
Abstract
Abstract
Introduction:
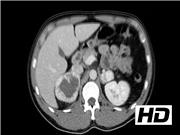
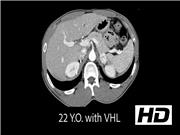
Von Hippel Lindau (VHL) disease has important genotype–phenotype correlations with respect to the risk of pheochromocytoma. The type II variant of VHL has a 50% lifetime incidence of pheochromocytoma and paraganglioma, which can present at a young age and bilaterally.1,2 Patients with VHL also have a 10% risk of developing pancreatic neuroendocrine tumors (PNETs). When >2 cm in the head or >3 cm in the tail or body, PNETs require surgical resection. 3
Materials and Methods:
We review several cases of patients with VHL at our institution treated for pheochromocytomas, including bilateral pheochromocytomas and extraadrenal paragangliomas, as well as PNET, to emphasize key clinical characteristics and optimal treatment.
Results:
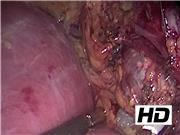
Three patients with VHL were treated surgically for pheochromocytoma and/or PNET. One patient underwent laparoscopic bilateral cortical-sparing adrenalectomy, and then required a pancreaticoduodenectomy to completely resect a PNET. Another patient was unable to undergo cortical-sparing adrenalectomy because of double pheochromocytomas present within the same adrenal gland. A third patient underwent laparoscopic bilateral cortical-sparing adrenalectomy, followed by retroperitoneoscopic resection of an extraadrenal paraganglioma.
Conclusions:
Type II variants of VHL have a high incidence of pheochromocytoma and paraganglioma. Patients can present at a young age with these manifestations. Surgery is the mainstay of treatment, and can often be completed in a minimally invasive manner. Cortical-sparing adrenalectomy is often possible but the feasibility is largely dependent on the size and location of the pheochromocytoma. PNETs are also common and can be monitored until they reach the size threshold for surgical intervention.
We have no commercial associations that would create a conflict of interest in connection with this video.
Runtime of video: 4 mins 59 secs
Get full access to this article
View all access options for this article.