
Abstract
The forearm vascular response to nitric oxide (NO) and calcitonin gene-related peptide (CGRP) was investigated in 10 migraine patients and 10 matched control subjects. Changes in forearm blood flow (FBF) during intrabrachial infusion of: (i) serotonin (releasing endogenous NO), (ii) sodium nitroprusside (SNP, exogenous NO-donor), and (iii) CGRP were measured using venous occlusion plethysmography. Flow-mediated dilation (FMD) of the brachial artery, a measure for the endogenous release of NO reactive to occlusion, was measured using ultrasound and expressed as percentage change vs. baseline diameter. FBF ratio (i.e. FBF in the infused over the control arm) at baseline (1.1 ± 0.1) did not differ between both populations. Serotonin, SNP and CGRP induced a dose-dependent increase (P < 0.001) in FBF ratio in controls (to 2.8 ± 0.3, 6.7 ± 1.4 and 6.9 ± 1.2 at the highest dose, respectively) and migraineurs (2.5 ± 0.4, 5.6 ± 0.8 and 6.5 ± 1.3, respectively); these ratios did not differ between both groups. FMD was comparable in control subjects (5.8 ± 1%) and migraine patients (5.2 ± 1%). Based on the forearm vascular response to NO and CGRP, migraine patients do not display generalized changes in vascular function.
Introduction
The pathophysiology of migraine has been much debated over the past decades. The idea that migraine is due to an inappropriate vasodilation of cranial blood vessels, as postulated by Graham and Wolff (1), has shifted towards a neurovascular theory regarding migraine as a complex disorder of the trigeminovascular system (2). Although this theory tends to highlight the activation of pain-producing perivascular sensory nerves more than the vascular changes accompanied by it, the concept of cranial vasodilation during a migraine attack is generally accepted. However, the trigger initiating the vascular and neuronal changes in migraine headache remains unknown.
One hypothesis, which has mainly been advocated by Olesen and coworkers (3–5), points to endothelial-derived relaxing factor or nitric oxide (NO) as the triggering molecule for migraine attacks. They demonstrated that, compared with healthy subjects, migraine patients suffer a more intense and more migraine-like headache after the administration of organic nitrates, acting as NO-donors, and histamine, stimulating the endogenous release of NO by the activation of endothelial H1-receptors (6, 7). Furthermore, an increased vasodilation of the middle cerebral artery was observed in migraine patients compared with control subjects after the administration of organic nitrates (8). In addition, nitric oxide synthase inhibition with L-NG-monomethyl-arginine (L-NMMA, 546C88) was shown to be a promising therapeutic principle in acute migraine (9). Based on these data, they concluded that migraine patients display an arterial supersensitivity to NO (8).
Apart from NO, calcitonin gene-related peptide (CGRP) has even more convincingly been shown to be a key player in the pathophysiology of migraine. Indeed, Goadsby and colleagues (10) provided evidence that during migraine headache the trigeminovascular system is turned on and CGRP, a potent vasodilator neuropeptide, is released into the cranial circulation. Moreover, successful treatment of acute migraine with sumatriptan normalizes cranial CGRP levels (11). Based on animal data, NO seems to play a regulatory role in neuropeptide release from perivascular sensory nerves (12, 13). Consequently, supersensitivity to NO might not only translate into an excessive vasodilation by the direct action of NO on the blood vessel wall of migraine patients, but also translate into an excessive release of CGRP from perivascular nerves induced by NO. Alternatively, we recently demonstrated that CGRP-induced vasodilation is, at least in part, dependent on the release of NO (14). Taken together, these data support an important role for both CGRP and NO in the vascular changes observed during an acute migraine attack.
The present study aims at a better understanding of the concept of supersensitivity to endogenous and exogenous NO and the vascular reactivity to CGRP in migraine patients.
Subjects and methods
Subjects
Ten migraine patients (as defined by the International Headache Society migraine criteria (15)) were compared with 10 healthy control subjects. Both groups were matched for gender, age, body mass index, blood pressure and cholesterol. Migraine patients were recruited from the Neurology Out-patient clinic and control subjects from among hospital and laboratory staff. Control subjects with first-degree relatives suffering from migraine were excluded. Baseline characteristics of both groups are presented in Table 1. Based on medical history, physical examination and routine laboratory tests, all participants were in good health. Major exclusion criteria were: smoking, history of cardiovascular disease, arterial hypertension (blood pressure > 95/160 mmHg), diabetes mellitus and hyperlipidaemia (total cholesterol > 6.4 mmol/l). Except for oral contraceptives, no regular medication was allowed. After approval by the Ethics Committee of the Academic Hospital of Maastricht, this research was carried out in accordance with the Declaration of Helsinki. All subjects gave written informed consent.
Baseline characteristics

Baseline characteristics recorded at screening or, when indicated with (∗) during the second study period (i.e. at the right arm using ultrasound).
Data presented as mean ± SEM. P-values by unpaired Student's t-test. AU, Arbitrary units.
Study design
After screening, eligible subjects were invited to return to the hospital for two study periods. Before each period, they were asked not to use any drugs for at least 3 days and to abstain from caffeine- or alcohol-containing beverages for at least 12 h. All measurements were performed by one investigator in a quiet, temperature-controlled room and with the subject lying comfortably in the supine position.
First study period
During this period, forearm vascular reactivity to serotonin (inducing an endogenous release of NO), sodium nitroprusside (SNP, an exogenous NO-donor) and CGRP was investigated invasively using the ‘perfused forearm model’ (16). After local anaesthesia (lidocaine 2%; B. Braun, Melsungen, Germany), the brachial artery of the non-dominant arm was cannulated with a 20-G catheter (Arterial cannula; Ohmeda, Hatfield, UK) for intra-arterial drug infusion (Graseby Syringe Pump 3200; Graseby Medical Ltd, Watford, UK) and invasive blood pressure monitoring (monitor type 78201B; Hewlett Packard GmbH, Bad Homburg, Germany). Forearm arterial blood flow (FBF) was measured by ECG-triggered venous occlusion plethysmography using mercury-in-Silastic strain gauges (Periflow; Janssen Scientific Instruments, Beerse, Belgium). Every five heart beats FBF was recorded simultaneously at both arms for three beats. Upper arm cuffs were intermittently and simultaneously inflated to a pressure of 45 mmHg. Heart rate, systolic and diastolic blood pressure were recorded with every FBF measurement.
After instrumentation, an equilibration period of at least 30 min was allowed. After equilibration, basal FBF was recorded for 5 min during infusion of saline (NaCl 0.9%). Baseline measurements were followed by incremental infusions of serotonin (1, 3.3 and 10 ng min−1 dl−1 forearm), SNP (0.04, 0.2 and 1 µg min−1 dl−1 forearm) and CGRP (3, 10 and 30 ng min−1 dl−1 forearm). Each dose of serotonin and SNP was infused for 6 min; each dose of CGRP for 12 min. The mean of the measurements made during the final 2 min of each infusion period (i.e. at steady state) was used in the analysis. After the highest dose of serotonin and SNP, saline was infused for at least 15 min to allow FBF to return to baseline. The rate of all intra-arterial infusions was kept constant at 0.1 ml min−1 dl−1 forearm volume and the dosages were normalized to forearm volume measured by water displacement. Before each experiment, all solutions were freshly prepared from commercially available vials (human αCGRP and serotonin purchased from Clinalfa, Läufelfingen, Switzerland) or from sterile stock solutions (SNP, provided by the hospital pharmacy). SNP was protected from light. In the analysis, vascular responses to serotonin, SNP and CGRP were expressed as the ratio in FBF of the infused over the control arm (FBF ratio).
Second study period
First, using ultrasound, flow-mediated dilation (FMD) of the brachial artery was assessed as a measure of endothelial NO release. Therefore, brachial artery diameter of the right arm was measured with a vessel wall movement detector system (Wall Track System®; Pie Medical, Maastricht, the Netherlands) (17). This system consists of an ultrasound device (Scanner 350; Pie Medical) equipped with a 7.5-MHz linear-array transducer connected to a data acquisition and processing unit. After 15 min of supine rest, the brachial artery was scanned over a longitudinal section about 5 cm proximal to the elbow. Both the position of the arm and the transducer remained fixed throughout the experiment. Baseline measurements of brachial artery diameter and velocity profiles were followed by inflation of a blood pressure cuff placed around the forearm to a pressure of 240 mmHg for 5 min. Thereafter, the cuff was released and the maximum velocity profile during the first 15 s was recorded, followed by a diameter measurement after 45 s of deflation. Finally, from 1 until 5 min after deflation brachial artery diameter was measured every 30 s. The velocity profile was measured with a pulsed-Doppler signal at a 70° angle to the vessel and a sampling volume (1.2 mm wide) in the centre of the artery. For each cardiac cycle the mean flow velocity was calculated electronically by multiplying the area under the velocity profile curve (corrected for angle) by the heart rate.
In the analysis the diameter change caused by FMD was expressed as the percentage change relative to the mean baseline diameter. Basal blood flow (ml/min) was calculated by multiplying the basal mean flow velocity by the cross-sectional area of the artery at rest. Maximal blood flow upon reperfusion was calculated by multiplying the mean flow velocity by the cross-sectional area of the artery 45 s after deflation of the cuff and was expressed as percentage change relative to the basal blood flow. From cuff deflation until 5 min after reperfusion the area under the time–diameter curve (AUC) was calculated.
Second, maximal post-ischaemic FBF of the left arm was measured using venous occlusion plethysmography. After 3 min of baseline FBF measurements, arterial occlusion of the upper arm was obtained for 13 min by inflating a blood pressure cuff to a pressure of 240 mmHg. During the last minute of ischaemia, dynamic exercise (10–20 hand contractions) was added. After release of the cuff, FBF was measured for 2 min. Both the maximum FBF and the time to reach maximum FBF were used in the analysis. During these measurements blood pressure was recorded non-invasively at the right upper arm using an oscillometric device (Dinamap®; Critikon Inc., FL, USA). Three blood pressure measurements were performed both at baseline and during reactive hyperaemia. Using maximal FBF, minimal forearm vascular resistance [defined as mean arterial pressure (MAP)/FBF] was calculated as a functional correlate of the morphological status of the forearm resistance vessels and expressed as arbitrary units (AU) (18).
Statistical analysis
Comparison of baseline characteristics and non-serial parameters between both groups was done with the unpaired Student's t-test. The haemodynamic responses [FBF ratio, MAP and heart rate (HR)] to incremental infusions of serotonin, SNP and CGRP within each group of subjects as well as the difference in forearm vascular response (FBF ratio) between migraine patients and control subjects, were evaluated by
Results
Baseline characteristics of migraine patients and control subjects did not differ (Table 1). In both groups two subjects were on oral contraceptives. Migraine patients had been suffering from migraine for an average of 13 ± 1 years and had an attack frequency of 2.2 ± 0.4 per month. In most cases migraine headache was one-sided (n = 8), pulsatile in nature (n = 7) and accompanied by nausea or vomiting (n = 9) and photo-/phonophobia (n = 10). Only one patient regularly experienced migraine with aura. As an abortive drug sumatriptan (n = 7) was most frequently used.
First study period
Baseline FBF of the infused arm and control arm was similar within each group and did not differ between migraine patients and control subjects (1.8 ± 0.3 vs. 1.7 ± 0.1 ml min−1 dl−1 forearm, respectively). A dose-dependent increase (P < 0.001) in FBF ratio was induced by serotonin, SNP and CGRP (Fig. 1) both in control subjects [from 1.1 ± 0.1 at baseline, to 2.8 ± 0.3 (serotonin), 6.7 ± 1.4 (SNP) and 6.9 ± 1.2 (CGRP) at the highest dose, respectively] and in migraine patients (from 1.1 ± 0.1 at baseline to 2.5 ± 0.4, 5.6 ± 0.8 and 6.5 ± 1.3, respectively). There were no significant differences in the responses to any of the drugs between both populations.

Comparison of forearm blood flow ratio (FBF ratio) between migraine patients (▪, solid line) and control subjects (□, dashed line) during the intrabrachial infusion of serotonin (top), sodium nitroprusside (SNP, middle) and calcitonin gene-related peptide (CGRP, bottom). FBF ratio: ratio in forearm blood flow in the infused arm over the control arm. Data are presented as mean ± SEM, n = 10 in each group. (NS, Not statistically significant.)
Second study period
FMD of the brachial artery (Fig. 2) averaged 5.2 ± 1.0% in migraine patients compared with 5.8 ± 1.0% in control subjects (P = 0.64). FMD expressed as AUC did not differ between migraineurs and controls (370 ± 150 µm/min vs. 570 ± 147 µm/min, respectively, P = 0.35).
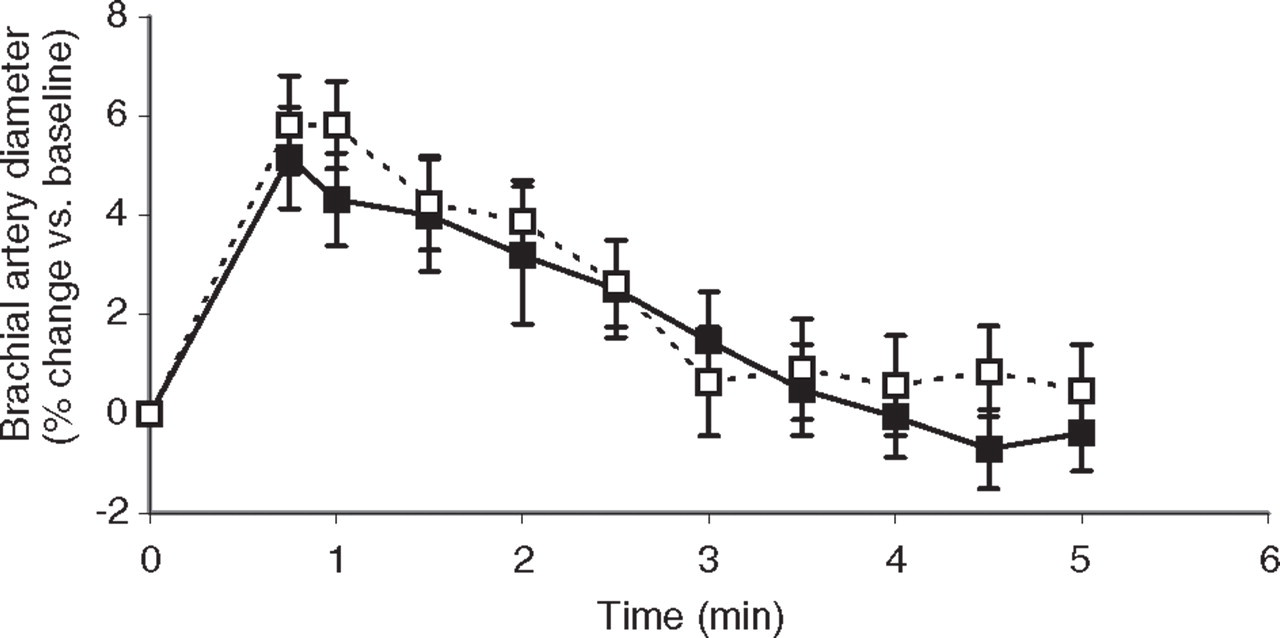
Comparison of flow-mediated dilation between migraine patients (▪, solid line) and control subjects (□, dashed line). Brachial artery diameter before occlusion (t = 0) and from 45 s until 5 min after reperfusion. Data presented as mean ± SEM, n = 10 in each group.
Maximal post-ischaemic FBF (Fig. 3) in migraine patients and control subjects did not differ (P = 0.42) with respect to basal FBF (2.2 ± 0.3 and 1.9 ± 0.2 ml min−1 dl−1 forearm, respectively) or maximal post-ischaemic FBF (15.0 ± 1.4 and 13.0 ± 1.8 ml min−1 dl−1 forearm, respectively). Furthermore, forearm vascular resistance at baseline (44.9 ± 5.4 vs. 48.0 ± 5.2 AU, respectively) and upon reperfusion (6.2 ± 0.6 vs. 7.5 ± 0.8 AU, respectively) was comparable (P = 0.68 and 0.22, respectively) between both groups. Time to reach maximal FBF or minimal forearm vascular resistance after reperfusion was similar (P = 0.62) for migraine patients and control subjects (86 ± 8 and 81 ± 7 s, respectively).
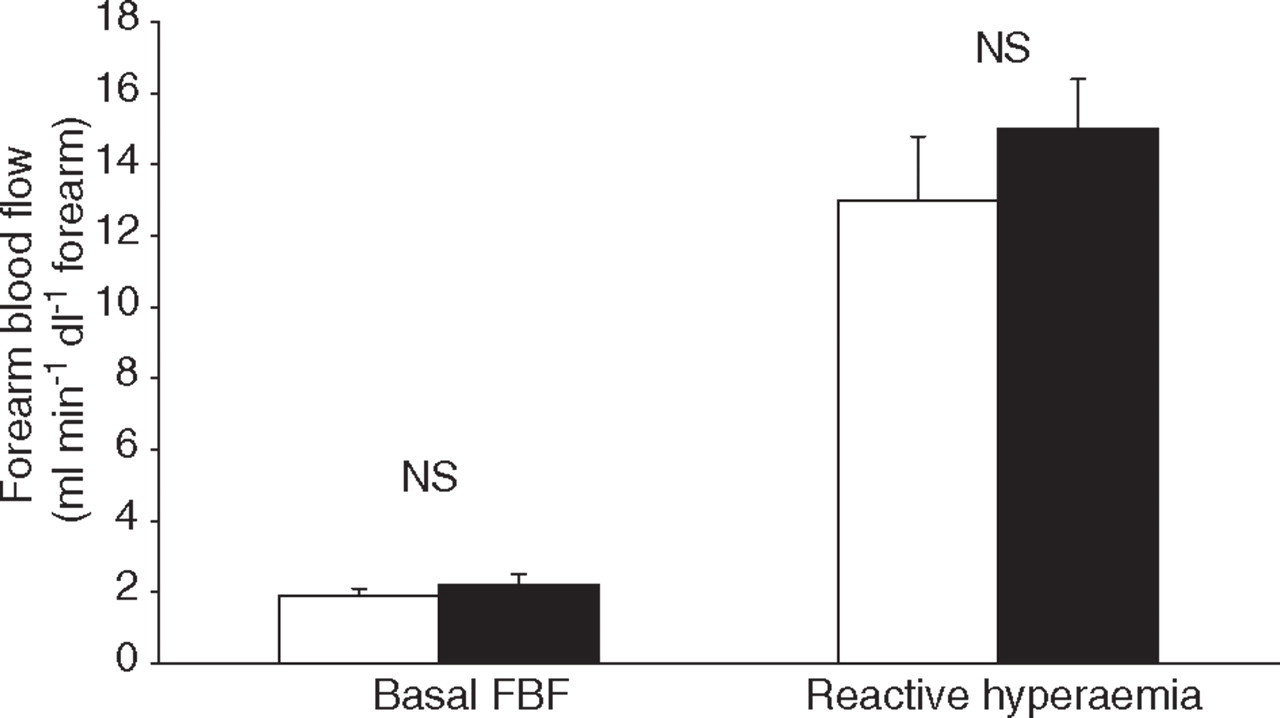
Comparison of baseline and maximal post-ischaemic forearm blood flow between migraine patients (▪) and control subjects (□). Data are presented as mean ± SEM, n = 10 in each group. (NS, Not statistically significant.)
Discussion
In vivo vascular research in migraine patients has been hampered by limited access to the cranial circulation. However, as migraine is associated with an increased incidence of Raynaud's syndrome (19) and with angina pectoris due to coronary artery vasospasm (i.e. Prinzmetal's angina) (20–22), it has been suggested to be part of a generalized vascular disorder (20). This was substantiated by the observation of an increased interictal peripheral arterial stiffness in migraineurs (23). Therefore, peripheral vascular research in migraine patients might offer an alternative approach to improve our understanding of the cranial vascular changes associated with it.
The main finding of the present study is that neither for NO nor for CGRP could an increased sensitivity be demonstrated for the resistance vessels of the human forearm in migraine patients. As minimal forearm vascular resistance was similar in both groups, there is no evidence suggesting structural differences of the resistance vessels either. In addition, flow-mediated dilation did not differ between migraine patients and matched control subjects, arguing against a supersensitivity to NO for conduit arteries as well. In summary, these data do not support the hypothesis of a systemic vascular supersensitivity to NO in migraine patients.
In the present study, the NO hypothesis of migraine was investigated by measuring the forearm vascular response to both a pharmacological and a physiological-induced endogenous release of NO and to the administration of an exogenous NO-donor. The vascular response to the pharmacological release of NO was investigated by the intra-arterial infusion of serotonin. It has previously been shown that at the dosages used in the present study, serotonin-induced vasodilation of the human forearm is endothelium dependent and accompanied by the release of NO (24, 25). The vascular sensitivity to an exogenous source of NO was investigated by the infusion of SNP. Like nitrates, SNP is known to induce an endothelium-independent vasodilation by the release of NO upon its degradation (26). As both endothelium-dependent and endothelium-independent NO-induced vasodilation were comparable between migraine patients and non-migraineurs, these findings argue against a vascular smooth muscle cell supersensitivity to NO in migraine patients. However, as venous occlusion plethysmography measures flow changes only at the level of resistance vessels, these findings do not allow to conclude the absence of NO supersensitivity at the level of conduit arteries. Therefore, flow-mediated dilation of the brachial artery was investigated. This non-invasive approach has been used frequently to demonstrate changes in endothelial function (27–30). It has been shown that in human peripheral conduit arteries vasodilation reactive to occlusion results from endothelial NO release in response to increased shear stress (31). In the present study, neither the peak increase in brachial artery diameter nor the duration of flow-mediated dilation was increased in migraine patients compared with control subjects. Therefore, at the level of the conduit artery of the human forearm our data do not support the hypothesis of supersensitivity to NO either.
What could be the explanation for the fact that the findings of the present study do not support the hypothesis of arterial supersensitivity in migraine patients as formulated by other investigators? First, different vascular territories are involved. The forearm vasculature, which was investigated in the present study, might well behave differently from the cranial vasculature, which is the territory of interest in migraine. Differences in receptor distribution and innervation could obviously result in a different pharmacology for both territories. Second, although investigated thoroughly, the earlier indications of increased arterial sensitivity to NO remain indirect (8). Vasodilation of large intracranial arteries can only be estimated by simultaneous transcranial Doppler (TCD) and single positron emission computed tomography (SPECT) measurements. Based on the observation that total cerebral blood flow (measured with SPECT) is not changed during nitroglycerine-induced headache while blood velocity (measured with TCD) is decreased, this rather suggests vasodilation than providing evidence for it. Finally, as only small numbers of subjects were used due to the complexity, duration and cost of the experiments, it can not be excluded that the present study was insufficiently powered to detect differences between migraineurs and control subjects. However, by applying the same methodological approach to comparable small numbers of subjects, changed peripheral vascular responses have repeatedly been reported before in other diseases (32–34).
Apart from the vascular smooth muscle cell, the endothelium has also been suggested to be a likely site of the primary abnormality in migraine (35). However, the findings of the present study indicate that endothelial function is unaffected. The preservation of the vasodilation resulting from both serotonin and shear-stress induced endothelial release of NO (as measured with FMD) makes it unlikely for migraine to be part of a generalized endotheliopathy. Of course, a local, cerebral endothelial abnormality cannot be excluded. It has also been hypothesized that one of the early steps in the generation of migraine headache is the activation of endothelial 5-HT2B-receptors on meningeal blood vessels inducing the release of NO (36, 37). Although serotonin does induce the endothelial release of NO (25), there is no evidence indicating an increased release in migraine patients at present.
Although we could not demonstrate an increased sensitivity to NO in terms of an increased vascular response, the migraine-like characteristics of headache induced by nitrates do make NO a likely candidate to be involved in the pathophysiology of migraine. One might wonder whether supersensitivity of migraine patients to NO should be interpreted in terms of an increased sensitivity of perivascular nerves to NO instead of an increased sensitivity of the blood vessel wall itself. If so, the low density of CGRP-containing perivascular nerve fibres in the peripheral circulation compared with the cranial circulation (38) may explain the lack of a difference in nitroprusside-induced changes in FBF between migraine patients and control subjects in the present study. According to this theory, in migraine patients NO might result in an overactivation of the trigeminovascular system and an excessive release of CGRP involved in neurogenic inflammation (6, 39). First, this hypothesis is supported by the observation that during a genuine migraine attack plasma levels of CGRP are increased in the cranial circulation (10). Although in healthy volunteers, nitroglycerine-induced headache was not accompanied by an increase in CGRP plasma levels (40), this does not exclude a role for trigeminal supersensitivity to NO in migraine patients. Second, nitrates induce typical migraine headache in migraine patients only after a surprisingly long latency period, suggesting that NO is involved in the initiation of a slow pathological process which eventually leads to the attack (5). The pathological process causing this delay could well be the activation of the trigeminovascular system. In addition, although nitrate-induced headache is accompanied by vasodilation, because of the delay between the fast vasodilation and the slow onset of migraine-like headache induced by nitrates, vasodilation as such is unlikely to be the cause of the headache. Finally, it should be noted that NO is involved in nociceptive processing and was shown to mediate vascular pain (41, 42). Thus, it cannot be exclude that an increased sensitivity to NO in migraine patients merely reflects a lower threshold for the noxious effects of this molecule compared with non-migraineurs.
In summary, neither peripheral resistance vessels nor conduit arteries display any differences in their vascular response to NO and the release of it between migraine patients and control subjects. Likewise, the peripheral vascular response to CGRP is not altered in migraine patients. In addition to the lack of a generalized difference in vascular function, no structural difference at the level of resistance vessels is present in migraine either. As these data do not exclude intracranial functional or structural vascular changes, it remains to be established whether trigeminovascular supersensitivity to NO contributes to the pathogenesis of migraine.
Footnotes
Acknowledgements
The expert technical assistance of Mr J. Willigers is greatly appreciated.