
Abstract
Migraine attacks can be provoked by administration of nitroglycerin, suggesting a role for nitric oxide (NO). The fact that release of the neuropeptide CGRP from trigeminal sensory nerves occurs during the pain phase of migraine and that NO can augment transmitter release prompted us to study CGRP release from the
Introduction
The neuropeptide CGRP is present in dura mater and cranial arteries together with other neuropeptides, e.g. substance P and neurokinin A in nociceptive sensory fibres, that have been implicated in migraine (1, 2). CGRP is released from the trigeminal sensory nerves to the venous outflow in migraine patients during the headache phase (3). The reason why the nerves that innervate dura mater and vessels are activated during the migraine attack is unknown but the release of neuropeptides may contribute to sensitization of the nociceptors and probably also to neurogenic inflammation. This type of aseptic inflammation has been implicated in the development of migraine and other vascular headaches (4) and is characterized by vasodi-latation, plasma protein extravasation and mast cell degranulation (4). It can be induced experimentally in the dura mater by antidromic electrical stimulation of the trigeminal ganglion or by chemical stimulation by i.v. injection of capsaicin (5–7).
The anti-migraine drug sumatriptan has been shown to counteract the headache as well as the elevated CGRP levels in cranial venous blood of migraine patients (8). Moreover sumatriptan attenuates the release of CGRP after trigeminal nerve stimulation in cats and rats (8, 9) and the stimulated CGRP release from cultured trigeminal neurones (10).
Whilst the pathogenesis of migraine remains unclear, it has been shown that nitroglycerin, i.e. glyceryltri-nitrate (GTN) and other nitric oxide (NO) donors are able to induced headache in normal persons and migraine attacks in migraineurs (11). It is thought that NO by its powerful vasodilatation of the cephalic arteries induces headache, but activation/sensitization of central and peripheral nociceptive pathways may also be involved. The fact that NO has been shown to augment transmitter release in central neurones (12–16) and the trigeminal cerebrovascular system (17) could also be of importance to understanding migraine pathophysiology.
In the present work we studied the release of CGRP from the
Materials and methods
Skull preparation
Male guinea pigs (200-350 g; obtained from Charles River, Sulzfeld, Germany) were stunned by a C02 atmosphere and decapitated using a guillotine. The lower jaw was removed. The skin of the skull was cut along the midline and retracted from the calvarium, which was then divided along the sagittal suture into two halves by a hand-held saw. After the brain halves were carefully removed without lesioning the dura mater or the trigeminal ganglion, the skulls were washed for 30 min in HEPES buffer (room temperature, pH 7.4) containing (in mM): 10.0 HEPES; 120.0 NaCl; 5.0 KC1; 0.62 MgS04, 7 H20; 1.0 CaCl2, 2 H20 and 6.1 glucose. Then the skulls were mounted in a mould of modelling wax with the lateral side downwards and placed in a closed chamber situated on a water bath which maintained the chamber at 37C, relative humidity at 100%, and which was supplied with a steady supply of oxygen. The skull cavities were filled with HEPES buffer (pH 7.4; 37C) and left untouched for 10 min before the experiment started. The essence was to measure the CGRP, which was released from the dura when exposed to a HEPES buffer with or without high potassium concentration/capsaicin added. The skull, without touching the dura, was repeatedly and carefully emptied and filled with the same volume of various solutions as described below. Between two exposures to HEPES buffer with capsaicin/high potassium the skull was rinsed with HEPES buffer during two 5-min periods. The relevant samples were immediately transferred to test tubes placed on ice before they were stored at - 20C for later analysis of the CGRP contents.
At the beginning of each experiment the skull was filled with HEPES buffer, which after 5 min was removed and the skull was refilled with the same volume of HEPES buffer. After an additional 5 min the second sample, the basal level, was collected. During the next periods the skull was repeatedly stimulated with HEPES buffers containing various compounds as indicated.
Compounds
The following drugs were used: capsaicin (Sigma Chemical Co., St Louis, MO), sumatriptan succinate (Glaxo Wellcome, Brondby, Denmark), GTN (The Dispensary at Glostrup Hospital), sodium nitroprusside (SNP) (Sigma), S-nitroso-N-acetylpenicillamine (SNAP) (Calbiochem, La Jolla, CA) and methiothepin (Sigma). Capsaicin (10 mM) was dissolved in ethanol (48%) and diluted with HEPES buffer. Potassium solutions were made by replacing an equal concentration of NaCl with KC1 in the HEPES buffer. The required amount of NO donor was weighted in test tubes and dissolved in the HEPES buffer immediately before use. In solutions of SNP>30mM, SNP replaced an equal concentration of NaCl in the HEPES buffer. Sumatriptan and methiothepin were dissolved in HEPES buffer.
Assay of CGRP
The samples from individual skulls were thawed at room temperature and then used directly to determine the CGRP content using a commercial Rat CGRP Enzyme Immunometric Assay (EIA) Kit (SPIbio Company, Paris, France) on a 96-well plate. This assay is based on a double-antibody sandwich technique. The wells of the microtitre plate are coated with a monoclonal antibody specific for CGRP. An acetylcholinesterase (AchE)-Fab’ conjugate, which binds selectively to a different epitope on the CGRP molecule, is also added to the wells. The two antibodies will form a sandwich by binding on different parts of the rat CGRP molecule. AchE catalyses the formation of a yellow dye from the reagents, which is measured photometrically on a microplate reader. Standard curves were made from CGRP dissolved in the HEPES buffer and included on each 96-well plate. The minimum detection limit of the assay is approximately 1 pg/ml. None of the tested drugs interfered with the present assay.
Statistical analysis
All data are expressed as means
Results
Stimulated release of CGRP from the dura mater by capsaicin and potassium

Release of CGRP from the
CGRP release is calcium-dependent
Figure 2 shows the CGRP concentration of the skull fluid when the skull preparation was stimulated twice, during periods of 5 min, with 50 mM potassium and finally with 1

Release of CGRP from the
Effect of sumatriptan on CGRP release
The ability of sumatriptan to interfere with CGRP release was tested by adding 1, 10 or 50
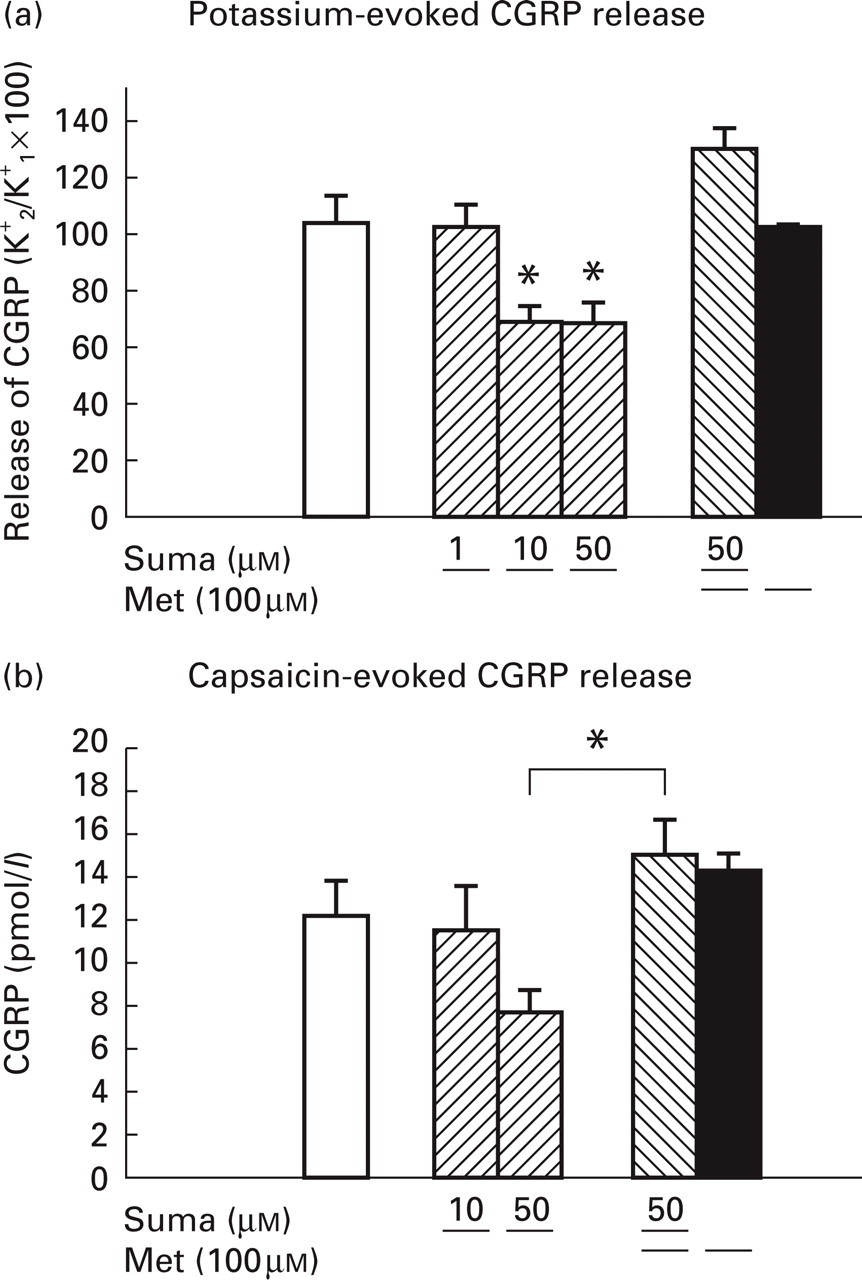
Effect of the 5-HTj-receptor agonist sumatriptan (suma) and the 5-HTj-receptor antagonist methiothepin (met) on potassium- or capsaicin-stimulated CGRP release from the
The effect of sumatriptan on capsaicin-evoked release was tested during four consecutive stimulations with increasing concentrations of capsaicin (as shown in Fig. la) with sumatriptan added. Before each stimulation period the skull was exposed to control HEPES buffer for 2X5 min, of which the latter contained sumatriptan. Five trials were performed and Fig. 3b show the results from using the highest concentration of capsaicin (1
Nitric oxide donors
The three different NO donors GTN, SNP and SNAP were used to evaluate the effect of NO. An augmentation of CGRP release from dura mater had been anticipated, but this was not the case. The donors were added to the control buffer in increasing concentrations during three consecutive exposures (Table 1). GTN from 0.01 to 1
Effect of nitric oxide donors on CGRP release from dura mater
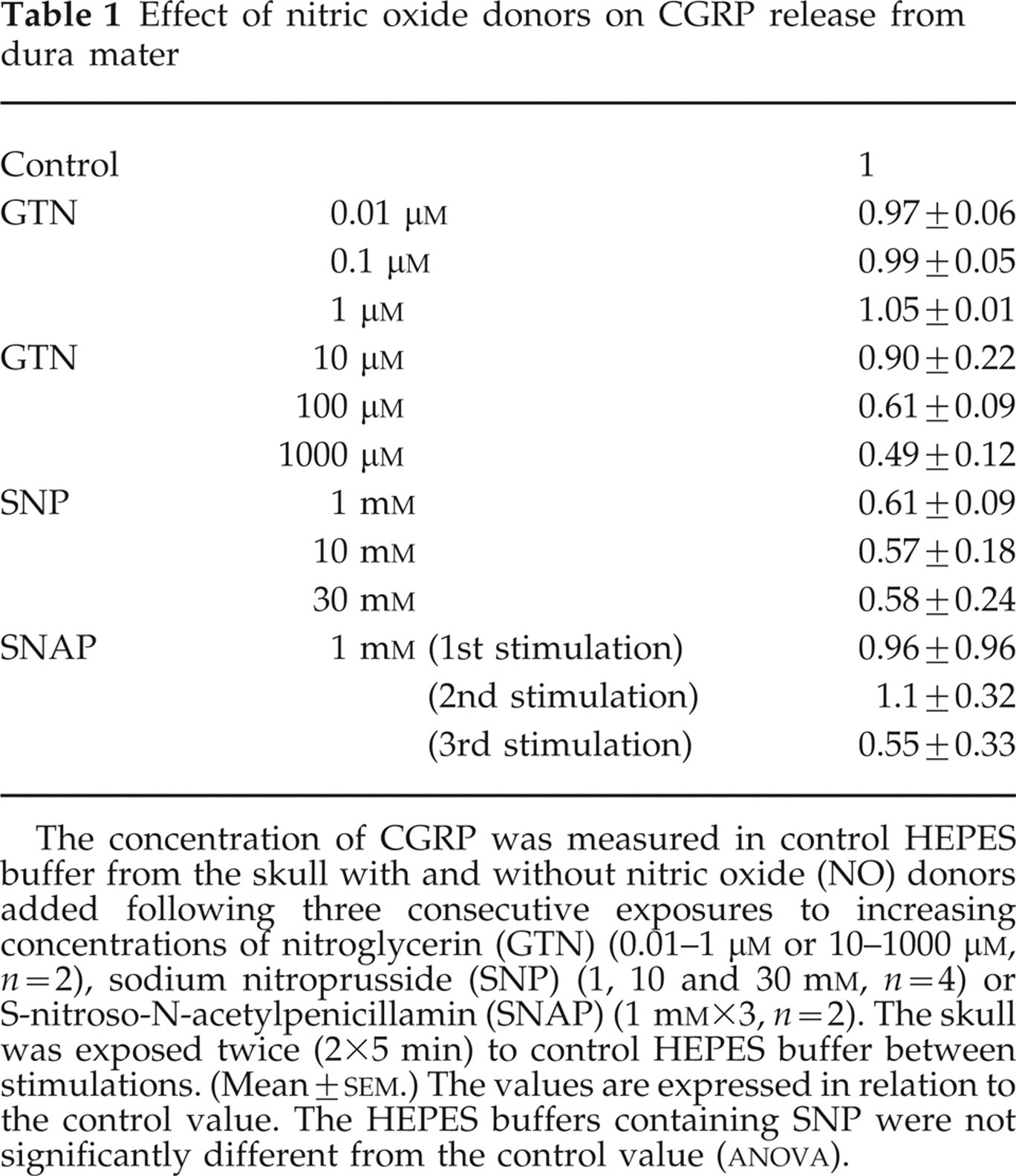
The concentration of CGRP was measured in control HEPES buffer from the skull with and without nitric oxide (NO) donors added following three consecutive exposures to increasing concentrations of nitroglycerin (GTN) (0.01-1
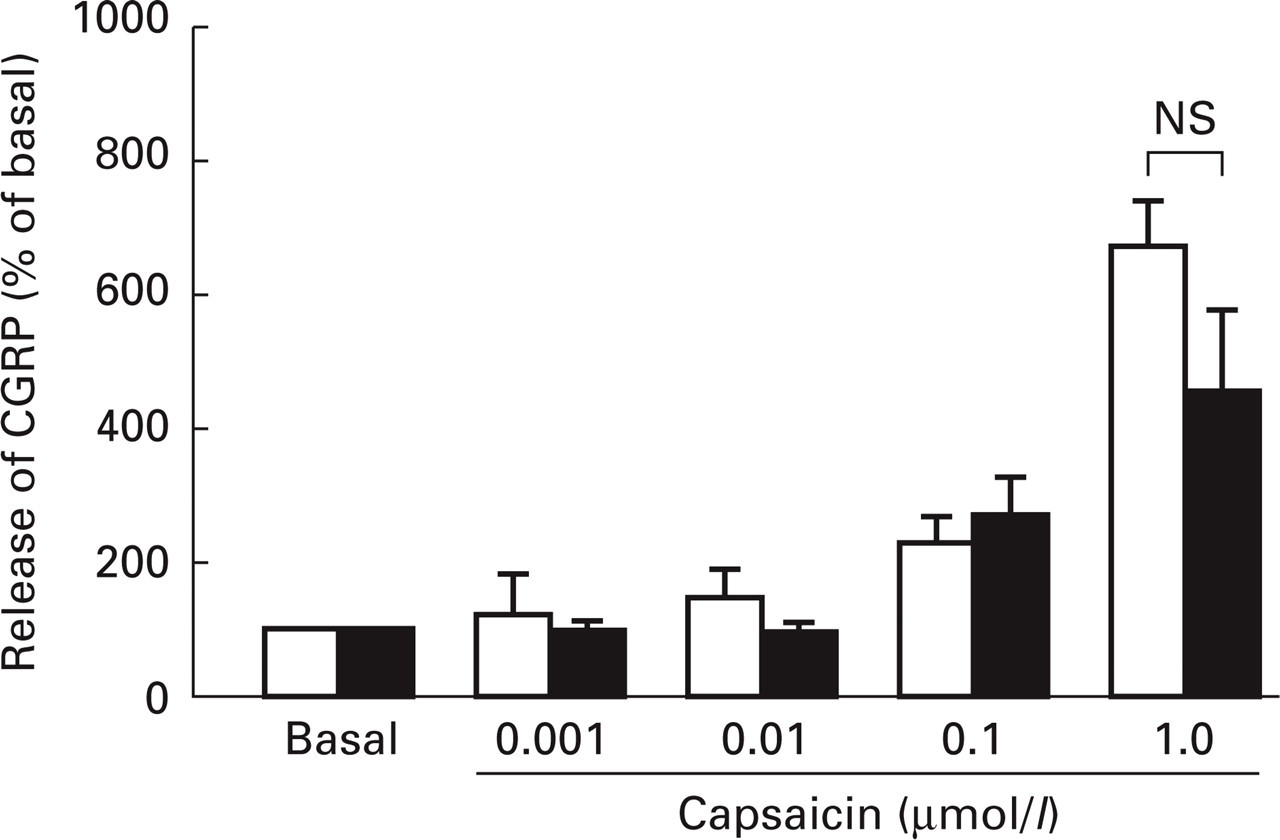
Effect of the nitric oxide donor nitroglycerine (GTN) on the capsaicin-stimulated release of CGRP from dura mater. The skull was stimulated four consecutive times with increasing concentrations of capsaicin with (?. n = 6) or without (?, n = 10) GTN (1
Discussion
The present study demonstrates that the intact dura mater encephali maintained in a skull preparation
Sumatriptan attenuates dural CGRP release
Sumatriptan was able to attenuate both potassium- and capsaicin-stimulated CGRP release in a dose-dependent manner. The findings are in agreement with those of Durham & Russo (10) who, in cultured trigeminal neurones, showed that sumatriptan at the same concentrations as used here inhibited the release of CGRP caused by potassium or a mixture of inflammatory mediators at low pH. The mechanism by which sumatriptan acts is not fully understood, but it involves 5-HTj-receptor subtypes. Sumatriptan, originally developed as a vasoconstrictor selectively acting as an agonist at 5-HT1B/1D-receptors, also inhibits CGRP release from trigeminal sensory nerves
The reason why 50
NO does not augment dural CGRP release
It is well established that NO by itself or together with other stimuli augments the release of neurotransmitters from brain slices, e.g. aspartate (12) and dopamine from striatum (13, 29, 30) and noradrenaline and glutamate from hippocampus (14, 31, 32). NO is believed to influence transmitter release via activation of guanylate cyclase, which leads to the formation of cGMP. The ensuing activation of cGMP-dependent protein kinase phosphorylates proteins that are involved in the neurotransmitter release process or in the activation, e.g. calcium channels. In the current study it was shown that none of the nitric oxide donors GTN, SNP or SNAP caused a release of CGRP from dura mater. In fact, there was a tendency towards a lowering of the release (Table 1), but the effect was not significant for SNP and for GTN and SNAP significance was not tested due to the limited number of experiments. In contrast to these observations, another study (33) indicated that SNP can stimulate CGRP release from dorsal horn slices from rat spinal cord. They, however, added SNP (30 mM) directly to the buffer, and it cannot be excluded that other mechanisms, e.g. hyperosmotic effects, could play a role (34). Also Dymshitz & Vasko (35) found in cultured DRG neurones that SNP was able to increase the release of CGRP provoked by capsaicin. However, only SNP, but not two other NO-donating compounds, SNAP and 3-morpholinosydnonimine, showed an effect, which again raises the possibility that NO is not responsible for the effect of SNP.
An explanation of why NO can augment the release of small transmitter substances while not affecting the release of CGRP could be the different storage conditions. Neuropeptides are presynaptically stored in large dense-cored vesicles, while the small neurotransmitters reside in clear small vesicles. While the release from both types of vesicles occurs by calcium-mediated exocytose, there are differences in the calcium requirement (36) and probably also in the cGMP-dependent processes.
There are several reasons why NO was expected to alter the release of CGRP. First, infusion of GTN, en exogenous source of NO, can induce migraine attacks, which are similar to spontaneous attacks (37). Second, GTN and SNP cause vasodilatation in the cerebral circulation in cats (17) and isolated rings of rat aorta (38). These effects are blocked by the CGRP antagonist CGRP8_37, suggesting that CGRP is responsible for at least a part of the vasodilatory effect of NO. In the study by Thomsen et al. (37) the concentrations of CGRP were not measured and it is therefore not known whether these migraine patients without aura experience an increased release of CGRP after GTN infusion. Increased levels of CGRP after GTN infusion have been shown in cluster headache patients (39), but only when the patient experienced a peak headache after the GTN infusion. Thus, it seems that GTN does not directly cause release of CGRP but triggers a cascade of events that subsequently leads to the release of CGRP. In contrast, in the two other studies (17, 38) performed
In conclusion, we have found that potassium depolarization and capsaicin, but not nitric oxide donors, cause the release of CGRP in the dura mater of isolated guinea pig skulls and that this release can be attenuated by the
Footnotes
Acknowledgements
We thank Dr Andrea Ebersberger for her valuable help in setting up the present method.