
Abstract
The effect of posttraumatic hypothermia (brain temperature controlled at 32°C for 4 h) on mortality after severe controlled cortical impact (CCI) was studied in rats. Four posttraumatic brain temperatures were compared: 37°C (n = 10), 36°C (n = 4), 32°C (n = 10), and uncontrolled (UC; n = 6). Rats were anesthetized and subjected to severe CCI (4.0-m/s velocity, 3.0-mm depth) to the exposed left parietal cortex. At 10 min posttrauma the rats were cooled or maintained at their target brain temperature, using external cooling or warming. Brain temperature in the UC group was recorded but not regulated, and rectal temperature was maintained at 37 ± 0.5°C. After 4 h, rats were rewarmed over a 1-h period to 37°C, extubated, and observed for 24 h. In the 37 and 36°C groups, 24-h mortality was 50% (37°C = 5/10, 36°C = 2/4). In the 32°C group, 24-h mortality was 10% (1/10). In the UC group, brain temperature was 35.4 ± 0.6°C during the 4-h treatment period and 24-h mortality was 0% (0/6). Mortality was higher in groups with brain temperatures ≥36°C versus those with brain temperatures <36°C (50 vs. 6%, respectively; p < 0.05). Additionally, electroencephalograms (EEG) were recorded in subsets of each temperature group and the percentage of time that the EEG was suppressed (isoelectric) was determined. Percentage of EEG suppression was greater in the hypothermic (32°C, n = 6; UC, n = 4) groups than in the normothermic(36°C, n = 3; 37°C, n = 6) groups (23.3 ± 14.3 vs. 1.2 ± 3.1%, respectively; p < 0.05). Posttraumatic hypothermia suppressed EEG during treatment and reduced mortality after severe CCI. The threshold for this protective effect appears to be a brain temperature <36°C. Thus, even mild hypothermia may be beneficial after severe brain trauma.
Hypothermia after cerebral trauma is a topic of renewed interest, with recent clinical trials reporting beneficial effects after severe traumatic brain injury (TBI) (Clifton et al., Marion et al., 1993; Shiozaki et al., 1993). The effects of hypothermia in experimental models of TBI have also been examined. Clifton et al. (1991) demonstrated that moderate hypothermia (30°C) instituted before trauma reduced mortality compared with mild hypothermia (33 and 36°C), normothermia (38°C), or hyperthermia (40°C) in rats after fluid-percussion injury. Hypothermia did not improve survival in several TBI models studied (Rosomoff et al., 1965; Bouzarth et al., 1967; Pomeranz et al., 1993). In these studies and others, however, hypothermia improved posttraumatic neurologic and/or histologic outcomes (Lyeth et al., 1993a; Palmer et al., 1993b; Dietrich et al., 1994). We know of no reports of improved survival using hypothermia initiated after brain trauma in experimental models.
Hypothermia produces beneficial effects in models of cerebral ischemia. These effects and the importance of brain temperature monitoring and regulation have been reviewed (Ginsberg et al., 1992; Maher and Hachinski, 1993). A reduction in brain temperature as small as 2°C can reduce ischemic cell damage, and a similar increase in brain temperature can produce deleterious effects including increased neuronal damage and impaired blood–brain barrier integrity. Despite careful monitoring of brain temperature in models of ischemia, brain temperature is not commonly monitored in cerebral trauma models. Several key studies characterizing various contemporary models in the rat including fluid percussion, controlled cortical impact (CCI), and weight drop have not reported brain temperature and/or have reported only the core (rectal) temperature (Dixon et al., 1987; McIntosh et al., 1989; Dixon et al., 1991; Sutton et al., 1993; Clark et al., 1994). Rectal temperature does not accurately reflect brain temperature, and general anesthesia alone reduces brain temperature by 1 to 2°C (Jiang et al., 1991). Despite clinical application, the details of hypothermic treatment in models of TBI (duration, degree) are less well characterized than in models of cerebral ischemia.
In this study, we tested the hypothesis that posttraumatic hypothermia (brain temperature controlled at 32°C for 4 h) reduces mortality after severe CCI in rats and assessed the effect of anesthesia and severe CCI on brain temperature. We also determined the threshold for the beneficial effect of hypothermia on mortality after severe TBI and evaluated the effect of hypothermia on posttraumatic electroencephalograms (EEG).
MATERIALS AND METHODS
Surgical procedures
Thirty-three virus-free, mature male Sprague–Dawley rats (280–400 g) were studied. The animals had free access to food and water until the time of the study. All studies were approved by the University of Pittsburgh Animal Care and Use Committee. AU surgical procedures were performed using aseptic technique.
Anesthesia was induced in a plastic jar with 4% isoflurane (Anaquest, Memphis, TN, U.S.A.) in O2. The trachea was intubated with a 14-gauge angiocatheter, and the lungs were mechanically ventilated with 2.0% isoflurane/66% N2O/balance O2. A femoral arterial catheter was inserted for continuous monitoring of blood pressure, arterial blood sampling, and administration of pancuronium bromide (0.1 mg/kg/h; Elkins-Sinn, Cherry Hill, NJ, U.S.A.). A rectal probe was inserted for temperature monitoring. Bicillin (100,000 U; Upjohn, Kalamazoo, MI, U.S.A.) and gentamicin (10 mg/kg; Elkins-Sinn) were administered intramuscularly.
In preparation for trauma, the head was fixed in a stereotactic device (David Kopf, Tujunga, CA, U.S.A.). After retraction of the scalp, a craniotomy was made over the left parietal cortex with a dental drill, using the coronal and interparietal sutures as margins. The intact dura and bone flap were left in place until immediately before trauma. A temperature probe (0.009-in. outside diameter; Physiotemp Corp., Clifton, NJ, U.S.A.) was inserted through a burr hole into the left parietal cortex 5 mm anterior to the bregma and 2.5 mm lateral to the sagittal suture (∼30 to 40 min before trauma). Anesthesia was reduced to 1.1% isoflurane/66% N2O/balance O2. Rats were warmed using a heated water blanket and heat lamp to a brain temperature of 37 ± 0.5°C and then allowed to equilibrate under anesthesia (1.1% isoflurane/66% N2O/balance O2) for 30 min. Fifteen minutes before trauma, an arterial blood sample (0.5 ml) was obtained to verify that arterial blood gas tensions, serum glucose, and hematocrit were within normal limits.
TBI was performed using the CCI method as described previously (Palmer et al., 1993a) with minor modification. After removal of the bone flap, injury was produced using the CCI device (Lighthall et al., 1990; Dixon et al., 1991). This device consists of a 5-mm metal impactor tip that is pneumatically driven at a predetermined velocity, depth, and duration of brain deformation. For all studies, a velocity of 4.0 ± 0.2 m/s and a duration of deformation of 50 ms was used. A depth of penetration of 3.0 mm was used to produce a severe cortical contusion. Following trauma the bone flap was replaced and sealed with dental cement (Koldmount, Vernon Benshoff Co., Albany, NY, U.S.A.), and the scalp incision was closed.
Hypothermia
Rats were randomized to one of three experimental groups: 37°C normothermia (n = 10), 36°C normothermia (n = 4), or 32°C hypothermia (n = 10). In the control group (n = 6), rectal temperature was maintained at 37°C but brain temperature was not controlled (uncontrolled; UC). At 10 min posttrauma, rats were maintained at, or cooled to, their target brain temperature over 30 min using external warming or cooling. Brain temperature was kept within 0.5°C of the target temperature throughout the treatment period. Brain temperature in the UC group was recorded but not regulated. Throughout the treatment period rats were maintained in the stereotactic frame under 1.1% isoflurane/66%N2O/balance O2 anesthesia. Pancuronium bromide was administered hourly to prevent skeletal muscle movement. Brain and rectal temperatures were continuously monitored and recorded every 15 min. Mean arterial pressure was continuously monitored and recorded every hour. Arterial blood gas tensions, hematocrit, and blood glucose levels were measured 15 min before and 1 and 3 h after trauma. The Paco2 was controlled at 35–45 mm Hg and the Pao2 was controlled at 100–200 mm Hg.
At 4 h after trauma rats in the 32°C group were rewarmed over 1 h to 37°C. Rats in the other groups were kept at their target temperatures for 1 h. Following the treatment period, vascular catheters were removed and anesthesia was discontinued. The rats were then weaned from mechanical ventilation within 1 h and extubated. They were maintained on supplemental O2 for 30 min after extubation, then returned to their cages, where they were allowed free access to food and water and observed for 24 h.
EEG/seizures
In initial experiments it appeared that some of the observed mortality was associated with clinical seizure activity; therefore, this protocol was modified to include electrographic and clinical monitoring for seizure activity. Biparietal scalp EEG electrodes were placed in 19 rats(37°C,n = 6;36°C,n =3; 32°C, n = 6; and UC, n = 4). A continuous EEG (Grass Instrument Co., Quincy, MA, U.S.A.) was obtained from 30 min before trauma until the end of the rewarming period. EEG were evaluated for epileptiform activity and duration of EEG suppression (isoelectricity) over 2-min intervals before trauma and at 1, 2, 3, 4, and 5 h after trauma. In addition, the percentage of EEG suppression during the 4-h treatment period was calculated. After extubation, rats were isolated and observed for clinical seizures over a 2-h period before being returned to their cages.
To determine the effect of brain temperature on EEG, three additional nontraumatized rats were anesthetized and monitored as described above (including brain temperature probe). Rats were cooled and EEG were recorded over 2-min intervals at 37, 36, 35, and 32°C and upon rewarming at each of these temperatures. EEG were evaluated for epileptiform activity and duration of EEG suppression at each of these temperatures.
Statistical analysis
All data are presented as mean ±SD. Comparisons of physiologic variables were made between groups using one-way analysis of variance (ANOVA). The duration of mechanical ventilation and the time until extubation were analyzed between normothermic and hypothermic groups using an unpaired t test. To achieve adequate statistical power for the comparison of mortality in normothermic and hypothermic rats, the 32°C and UC groups were combined into a hypothermic group, and the 36 and 37°C groups were combined into a normothermic group. Mortality data were analyzed using the Fisher exact test. The percentage of EEG suppression between the normothermic and the hypothermic groups was analyzed using the Mann–Whitney U test. The effect of brain temperature on EEG suppression in nontraumatized rats was analyzed using repeated-measures ANOVA and the Bonferroni test; p < 0.05 was considered significant.
RESULTS
There was no difference between groups in any physiologic variable measured before trauma or during the treatment period (excluding brain and rectal temperature, Table 1). Brain temperature in the UC group decreased from 36.9 ± 0.1°C immediately before trauma to 35.7 ± 0.6°C 15 min after trauma and averaged 35.4 ± 0.6°C throughout the 4-h treatment period (Fig. 1). Rectal temperature was maintained at 37 ± 0.5°C in this group.
Physiologic data at baseline and 1 and 3 h after trauma
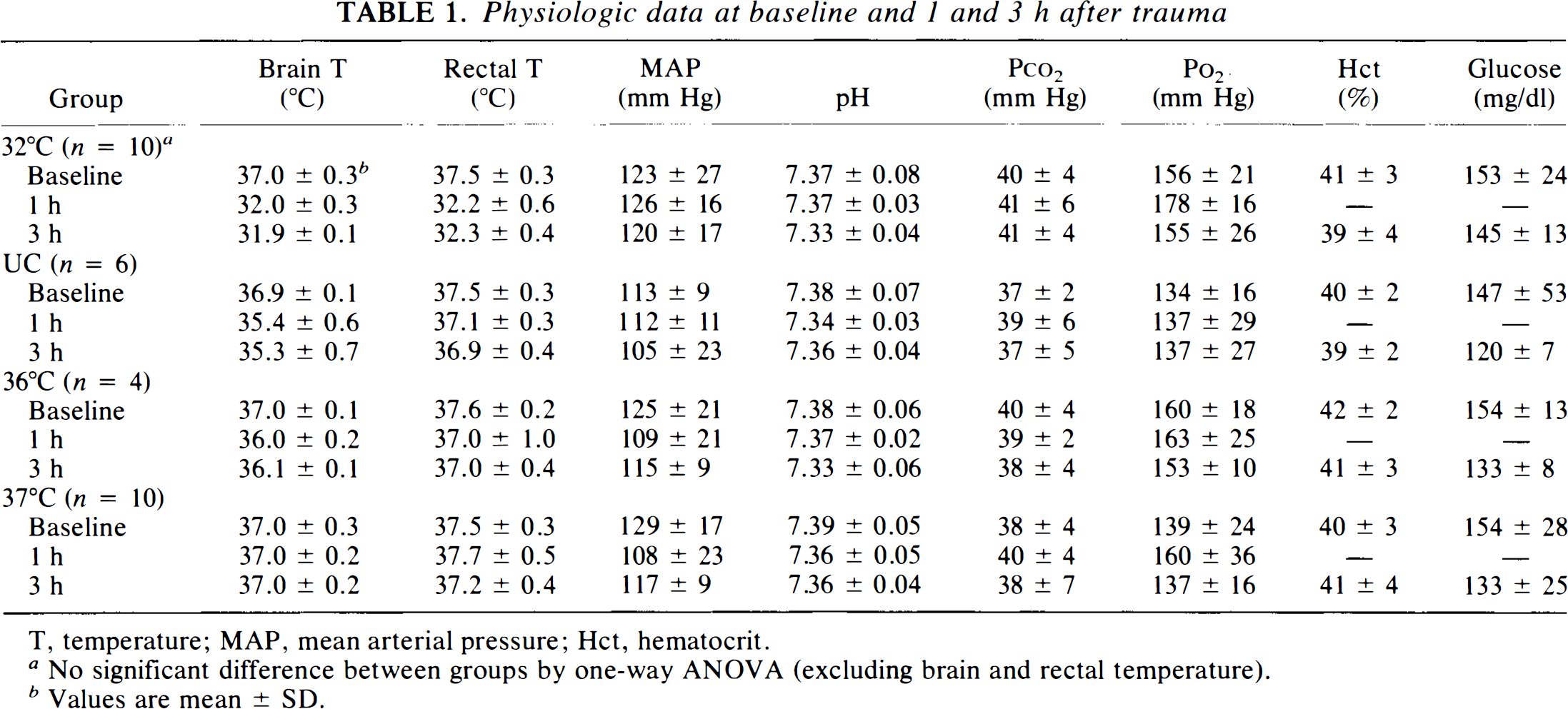
T, temperature; MAP, mean arterial pressure; Hct, hematocrit.
No significant difference between groups by one-way ANOVA (excluding brain and rectal temperature).
Values are mean ± SD.
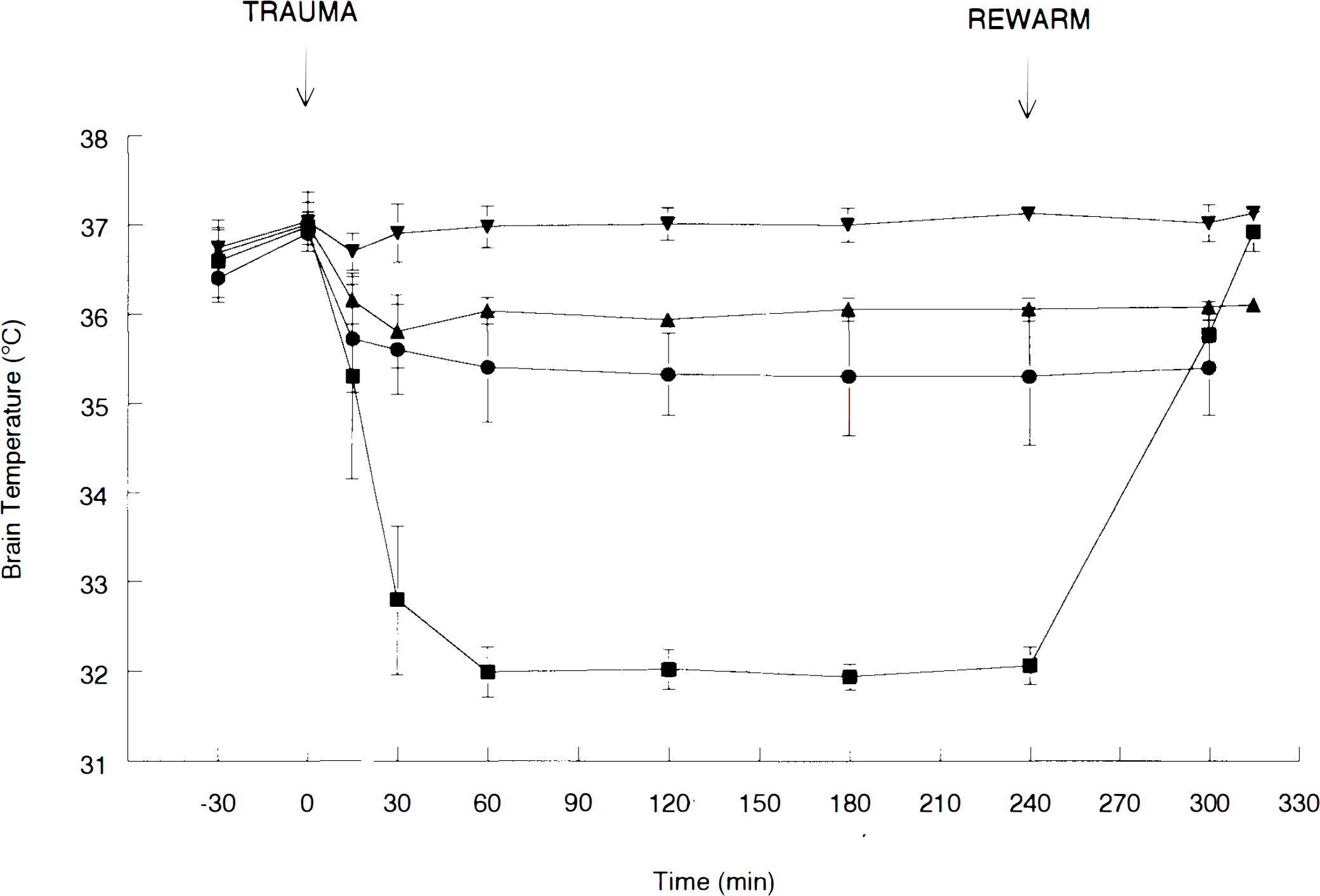
Temperature vs. time monitored in the 37°C (▾ n = 10), 36°C (▴ n = 4), UC (• n = 6), and 32°C (▪ n = 10) groups.
All rats survived the trauma, the 4-h treatment period, and the rewarming period. For nonsurvivors, death typically occurred within 2 h after extubation (6 to 8 h after trauma). Death was associated with respiratory failure (defined as labored breathing, progressing to agonal respiration, apnea, and death), seizures, and isolated apnea (Table 2). In the 37 and 36°C groups, 24-h mortality was 50% (37°C = 5/10, 36°C = 2/4). In the 32°C group, 24-h mortality was 10% (1/10). In the UC group, 24-h mortality was 0% (0/6). There was no difference in mortality between individual groups, however, when the 32°C and UC groups were combined (hypothermic) and the 36 and 37°C groups were combined (normothermic), there was a reduction in mortality in the hypothermic compared to the normothermic groups (6 vs. 50%, respectively; p < 0.05, Fisher exact test). There was no difference between the normothermic and the hypothermic groups in the duration of mechanical ventilation (345 ± 21 vs. 347 ± 38 min, respectively; p = 0.85, t test) or in the time until extubation after discontinuation of anesthesia (30 ± 22 vs. 25 ± 20 min, respectively; p = 0.58).
Mortality in rats after severe CCI
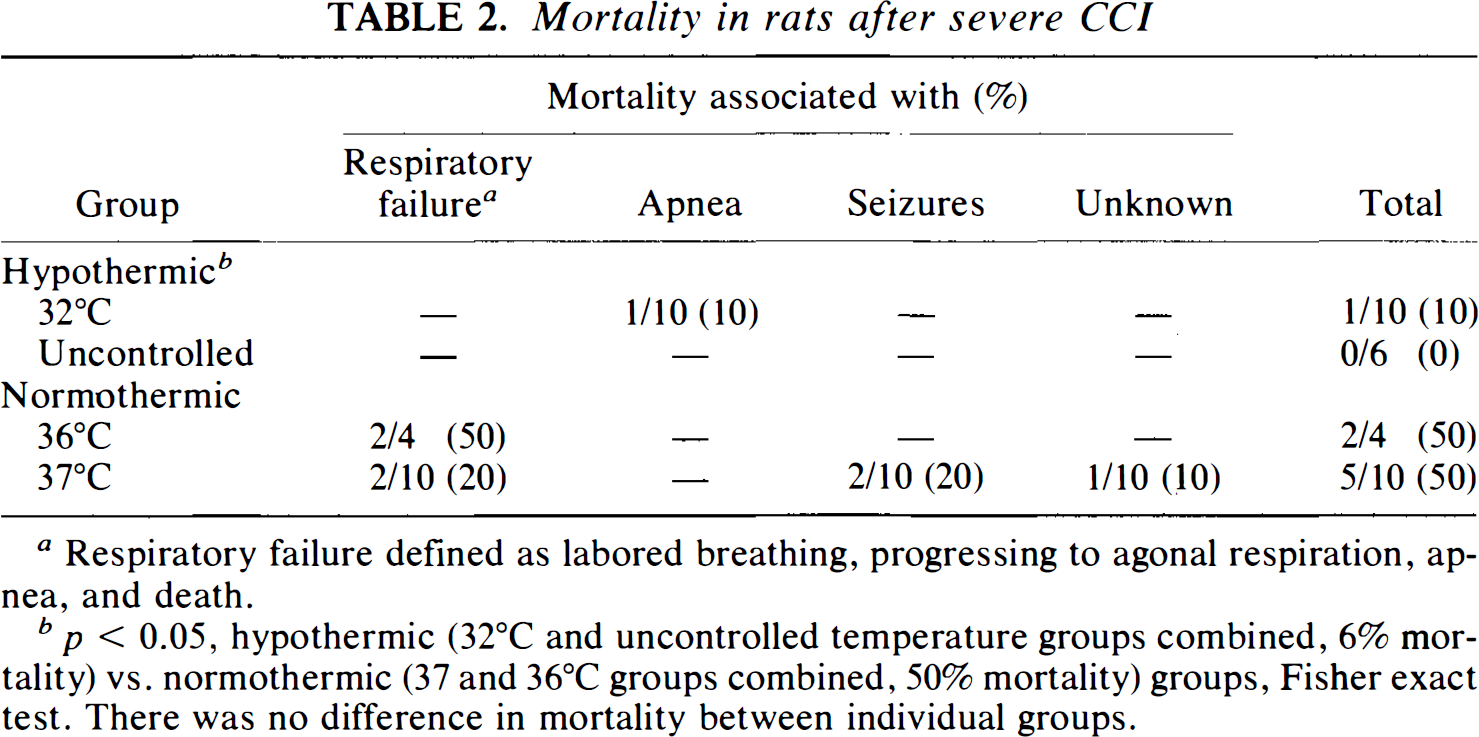
Respiratory failure defined as labored breathing, progressing to agonal respiration, apnea, and death.
p < 0.05, hypothermic (32°C and uncontrolled temperature groups combined, 6% mortality) vs. normothermic (37 and 36°C groups combined, 50% mortality) groups, Fisher exact test. There was no difference in mortality between individual groups.
Figure 2 shows representative EEG from rats in each group. Hypothermia produced a burst suppression pattern (isoelectricity) in the 32°C treatment group, as well as in the UC group. In the 32°C treatment group the EEG frequency returned to baseline during rewarming. Electrographic seizures were seen in five rats: two from the 32°C, two from the 36°C (Fig. 2C), and one from the 37°C group. In the 32°C group, electrographic seizures were observed only during the rewarming period, when the brain temperature had reached 35.8 ± 0.4°C (approximately 5 h posttrauma). Clinical generalized seizures were observed in two rats from the 37°C group. Electrographic seizures did not correlate with clinical seizures or mortality in this study, although the sample size was small, and the ability to detect seizures using scalp EEG electrodes may be limited.
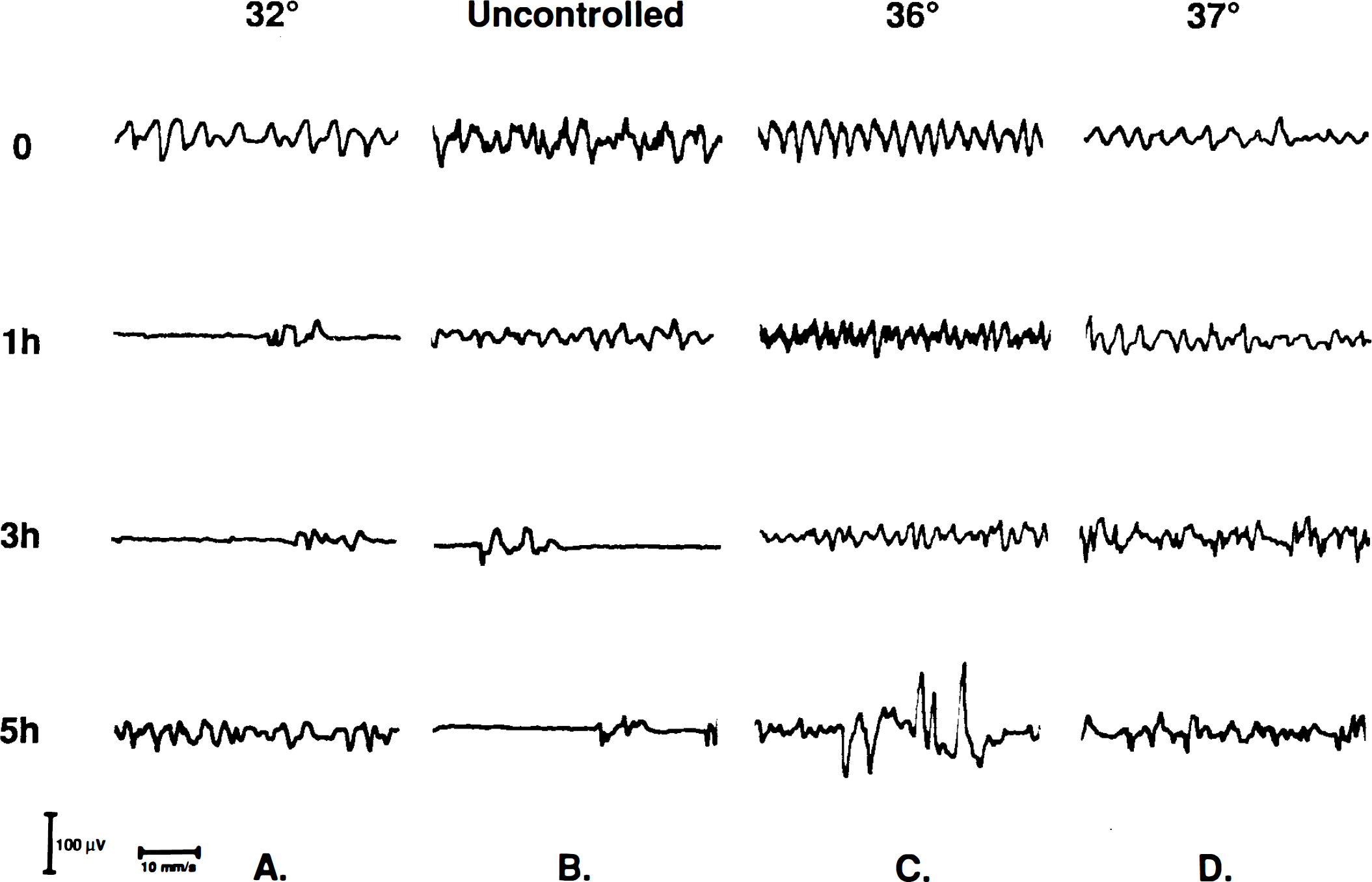
Representative EEG recordings at designated times posttrauma from rats in each brain temperature group:
The percentage of time that the EEG was suppressed (isoelectric) was determined in each group during the 4-h treatment period (Fig. 3). In the hypothermic (32°C, n = 6; UC, n = 4) groups, there was an increase in EEG suppression compared to the normothermic (36°C, n = 3; 37°C, n = 6) groups (23.3 ± 14.3 vs. 1.2 ± 3.1%, respectively; p < 0.05, Mann–Whitney U test).
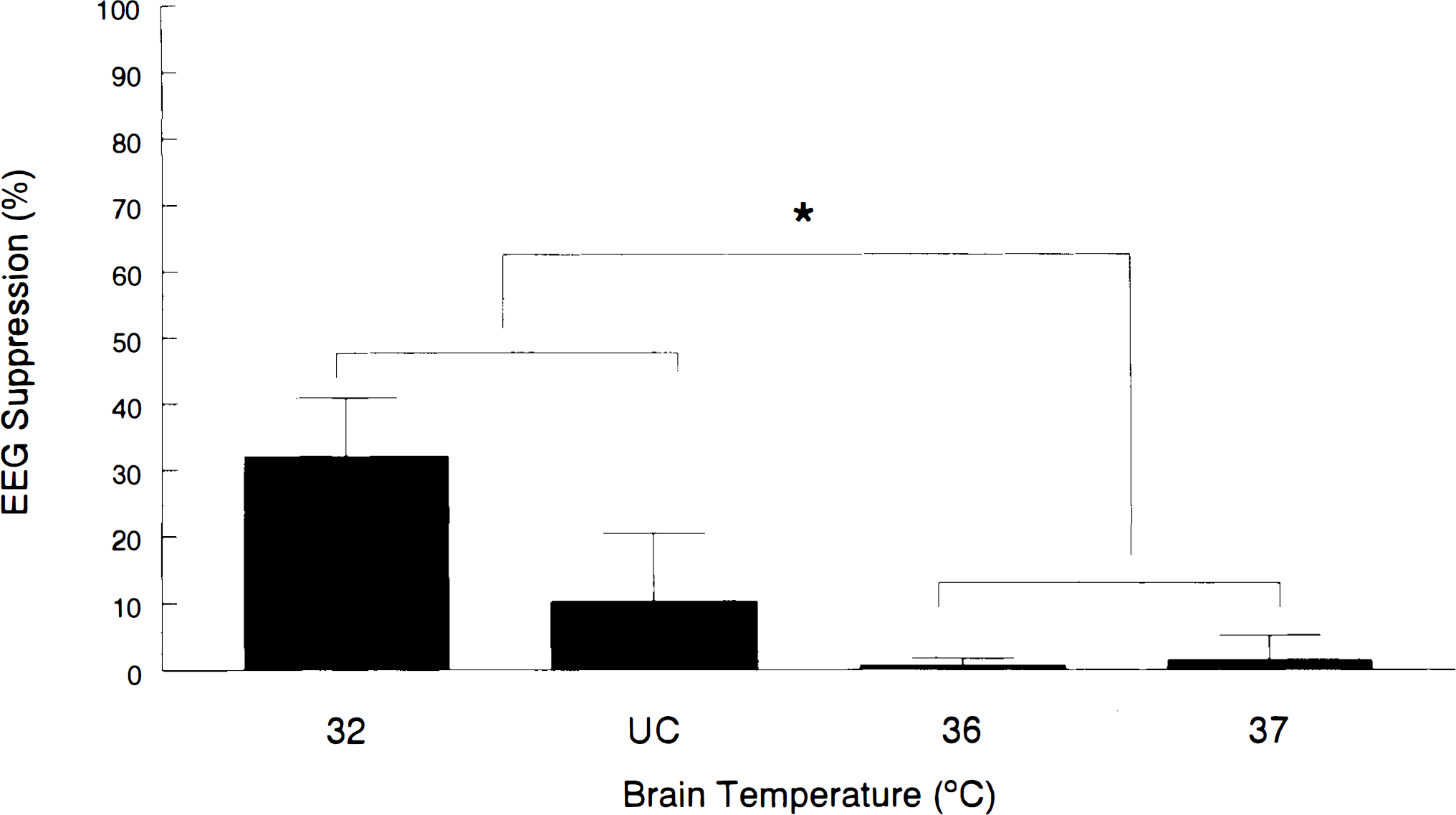
EEG suppression (%) in each temperature group. In the hypothermic (32°C, n = 6; UC, n = 4) groups there was an increase in EEG suppression compared to the normothermic (36°C, n = 3; 37°C, n = 6) groups (p < 0.05, Mann–Whitney U test).
In the nontraumatized, anesthetized rats, EEG suppression was observed at 36, 35, and 32°C during cooling. Suppression was not observed at 35 or 36°C during rewarming (Fig. 4). In the anesthetized, nontraumatized rats, the percentage of EEG suppression at a brain temperature of 32°C was similar to that in the traumatized rats at 32°C (40.4 ±21.1 vs. 32.0 ± 8.9, respectively). There was no seizure activity seen in these nontraumatized rats and all rats survived.
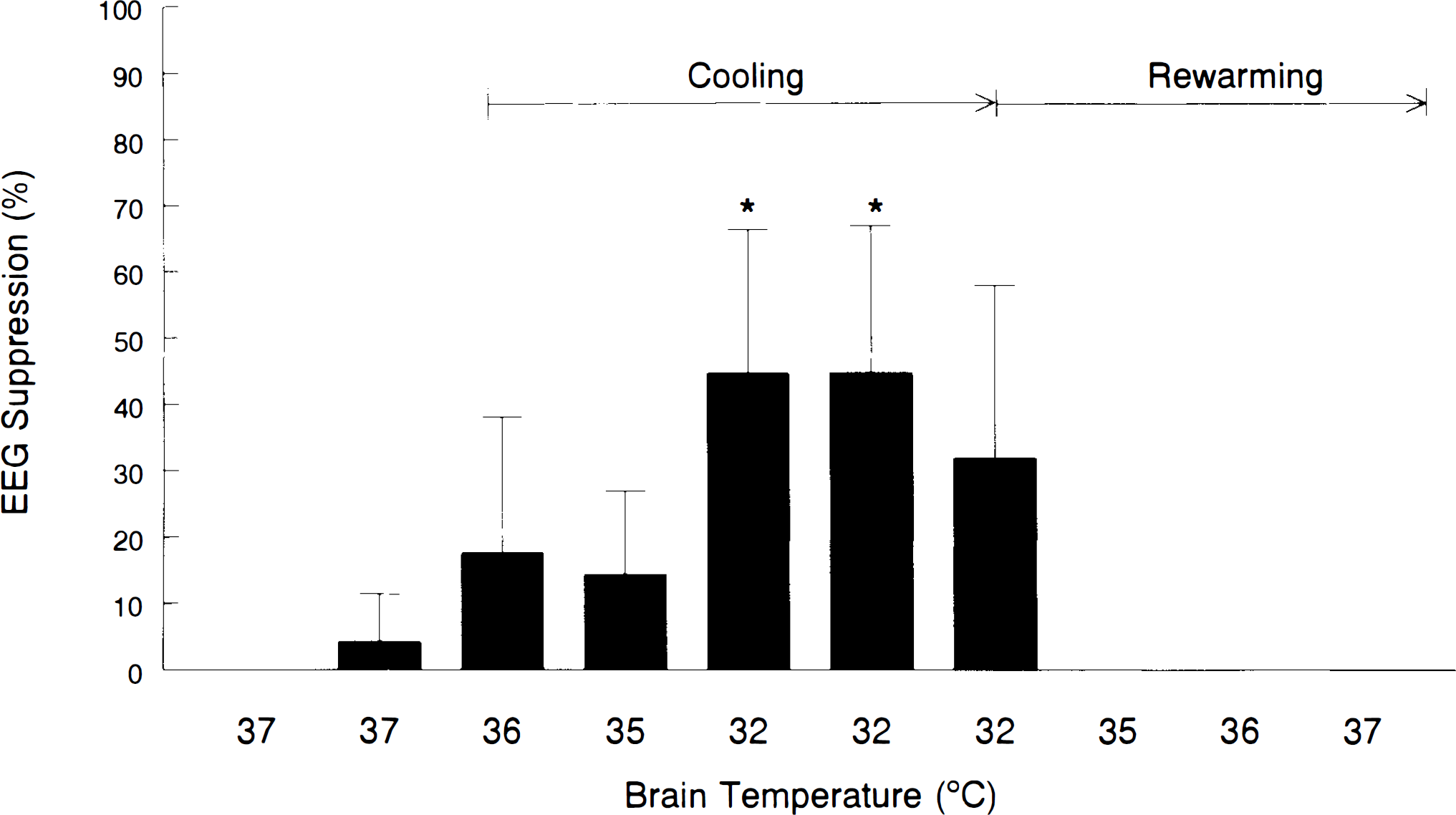
EEG suppression (%) in anesthetized, nontraumatized rats (n = 3) at various brain temperatures during cooling and rewarming. *p < 0.05 vs. baseline by repeated-measures ANOVA and Bonferroni test.
DISCUSSION
To our knowledge this is the first study to demonstrate a reduction in mortality when hypothermia is instituted after severe experimental TBI. A posttraumatic reduction in brain temperature to 32°C by active cooling, or to 35.4 ± 0.6°C by exposing the brain without controlling brain temperature, reduced mortality by 44% compared to groups with brain temperature controlled at 36 or 37°C. The temperature threshold for this effect appears to be below 36°C, although the numbers of rats in the UC and 36°C groups were not large enough to detect statistical differences between these intermediate temperature groups. This apparent therapeutic threshold, a reduction in brain temperature by ∼2°C, is consistent with that seen in models of cerebral ischemia (Berntman et al., 1981; Ginsberg et al., 1992).
The mechanism(s) by which hypothermia imparts its beneficial effects is multifactorial. In models of ischemia, hypothermia reduces neuronal injury, promotes postischemic metabolic recovery, attenuates excitatory amino acid release, reduces blood–brain barrier disruption, and improves functional outcome (Dietrich, 1992; Ginsberg et al., 1992; Maher and Hachinski, 1993). After moderate fluid percussion injury, hypothermia reduces blood–brain barrier disruption (Jiang et al., 1992) and histopathologic damage (Dietrich et al., 1994) and improves neurologic outcome (Clifton et al., 1991; Lyeth et al., 1993a). Hypothermia attenuates the loss of hippocampal microtubule-associated protein 2, a protein important in neuronal cytoskeletal cross-linkage (Taft et al., 1993), and blunts the increase in acetylcholine concentration in cerebrospinal fluid that occurs after trauma (Lyeth et al., 1993b). Palmer et al. (1993b) used a CCI trauma less severe than that used in this study (2.5- vs. 3.0-mm depth) to demonstrate that hypothermia (applied beginning 30 min before and continuing for 2 h after trauma) reduced infarct size at 2 weeks. In that study, an attenuation of excitatory amino acid elevations after trauma was not seen. This is in contrast to what is reported in cerebral ischemia models. In canine models, systemic hypothermia reduces histopathologic damage (Pomeranz et al., 1993), delays leukocyte infiltration (Rosomoff et al., 1965), and reduces cerebral O2 consumption and cerebral blood flow (CBF) (Rosomoff and Holaday, 1954; Michenfelder and Milde, 1991).
In contrast to systemic hypothermia, Kuluz et al. (1993) reported that CBF increases with selective brain hypothermia in rats. In that study, CBF increased when brain temperature was reduced below 36°C and plateaued at −200% of normal when brain temperatures reached 34°C. Rectal temperatures were maintained at 37.5°C. This cerebrovasodilatory response may explain some of the protective effects of hypothermia. It is possible that in our model different mechanisms of protection were involved in the 32°C and the UC hypothermic groups. In all controlled-temperature groups, the rectal temperature was only between 0.2 and 1.0°C higher than the target brain temperature. Thus, the 32°C group was systemically hypothermic and both CBF and cerebral metabolism were probably reduced. The UC group, however, was essentially a selective brain hypothermia model, with brain temperatures of 35.4 ± 0.6°C and rectal temperatures of 37.0 ± 0.5°C. Thus, an additional effect of hypothermia involved in the reduction in mortality in this group could be that CBF was increased during the 4-h treatment period, a time when CBF is reduced in this model (Kochanek et al., 1995). The present work and the study by Kuluz et al. (1993) further accentuate the importance of careful monitoring and reporting of both brain and core temperature in the standardization of trauma models and therapeutic trials (Dietrich, 1992; Ginsberg et al., 1992).
To evaluate the effects of hypothermia on survival after TBI, we specifically chose to use a severe injury, where mortality would be sufficient to detect a difference between groups. Thus, mortality in our study was higher than that reported by others using the CCI model (Dixon et al., 1991; Palmer et al., 1993a; Sutton et al., 1993), however, these studies used a less severe impact in most cases, and brain temperature was not controlled. Dixon et al. (1991) reported a 17% mortality after a CCI injury that was more severe (6-m/s velocity, 3.0-mm depth) but without brain temperature control. Previously, we used the same CCI injury as in the present study (4-m/s velocity, 3.0-mm depth) and reported a very low (<5%) mortality (Clark et al., 1994), but brain temperature was also not controlled. Based on the present work, brain temperature in this model, if not controlled, decreases to ∼35°C and improves survival. This again stresses the importance of monitoring brain temperature in models of TBI, particularly those models using an open craniotomy. Because of the high mortality associated with this injury when brain temperature is maintained at 37°C, it was not possible to examine accurately other end points, i.e., morphologic effects and functional outcome. With the degree of trauma used in this model, presumably the most severely injured rats (in the normothermic groups) would have died before a standardized functional or morphologic outcome determination could be made, potentially biasing the data.
It is important to clarify that brain temperature probes inserted into the cortex for monitoring during the study period were present at the time of impact. Thus, it is possible that the probe may have led to additional tissue damage at the time of trauma. We have not examined for histologic changes around the temperature probe tract with and without injury in this model. It is possible that the presence of the probe at the time of injury affected mortality, but all groups had brain temperature probes placed at the same time in the study protocol. In addition, although nontraumatized rats were used to determine the effect of hypothermia on EEG, true sham controls (including craniotomy) were not employed in this study. We have previously reported that there is no infarction or cerebral edema as a result of the craniotomy used for this model (Schoettle et al., 1990); however, brain temperature was not measured in these studies, and different anesthetics (halothane vs. isoflurane) and rat strains (Wistar vs. Sprague–Dawley) were used. Although craniotomy can produce local effects on underlying brain, it is unlikely that maintenance of brian temperature at 37°C for 4 h after craniotomy would be associated with mortality.
EEG suppression
In our model, hypothermia produced a predictable pattern of EEG suppression. This suppression began when brain temperature fell below 36°C in traumatized rats and when brain temperature reached 36°C in nontraumatized rats (Figs. 2–4). At 32°C, EEG suppression reached 32 ± 8.9% isoelectricity in traumatized rats and 40.4 ± 21.1% in nontraumatized rats (Figs. 3 and 4). Thus, only mild hypothermia is required to produce EEG suppression in anesthetized rats. Increasing degrees of hypothermia lead to increased EEG suppression. This effect has been also reported in both clinical (Hicks and Poole, 1981; Hickey and Andersen, 1987) and laboratory (Michenfelder and Milde, 1991; Gillinov et al., 1993) cardiopulmonary bypass. The additive effects of hypothermia in combination with anesthesia on EEG suppression have also been demonstrated (Loomis et al., 1986; Woodcock et al., 1987). It is suggested that hypothermia-induced cerebral protection occurs at the point of cerebral inactivity in patients undergoing anesthesia and cardiopulmonary bypass (Hickey and Andersen, 1987; Mizrahi et al., 1989) and that EEG is more reliable than systemic body temperature in determining this threshold. In our study, an increase in EEG suppression was seen in the hypothermic group compared to the normothermic group, and this was associated with reduced mortality.
Recently, trials of moderate systemic hypothermia have shown beneficial effects in humans after severe TBI (Clifton et al., 1993; Marion et al., 1993; Shiozaki et al., 1993). In the clinical setting, one therapeutic goal of treatment with hypothermia is to reduce brain temperature in order to reduce cerebral metabolism; to this end, EEG may be helpful in determining what degree of hypothermia is required. Caution is in order, however, since clinical trials of pentobarbital coma with pharmacologically induced burst suppression of EEG did not lead to improved outcome (Ward et al., 1985). Also important is the question of the optimal duration of posttraumatic hypothermia. We have shown a reduction in mortality in rats actively or passively cooled to a brain temperature of <35.5°C for 4 h after CCI, compared with normothermic controls. Clifton et al. (1991) demonstrated improved functional outcome in rats cooled to 30°C for 1 to 2 h after fluid-percussion injury compared to normothermic and hyperthermic controls. Future investigation aimed at determining the optimal duration and degree of hypothermia, using functional and morphological outcomes, would be valuable.
In conclusion, using a rat model of severe controlled cortical impact we have shown that (a) transient, mild, posttraumatic hypothermia reduces mortality; (b) brain temperature decreases by 1 to 2°C despite rectal temperature control, and this reduction in temperature is protective; and (c) EEG suppression is seen at brain temperatures associated with decreased mortality. Finally, the effect of local hypothermia during exposure of the brian for trauma should be considered in the interpretation of previous studies involving models of TBI, and brain and core temperature should be monitored and reported in future studies.
Footnotes
Acknowledgment:
The authors would like to thank Dr. Peter Safar for critical review of this manuscript, Dr. Edwin Nemoto for helpful suggestions, and Francie Siegfried for editorial assistance. This work was previously presented in part at the 12th Meeting of the Neurotrauma Society, November 12–13, 1994, Miami Beach, Florida. It was supported in part by a seed grant from the University of Pittsburgh Department of Anesthesiology and Critical Care Medicine and by Grant NS 30318 from the National Institute for Neurologic Disorders and Stroke. Dr. Kochanek is supported in part by an Established Investigator Grant from the Society of Critical Care Medicine. Dr. Clark is supported in part by a Fellowship Grant from the American Heart Association, Pennsylvania Affiliate.