptic transmission, as well as voltage-gated and ligand-gated ion currents in neurons (Andrew, 1991; Somjen et al., 1993; Huang and Somjen, 1995a, b ; Vreugdenhil et al., 1995). Hypertonia seems to be more effective than the previously examined treatments in actually forestalling hypoxic SD, but it may act through a similar nonspecific mechanism.
Finally we wonder whether raising osmotic pressure could be used to prevent hypoxic brain damage in human patients who are at acute risk of global cerebral ischemia. Mannitol is used in clinical practice to reduce intracranial pressure and to prevent or to reverse cerebral edema (Wise and Chater, 1962; Feig and McCurdy, 1977). It has no known toxic side effects, but its prolonged administration may be limited by the hazard of rebound brain swelling after withdrawal, which could occur as an aftermath of the regulatory volume increase (RVI) taking place during the treatment (Cserr and Patlak, 1991; Cserr et al., 1991). RVI is believed to occur in two phases, an initial, readily reversible phase mediated by the cellular uptake of electrolytes, followed by uptake of and exchange with organic osmolytes, which is only slowly reversed after restoration of πo (McManus and Churchwell, 1994).
Footnotes
Acknowledgment:
We thank the Heart and Stroke Foundation of Canada for a Research Fellowship to Dr. Rong Huang. The work was supported by grants NS 18670 and NS 17771 of the NIH, USPHS.