
Abstract
Previously, we have shown that the selective mitochondrial ATP-sensitive potassium (mitoKATP) channel opener BMS-191095 (BMS) induces neuronal preconditioning (PC); however, the exact mechanism of BMS-induced neuroprotection remains unclear. In this study, we have identified key components of the cascade resulting in delayed neuronal PC with BMS using isolated rat brain mitochondria and primary cultures of rat cortical neurons. BMS depolarized isolated mitochondria without an increase in reactive oxygen species (ROS) generation and induced rapid phosphorylation of Akt and glycogen synthase kinase-3β. Long-term (3 days) treatment of neurons with BMS resulted in sustained mitochondrial depolarization, decreased basal ROS generation, and elevated ATP levels. This treatment also elicited almost complete protection against glutamate excitotoxicity, which could be abolished using the phosphoinositide 3-kinase (PI3K) inhibitor wortmannin, but not with the superoxide dismutase (SOD) mimetic M40401. Long-term BMS treatment induced a PI3K-dependent increase in the expression and activity of catalase without affecting manganese SOD and copper/zinc-dependent SOD. Finally, the catalase inhibitor 3-aminotriazole dose-dependently antagonized the neuroprotective effect of BMS-induced PC. In summary, BMS depolarizes mitochondria without ROS generation, activates the PI3K—Akt pathway, improves ATP content, and increases catalase expression. These mechanisms appear to play important roles in the neuroprotective effect of BMS.
Introduction
Stroke is a leading cause of death and permanent disability worldwide. Therefore, strategies to prevent or decrease the damage caused by prolonged ischemia are of great importance. One of the promising protective approaches is preconditioning (PC). This ubiquitous natural phenomenon was first described in the brain more than 40 years ago by Dahl and Balfour (1964) and was defined by Murry et al (1986) as an increased tolerance induced by a subtoxic stimulus (e.g., a short period of anoxia) against a subsequent, otherwise lethal insult such as prolonged ischemia. Preconditioning can be induced by several different approaches, including the selective pharmacological activation of the ATP-sensitive potassium (KATP) channel in the inner mitochondrial membrane (mitoKATP; reviewed by Busija et al, 2004). Diazoxide (DZ) is the prototypical activator of the potassium channel and it has been used in most pharmacological PC studies. However, DZ is known to have effects unrelated to the mitoKATP channels, such as the inhibition of the mitochondrial electron transport chain (Andrukhiv et al, 2006; Kis et al, 2003; Minners et al, 2007). Furthermore, in higher doses, DZ also opens plasma membrane KATP channels (Garlid et al, 1996). Therefore, new and more selective activators are needed to further explore the protection mediated by mitoKATP channels. BMS-191095 (BMS) is a novel opener of the mitoKATP channels and it has shown itself to be more potent and selective than DZ (Grover et al, 2001; Mayanagi et al, 2007). The effective dose of this compound for PC has no vasoactivity and does not influence whole-cell K+ currents or plasma membrane potential. In previous reports from our laboratory, we have shown that acute and delayed PC with BMS protected rat cortical neuronal cultures in vitro against toxic stimuli (Kis et al, 2004) and induced delayed tolerance against transient middle cerebral artery occlusion in vivo (Mayanagi et al, 2007). In addition, BMS PC abolished the reactive oxygen species (ROS) surge that was induced by glutamate exposure (Kis et al, 2004). We have also reported that the application of BMS depolarized isolated piglet brain mitochondria without induction of ROS generation (Busija et al, 2005). BMS was also shown to induce both acute and delayed cardioprotection against left anterior descending coronary artery occlusion in the rat heart (Gross et al, 2007; Wang et al, 2007b) by the activation of the phosphoinositide 3-kinase (PI3K) signaling pathway. Despite these results, the exact mechanism of BMS-induced neuroprotection has not yet been clarified. Therefore, further studies are needed to explore the details of immediate and delayed PC induction by BMS.
The purpose of this study was to identify key components of the cytoprotective cascade activated by BMS, ultimately resulting in delayed neuronal PC. We analyzed the immediate and long-term effects of BMS on isolated rat mitochondria and cultured rat cortical neurons, respectively. Furthermore, we examined the role of the PI3K signaling pathway and the expression of several cytoprotective proteins in the mediation of BMS-induced delayed neuronal PC. Through these experiments, we have also shown that the PC due to activation of mitoKATP channels by BMS is independent of ROS production.
Materials and methods
Materials
Cell culture plastics were purchased from Becton Dickinson (San Jose, CA, USA). Dulbecco's modified Eagle medium, Neurobasal medium, B27 Supplement, 2-mercaptoethanol, and horse serum were obtained from Gibco BRL (Grand Island, NY, USA). Percoll was purchased from Amersham Biosciences (Uppsala, Sweden), dispase I from Roche (Mannheim, Germany), and M40401 from Metaphore Pharmaceuticals (St Louis, MO, USA). CellTiter-Glo Luminescent Assay, CellTiter 96 AQueous One Solution Assay, and SV Total RNA Isolation Kit were procured from Promega (Madison, WI, USA). Hydroethidine (HEt), tetramethylrhodamine ethyl ester (TMRE), and Amplex Red Catalase Assay Kit were purchased from Molecular Probes (Eugene, OR, USA). The SOD Assay Kit was purchased from Fluka (Buchs, Switzerland). Taqman Gene Expression Assays for catalase (assay identification no. Rn00560930_m1), manganese-dependent superoxide dismutase (MnSOD; Rn00566942_g1), and β-actin (4352340E) were procured from Applied Biosystems (Foster City, CA, USA). Antibodies were obtained from the following sources: anti-glial fibrillary acidic protein antibody from Chemicon (Temecula, CA, USA); anti-microtubule-associated protein-2, monoclonal anti-MnSOD, monoclonal protein kinase B (PKB)α/Akt antibodies from Becton Dickinson; polyclonal anti-catalase, and polyclonal anti-copper and zinc-dependent SOD antibodies from Calbiochem (San Diego, CA, USA); polyclonal anti-pS473 Akt antibody from Promega; polyclonal anti-phospho-glycogen synthase kinase 3α/β (Gsk3α/β) (Ser 21/9) antibody and monoclonal anti-Gsk3β antibody from Cell Signaling Technology (Danvers, MA, USA); and anti-rabbit IgG and anti-mouse IgG from Jackson ImmunoResearch (West Grove, PA, USA). All other chemicals were from Sigma (St Louis, MO, USA).
Primary Rat Cortical Neuronal Culture
Timed pregnant Sprague—Dawley (SD) rats were obtained from Harlan (Indianapolis, IN, USA) and were maintained and used in compliance with the principles set forth by the Animal Care and Use Committee of Wake Forest University Health Sciences. Primary rat cortical neurons were isolated from E18 SD fetuses as described in Kis et al (2003). The cells were plated at a density of 2 × 105 cells/cm2 onto poly-
Treatment with BMS
The time line of the experiments is shown in Figure 1A. To induce delayed PC, 7-day-old neuronal cultures were treated with BMS (30 μmol/L) in the regular culture medium once a day for three consecutive days. The dose of BMS was based on that used in previous studies from our laboratory in which the dose-dependency of BMS-induced neuroprotection was examined (Kis et al, 2004). In some of the experiments, the cells were cotreated with BMS (30 μmol/L) and the putative selective mitoKATP blocker 5-hydroxydecanoic acid (5HD), the protein kinase C (PKC) inhibitor chelerythrine (Chel), the SOD mimetic M40401, the large conductance Ca2+-activated K+ (KCa) channel antagonist paxilline (Pax), or the PI3K inhibitor wortmannin (Wrt). At the end of the treatments, all drugs were washed out thoroughly with phosphate-buffered saline (PBS), and the cells were then maintained in the regular cell culture medium.
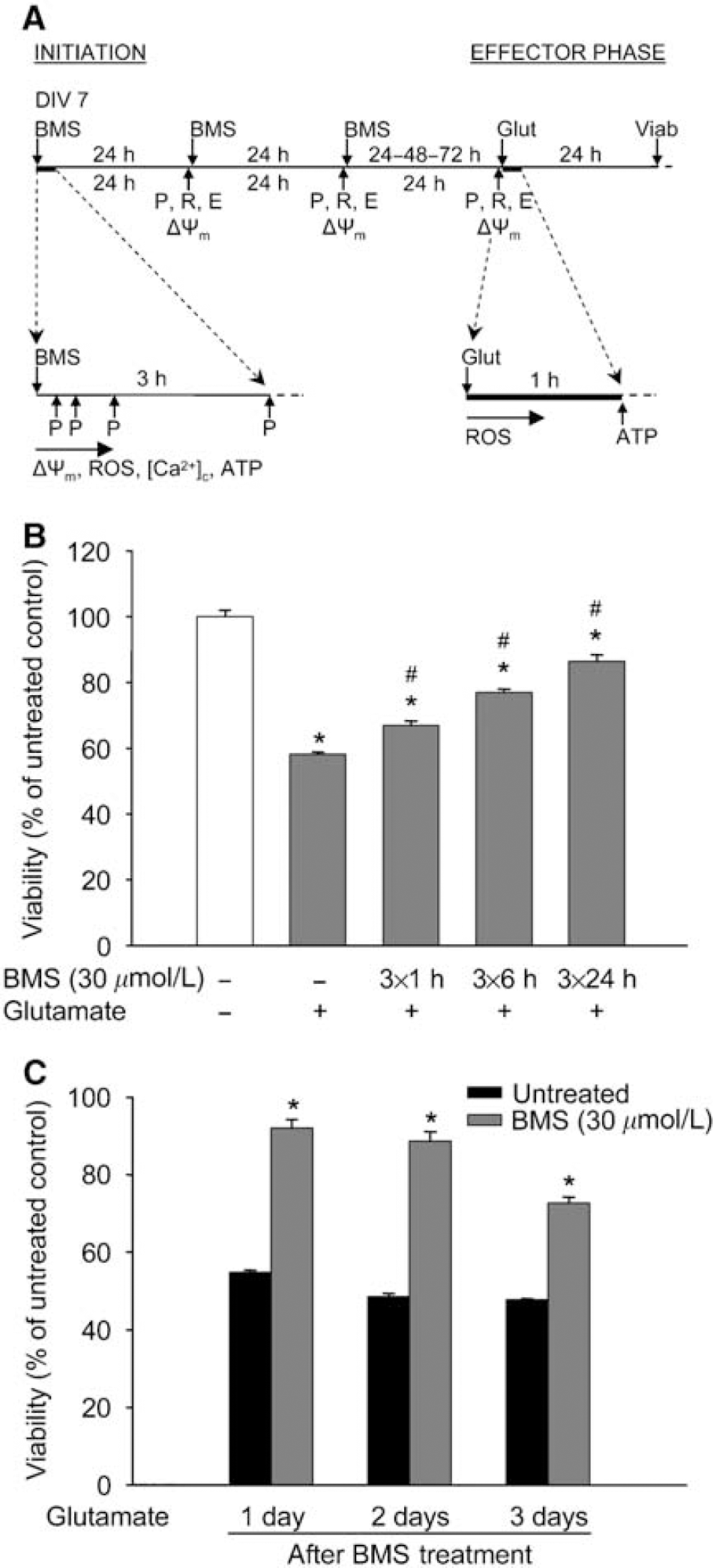
The time line of the experiments and the protective effect of long-term BMS treatment. (
Glutamate Excitotoxicity
Cell cultures in 96-well plates were exposed to glutamate (200 μmol/L) 24 h after the initiation of the last BMS treatment in the regular culture medium for 60 mins at 37°C in the 5% CO2 incubator. Afterward, the cells were rinsed and returned to the 5% CO2 incubator in regular culture medium. In some experiments, different concentrations of the catalase inhibitor 3-aminotriazole (3AT; 1, 10, 100 mmol/L) were applied to the cultures during and for 3 h after glutamate excitotoxicity.
Quantification of Neuronal Survival
The cell viability in neuronal cultures was determined 24 h after the neurotoxic insults using the tetrazolium-based CellTiter 96 AQueous One Solution Assay, as described previously (Kis et al, 2003). Absorbance at 492 nm was measured with a microplate reader (FLUOstar OPTIMA, BMG Labtech GmbH, Offenburg, Germany). Comparisons were always made in the same manner, between sister cultures exposed to the neurotoxic stimulus on the same day, and cell viability was expressed as a percentage of the corresponding control culture (untreated and not exposed to the lethal insult) as follows: % viability=(absorbanceSAMPLE−absorbanceBACKGROUND) × 100/(absorbanceCONTROL−absorbanceBACKGROUND).
Isolation of Rat Brain Mitochondria
Brain mitochondria were isolated using a discontinuous Percoll gradient as described previously, with some modifications (Rajapakse et al, 2001; Sims, 1990). All the instruments and buffers were kept ice-cold during the procedure. Male SD rats (280 to 320 g; Harlan) were overanesthetized with isoflurane and decapitated. The brain, without the cerebellum, was removed, weighed, and then homogenized in mitochondrial isolation buffer (MIB; sucrose, 250 mmol/L; K+-EDTA (ethylenediaminetetraacetic acid), 0.5 mmol/L; Tris-HCl (pH 7.4), 10 mmol/L; bovine serum albumin, 1%) using Dounce homogenizers and glass pestles (Kontes Glass Co., Vineland, NJ, USA). The homogenate was centrifuged for 3 mins at 500g and then the supernatant was collected and resuspended in equal amounts of 24% Percoll in MIB. This suspension was layered onto a discontinuous Percoll gradient (24% and 40% in MIB). The gradient was centrifuged for 5 mins at 28,000g, and the layer between the 24% and 40% Percoll suspensions containing the purified mitochondria was collected. The preparation was washed in MIB and centrifuged for 12 mins at 15,000g. For fluorescent plate reader measurements, the protein content was determined and then equal amounts of mitochondria were transferred in MIB into black-walled, clear-bottomed 96-well plates.
Analysis of Reactive Oxygen Species Formation
Reactive oxygen species generation was assessed in black-walled 96-well plates with HEt using the same microplate reader used for the viability measurements (λex=510, λem=590 nm). Isolated mitochondria were washed and then loaded with HEt (5 μmol/L) and BMS (10 to 40 μmol/L) or DZ (500 μmol/L) in MIB 1 min before the assay. Hydroethidine fluorescence in each well was measured at 37°C every minute for 30 mins. Data were expressed as a percentage of the starting intensity of the untreated control: % HEt fluorescenceSAMPLE=(HEt fluorescenceSAMPLE−HEt fluorescenceBACKGROUND) × 100/(HEt fluorescenceCONTROL AT TIME 0−HEt fluorescenceBACKGROUND).
To assess the delayed effects of BMS treatment, neuronal cultures were treated with BMS for 3 days. On the day after the last treatment, the cells were loaded with HEt (5 μmol/L) and subjected to glutamate (200 μmol/L). The intensity of HEt fluorescence was measured at 37°C every minute for 30 mins. The change of HEt fluorescence (steepness of the curves) was calculated using the following equation: HEt fluorescenceSAMPLE in relative fluorescent units/min (ΔRFU/min)=(HEt fluorescenceSAMPLE at 30 mins−HEt fluorescenceSAMPLE at 0 min)/30.
Determination of Mitochondrial Membrane Potential
The changes of mitochondrial membrane potential (Δψm) were analyzed using the Δψm-sensitive dye TMRE. Measurements were performed in PBS containing 1 mg/mL glucose (neurons) or in MIB (mitochondria) at 37°C. To measure immediate changes, isolated mitochondria in MIB were loaded with BMS (10 to 40 μmol/L) or with DZ (500 μmol/L) and TMRE (0.5 μmol/L) for 20 mins at 4°C in the dark after which the drugs were washed out thoroughly. To analyze the delayed effects of BMS on Δψm, neuronal cultures in black-walled, clear-bottomed 96-well plates were treated with BMS (30 μmol/L) and 5HD (1, 5 mmol/L) once a day for 1 to 3 days. Twenty-four hours after the last treatment, the cells were loaded with TMRE (0.5 μmol/L) at 37°C in a 5% CO2 incubator. After 20 mins, the neurons were washed with PBS, and TMRE fluorescence was measured in each well using the same microplate reader, which was used for viability measurements (λex=510 nm, λem=590 nm). Data were expressed as a percentage of the intensity of the untreated control culture: % TMRE fluorescenceSAMPLE=(TMRE fluorescenceSAMPLE−TMRE fluorescenceBACKGROUND) × 100/(TMRE fluorescenceCONTROL−TMRE fluorescenceBACKGROUND).
Monitoring Free Cytosolic Calcium Levels
Changes of free cytosolic calcium levels ([Ca2+]c) were monitored using the Ca indicator dye Fluo-4 AM in glucose containing (1 mg/ml) PBS. Neuronal cultures were loaded with 2 μmol/L Fluo-4 AM and 1 μmol/L Pluronic F-127 in PBS in the dark for 60 mins at room temperature (21°C) and then washed three times. BMS (30 μmol/L) was mixed with the buffer, and confocal images of cellular Fluo-4 AM fluorescence (λex=488 nm, λem=520 nm) were acquired using a laser scanning microscope (LSM 510; Zeiss, Jena, Germany) with a × 63 water immersion objective (Zeiss). Images were recorded every 20 secs for 10 mins, and the average pixel intensity of individual cell bodies was determined using the software supplied by the manufacturer (Zeiss). Data were expressed as a percentage of the starting intensity of the untreated control culture: % Fluo-4 fluorescenceSAMPLE=(Fluo-4 fluorescenceSAMPLE−Fluo-4 fluorescenceBACKGROUND) × 100/(Fluo-4 fluorescenceCONTROL AT TIME 0−Fluo-4 fluorescenceBACKGROUND).
ATP Assay
The ATP level of neurons was measured with the glow-type CellTiter-Glo Luminescent Assay, as directed by the manufacturer. Cortical neurons cultured in opaque-walled 96-well plates were equilibrated to room temperature (21°C) for 30 mins. CellTiter-Glo was added to each well, and the plates were incubated at room temperature for 10 mins to stabilize the luminescent signal, which was then measured with a FLUOstar OPTIMA microplate reader. An ATP standard curve was generated for each measurement to calculate the ATP contents of wells.
Western Blotting for Catalase, Manganese-Dependent Superoxide Dismutase, Copper and Zinc-Dependent Superoxide Dismutase, Total and Phosphorylated Akt, and Glycogen Synthase Kinase-3β
Cultured cells were washed twice in ice-cold PBS and then harvested by scraping in ice-cold Nonidet P40 lysis buffer supplemented with proteinase inhibitors (1 μg/mL aprotinin, 50 μg/mL phenylmethylsulfonyl fluoride, and 1 μg/mL leupeptin) and a phosphatase inhibitor cocktail (1 mmol/L EDTA, 1 mmol/L sodium orthovanadate, 10 μg/mL benzamidine, 1 mmol/L sodium pyrophosphate, and 1 mmol/L sodium fluoride). Equal amounts of protein for each sample were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a polyvinylidine difluoride sheet (Polyscreen PVDF; Perkin Elmer Life Sciences, Boston, MA, USA). Membranes were incubated in a blocking buffer (Tris-buffered saline, 0.1% Tween 20, and 5% skimmed milk powder) for 1 h at room temperature after which the blots were incubated with monoclonal anti-MnSOD (1:1,000), polyclonal anti-copper and zinc-dependent superoxide dismutase (CuZnSOD) (1:1,000), polyclonal anti-catalase (1:4,000), monoclonal anti-Akt (1:1,000), polyclonal anti-pS473 Akt (1:2,500), monoclonal anti-Gsk3β (1:1,000), and polyclonal anti-phospho-Gsk3α/β (1:1,000) antibodies overnight at 4°C. The membranes were then washed three times in Tris-buffered saline with 0.1% Tween 20 and incubated for 1 h in the blocking buffer with anti-rabbit IgG (1:50,000) or anti-mouse IgG (1:5,000) conjugated to horseradish peroxidase. The final reaction products were visualized using enhanced chemiluminescence (SuperSignal West Pico; Pierce, Rockford, IL, USA) and recorded on X-ray film.
Real-Time Reverse Transcription-PCR for Catalase and Manganese-Dependent Superoxide Dismutase
RNA was isolated from neurons using the SV Total RNA Isolation Kit as directed by the manufacturer. The RNA was incubated with DNase to eliminate any residual DNA that might amplify during PCR. Real-time quantitative reverse transcription-PCR (RT-PCR) experiments were performed in the ABI/Prism 7000 Sequence Detection System (Applied Biosystems). From each sample, 25 pg total RNA was reverse transcribed and amplified using QuantiTect Probe RT-PCR Kit (Qiagen, Valencia, CA, USA) and cycle program as follows: 30 mins at 50°C, 15 mins at 95°C, and then 40 cycles each at 94°C for 15 secs and 60°C for 60 secs. All amplifications were conducted with the Pre-Developed TaqMan Gene Expression Assays (Applied Biosystems). All reactions were performed in triplicate with β-actin serving as an internal control. Relative quantification of the mRNA expression levels of target genes was calculated using the 2−ΔΔCT method (Schmittgen et al, 2000).
Catalase Assay
Catalase activity was assessed with the Amplex Red Catalase Activity Assay Kit. Neurons on 96-well plates were pretreated with BMS (30 μmol/L) for 1, 2, or 3 days and then, 24 h after the last treatment, the plates were rinsed three times with PBS and enzyme activity was measured with the same microplate reader that was used for other fluorescent measurements (λex=555 nm and λem=590 nm). A standard curve was generated for each measurement to calculate the catalase activity of wells. Data were expressed as a percentage of the activity of the untreated control culture using the following equation: % catalase activitySAMPLE=catalase activitySAMPLE × 100/catalase activityCONTROL.
Enzyme Activity Assay for Manganese-Dependent Superoxide Dismutase
Manganese-dependent superoxide dismutase activity was measured using the tetrazolium-based SOD Assay Kit as directed by the manufacturer. Neurons on 96-well plates were pretreated with BMS (30 μmol/L) for 1, 2, or 3 days, then 24 h after the last treatment, the cells were incubated with NaCN (5 mmol/L) for 10 mins to inhibit CuZnSOD activity. Subsequently, the plates were rinsed three times with PBS and enzyme activity was measured with the same microplate reader that was used for the viability measurements (λabs=460 nm). Data were expressed as a percentage of the activity of the untreated control culture using the following equations:

and
Statistical Analysis
Statistical analyses were performed with SigmaStat (SPSS, Chicago, IL, USA). Data are presented as means±s.e.m. Differences between groups were assessed by one-way analysis of variance followed by Tukey comparison tests. A value of P<0.05 was considered to be statistically significant.
Results
Long-Term Treatment with BMS Induced Prolonged Protection against Glutamate Excitotoxicity
Treatment of neurons with BMS during glutamate exposure did not provide protection (viability: untreated, 64.00%±0.83%; BMS 30 μmol/L, 65.4%±0.70%, data expressed as mean±s.e.m., n=8 to 16). One, six, and twenty-four hours daily treatments for 3 days with 30 μmol/L BMS induced increasing tolerance against glutamate excitotoxicity with the best protection after the longest daily treatment (Figure 1B). Therefore, the 24-h protocol was chosen as a long-term treatment paradigm for the further experiments. This paradigm did not influence the viability of quiescent neurons (untreated, 100.00%±1.40%; BMS 30 μmol/L, 102.99%±0.69%, data expressed as mean±s.e.m., n=32 in each group), but the protection against glutamate exposure was still apparent 3 days after the last treatment (Figure 1C).
BMS Depolarized Mitochondria
Exposure of isolated rat brain mitochondria to increasing concentrations of BMS resulted in an immediate dose-dependent depolarization, as shown by the decrease of TMRE fluorescence intensity (Figure 2A). DZ (500 μmol/L) was used as a positive control.
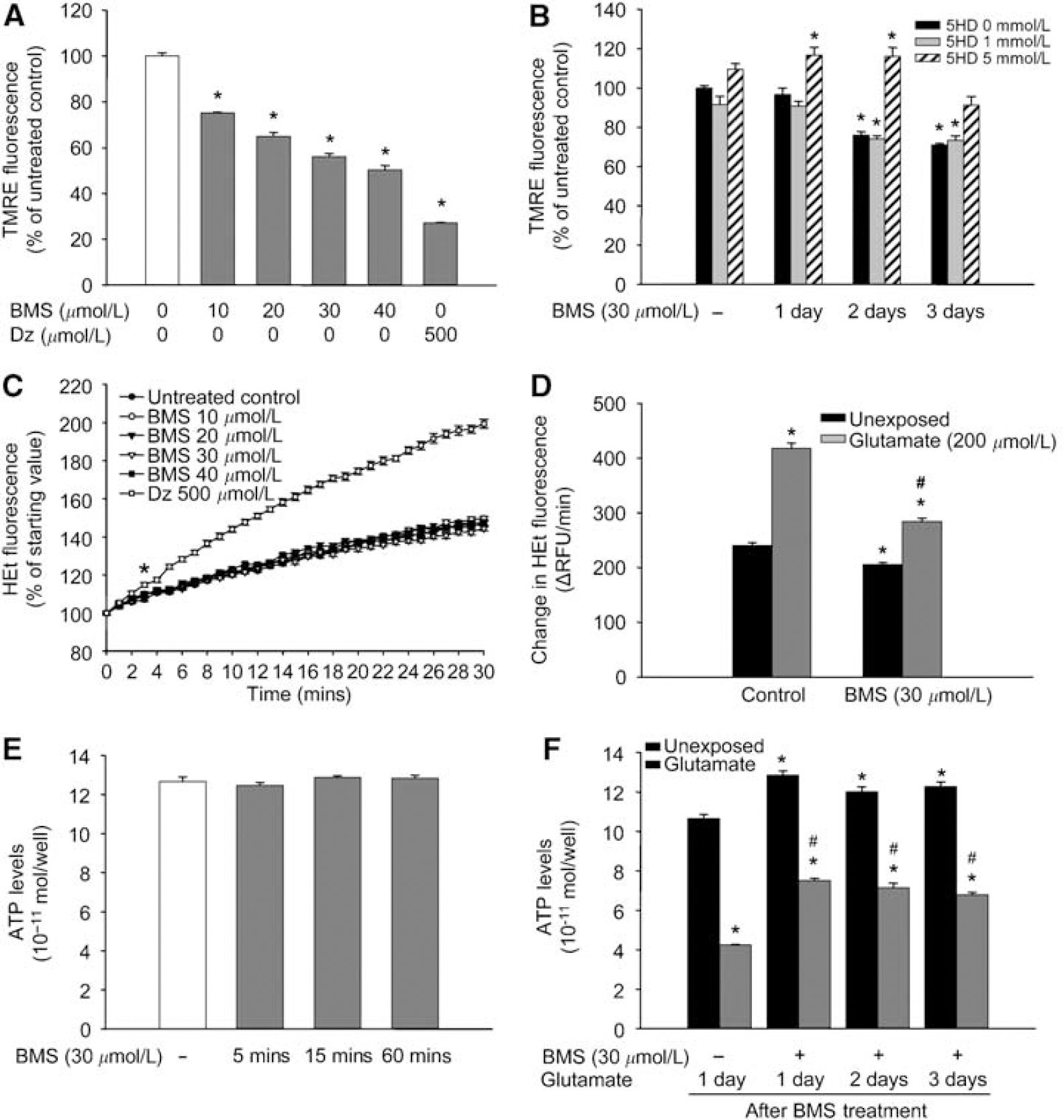
Acute and long-term effects of BMS-191095 (BMS) on mitochondrial membrane potential (Δψm), reactive oxygen species (ROS) generation, and ATP-levels. Isolated rat brain mitochondria were subjected to different doses of BMS, and the intensity of tetramethylrhodamine ethyl ester (TMRE) fluorescence was recorded using a fluorescent microplate reader (
Twenty-four hours after a single treatment with BMS, the membrane potential of mitochondria in neurons returned to the normal level (Figure 2B). However, after 2- and 3-day treatments, mitochondria remained in a depolarized state. The depolarizing effect of BMS was counteracted by 5 mmol/L 5HD (Figure 2B). Mitochondrial depolarization was still detectable 72 h after the third treatment (Δψm, days after the third treatment: 1 day, 73.97%±0.62%∗; 2 days, 78.63%±0.92%∗; 3 days, 81.12%±1.38%∗; data expressed as mean±s.e.m.; n=8 to 16; ∗P<0.05 versus untreated control).
The Effect of BMS on Reactive Oxygen Species Generation
The moderately increasing baseline curve of HEt fluorescence showed continuous basal ROS generation in mitochondria (Figure 2C). Administration of increasing doses of BMS (10 to 40 μmol/L) did not change the kinetics of ROS production of mitochondria. DZ (500 μmol/L), which is known to induce ROS generation, was used as a positive control to show the viability of mitochondrial preparations.
Compared with the untreated control group, long-term BMS treatment resulted in significantly reduced basal ROS generation rate (Figure 2D) in cultured neurons. Exposure to glutamate (200 μmol/L, 30 mins) induced a ROS surge in both untreated and BMS-treated neurons. However, although the rate of ROS generation doubled in control neurons, the increase in the BMS-treated group was markedly less (Figure 2D).
BMS Treatment Improved ATP Homeostasis and Utilization in Neurons
Treatment with BMS (30 μmol/L) for 5, 15, or 60 mins did not change ATP levels of neurons (Figure 2E). Conversely, neurons treated with BMS for three consecutive days showed significantly higher ATP content than control cells. Moreover, increased ATP content of BMS-treated cells was preserved for 3 days (Figure 2F). The ATP content of control groups showed a significant decrease after glutamate challenge (200 μmol/L, 60 mins). In the BMS-treated group, the decrease was markedly less and BMS-treated neurons contained almost twice as much ATP as the control neurons at the end of the 60-min glutamate exposure.
BMS Application Induced an Increase in the Free Cytosolic Ca2+ Level of Neurons
In quiescent cells, the intensity of Fluo-4 fluorescence did not change over time (Figure 3). Administration of 30 μmol/L BMS resulted in an approximate two-fold increase in the signal. Ten minutes later, at the end of the measurement, however, [Ca2+]c of neurons almost reached the resting level (Figure 3). A similar response in [Ca2+]c levels was seen using DZ and a more robust increase with the KCa channel opener NS1619 (data not shown).
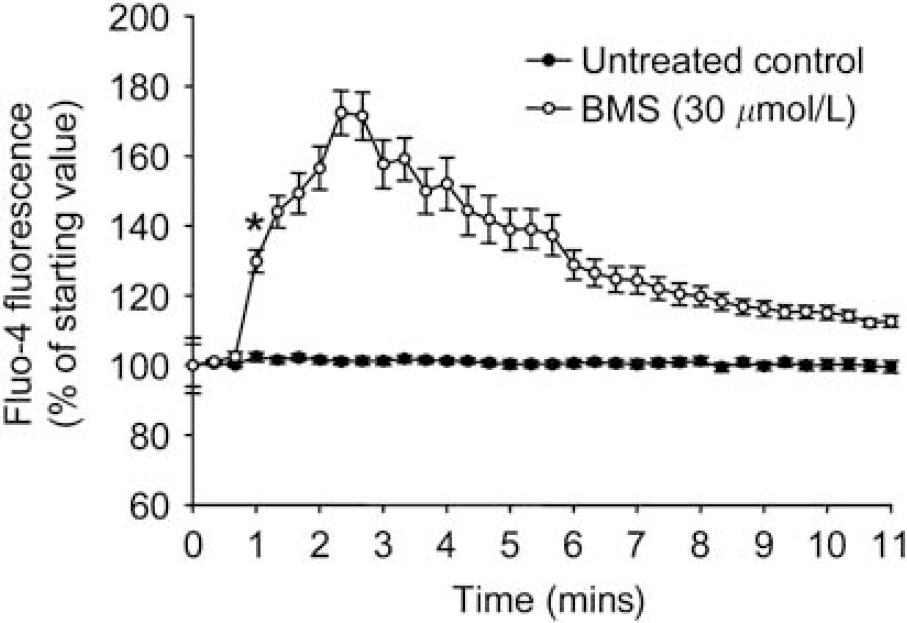
Administration of BMS-191095 (BMS) induces transient increase in cytosolic free Ca2+ [Ca2+]c levels in neurons. Neuronal cultures on poly-
BMS Increased Akt and Glycogen Synthase Kinase-3β Phosphorylation
Western blot analysis with the anti-pS473 Akt antibody revealed increased phosphorylation of Akt within 15 mins after the initiation of BMS treatment (Figure 4A). However, 3 h after the beginning of the incubation, the level of phosphorylated Akt returned to the baseline. Phosphorylation (i.e., inhibition) of Gsk3β showed a significant increase 15 mins after BMS treatment, and it reached its peak at 30 mins. At the end of the measurement (3 h after the application of BMS), the level of the phosphorylated protein was still very high (Figure 4B). One day after a single treatment with BMS, phosphorylation levels of both Akt and Gsk3β returned to baseline, and no further changes were detected 24 h after 3-day treatments, at the time of glutamate challenge (data not shown).
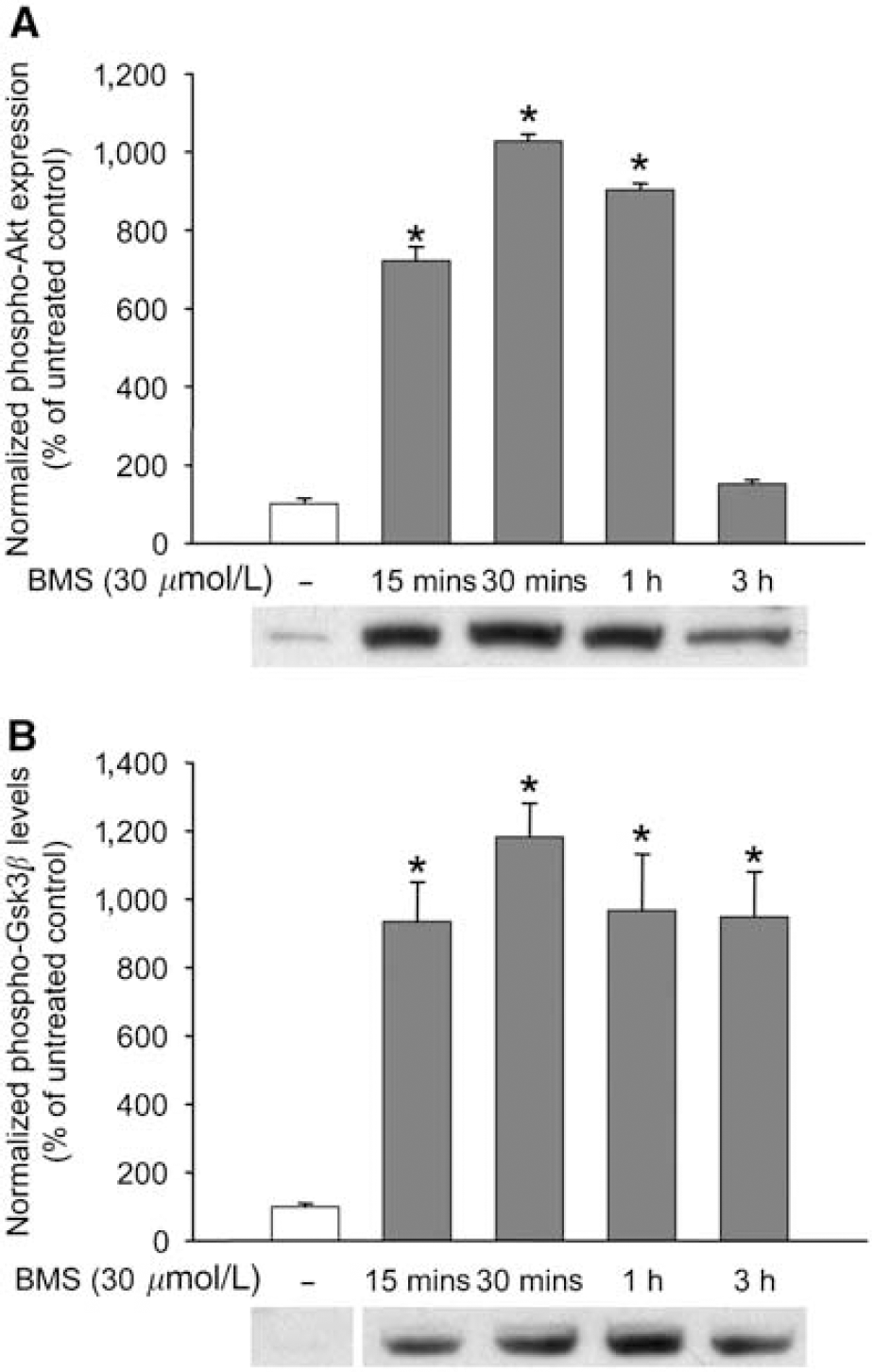
BMS-191095 (BMS) induces rapid phosphorylation of Akt and glycogen synthase kinase 3β (Gsk3β). Neuronal cultures in 35 mm dishes were treated with BMS for 15 mins, 30 mins, 1 h, and 3 h, and then the proteins were extracted and subjected to Western blot analysis for total and phosphorylated Akt (
Phosphoinositide 3-Kinase Inhibition did not Antagonize Mitochondrial Depolarization
To determine whether PI3K activation is a cause or a consequence of the mitoKATP opening effect of BMS, we loaded neuronal cultures with TMRE in the presence of BMS (30 μmol/L) and Wrt (30, 100 nmol/L) for 20 mins. Wrt did not change Δψm and did not alter the depolarizing effect of BMS (Δψm: control, 100.0%±1.65%; Wrt 30 nmol/L, 100.9%±1.09%, Wrt 100 nmol/L, 101.4%±1.34%, BMS 30 μmol/L, 65.6%±0.94%∗; BMS 30 μmol/L+Wrt 30 nmol/L, 63.9%±0.82%∗; BMS 30 μmol/L+Wrt 100 nmol/L, 66.4%±0.89%∗; data expressed as mean±s.e.m.; n=16 in each group; ∗P<0.05 versus control).
Wortmannin, but not M40401, and Paxilline Antagonized the Neuroprotective Effect of BMS
The PI3K inhibitor Wrt (10 to 100 nmol/L) had no effect on the survival of neurons when used once a day for 3 days (Figure 5A). Furthermore, Wrt did not influence cell survival after exposure to glutamate (200 μmol/L, 60 mins). Wrt dose-dependently abolished the neuroprotective effect of BMS against glutamate, however, resulting in the total loss of protection at the highest dose (Figure 5A). In contrast, the SOD mimetic M40401 (Figure 5B) did not change viability of neuronal cultures and had no effect on the neuroprotection induced by BMS. Moreover, the calcium-activated potassium channel inhibitor Pax proved to be ineffective in terms of antagonizing the delayed PC effect of BMS (viability after exposure to glutamate: untreated, 51.39%±0.31%, Pax 10 μmol/L, 53.80%±0.73%, BMS 30 μmol/L, 89.69%±1.34%∗; BMS 30 μmol/L+Pax 10 μmol/L, 90.97%±1.64%∗; data expressed as mean±s.e.m.; n=16 in each group; ∗P<0.05 versus untreated). Interestingly, the PKC inhibitor Chel by itself induced some protection against glutamate excitotoxicity, but it could not block the beneficial effect of BMS (viability after exposure to glutamate: untreated, 55.37%±0.55%, Chel 2.5 μmol/L, 73.04%±1.49%∗, BMS 30 μmol/L, 91.00%±1.76%∗; BMS 30 μmol/L+Chel 2.5 μmol/L, 87.37%±2.34%∗; data expressed as mean±s.e.m.; n=16 in each group; ∗P<0.05 versus untreated).
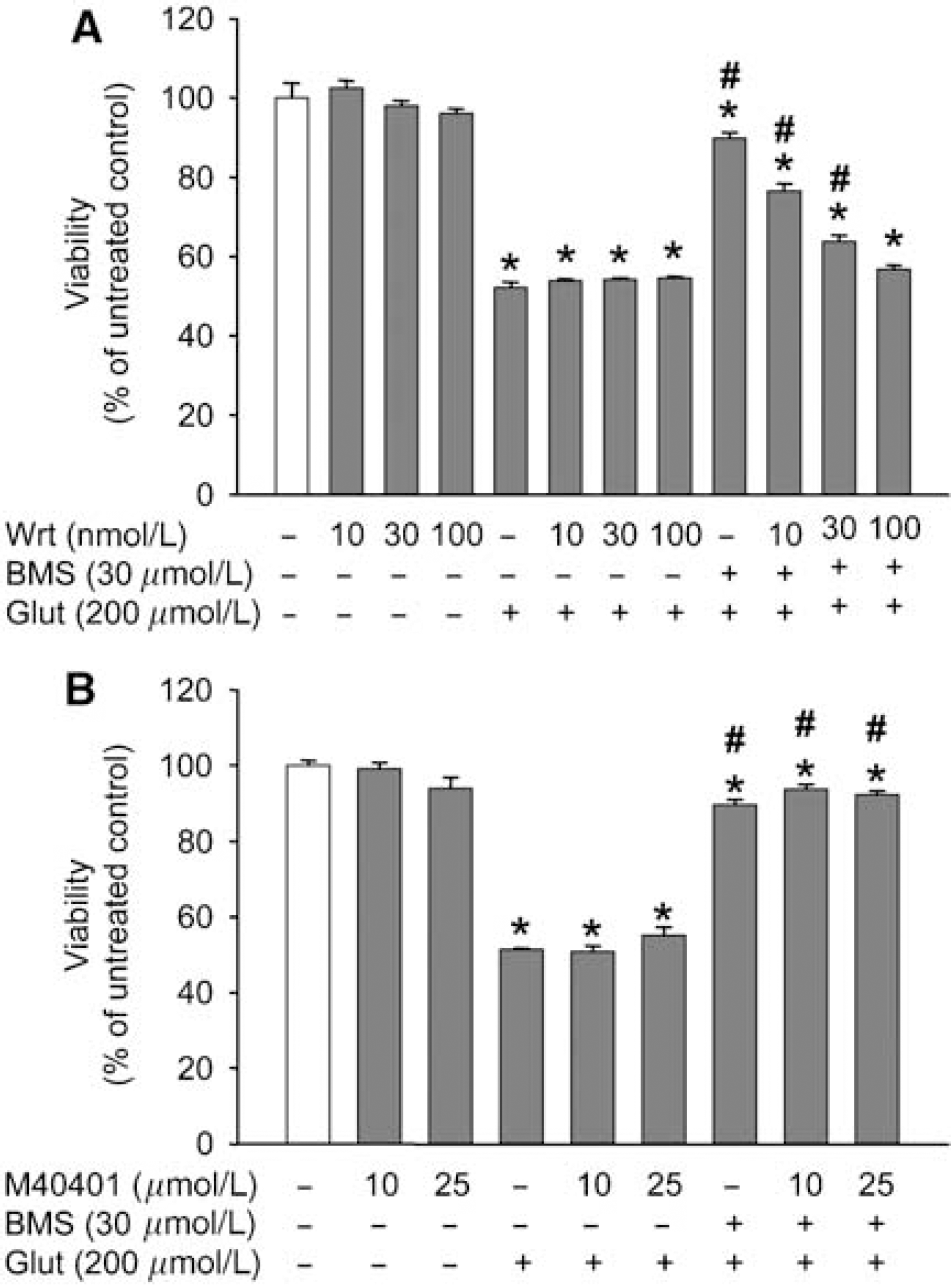
The delayed preconditioning effect of BMS-191095 (BMS) is dependent on the activation of the phosphoinositide 3-kinase (PI3K) pathway but not on the reactive oxygen species generation. Cortical neurons in 96-well plates were treated with BMS and with the PI3K inhibitor wortmannin (Wrt) (
Preconditioning with BMS Resulted in Phosphoinositide 3-Kinase-Dependent Overexpression of Catalase
Real-time RT-PCR showed a gradual increase in catalase mRNA, whereas no change was detected in the mRNA level of MnSOD (Figure 6A). Western blotting also revealed a dose-dependent increase in the expression of catalase after 1, 2, and 3 days of BMS treatment (Figure 6B). The same treatment, however, did not induce any changes in the protein levels of MnSOD and CuZnSOD (data not shown). Similar to its mRNA and protein levels, the activity of catalase increased as well (untreated control, 100.00%±3.73%; BMS 30 μmol/L for 3 days, 169.23%±2.67%∗, data expressed as mean±s.e.m.; n=24 in each group; ∗P<0.05 versus untreated control). In contrast, enzymatic activity of MnSOD was not altered by BMS treatment (data not shown). Finally, the overexpression of catalase could be antagonized by cotreatment of neurons with 30 μmol/L BMS and 100 nmol/L Wrt for 3 days (Figure 6C).
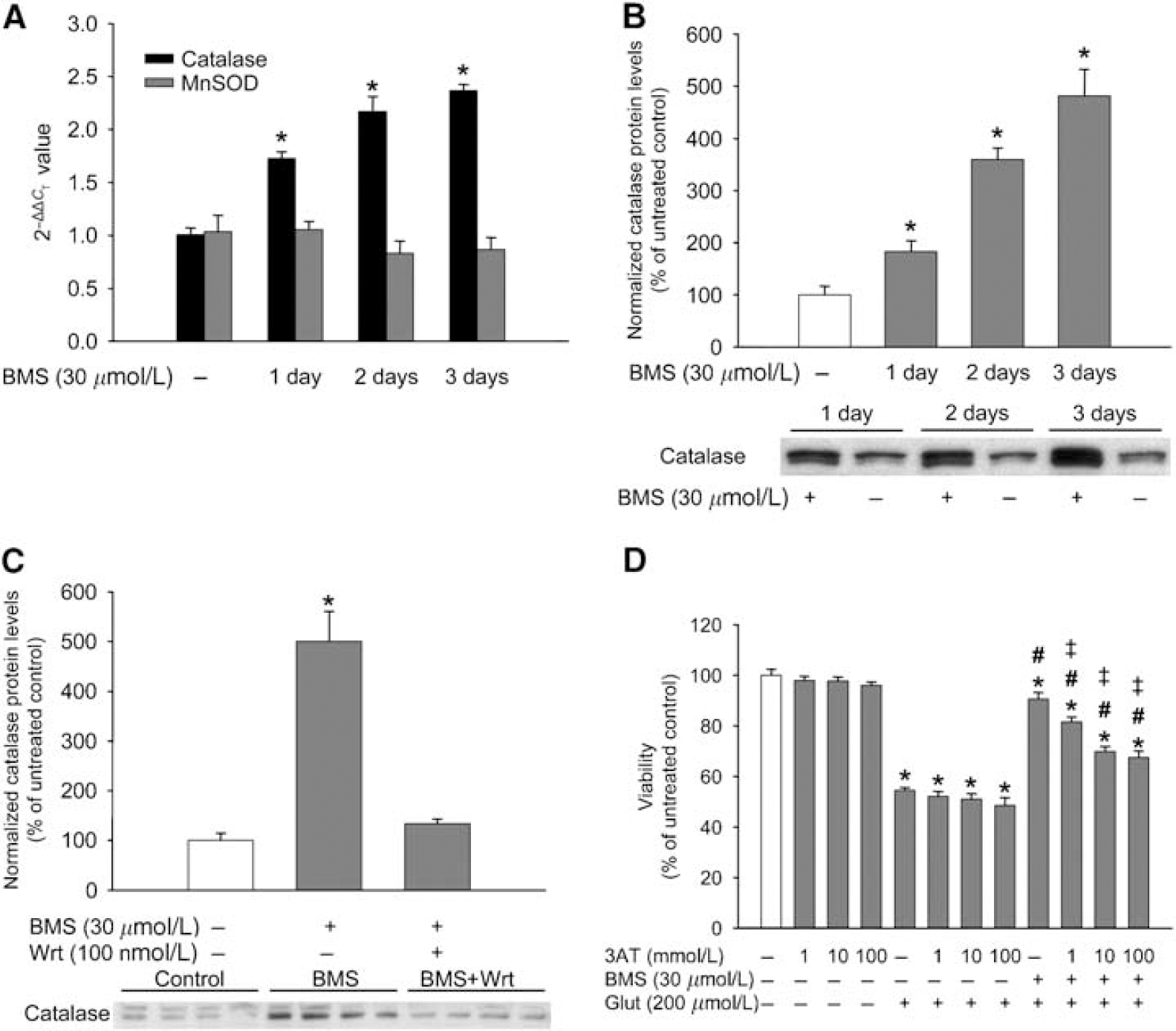
Delayed preconditioning with BMS-191095 (BMS) increases the expression and activity of catalase through the phosphoinositide 3-kinase (PI3K) pathway. Cultured neurons in 35 mm dishes were treated with BMS once a day for 1, 2, or 3 days, and then total RNA was isolated and subjected to real-time reverse transcription-PCR for catalase and manganese-dependent superoxide dismutase (MnSOD) (
The Catalase Inhibitor 3AT Dose-Dependently Antagonized the Protective Effect of BMS
In untreated cells, the high doses of 3AT that were used had no toxic effect on neuronal survival (Figure 6D). In addition, 3AT did not influence cell survival after exposure to glutamate (200 μmol/L) for 60 mins. However, 3AT dose-dependently decreased the neuroprotective effect of BMS. The maximal reduction in the protection was observed using 10 mmol/L 3AT, which is known to effectively inhibit catalase, and the highest dose (100 mmol/L) had no additional effect on cell survival.
Discussion
Several important findings of our study are as follows: (1) the selective mitoKATP channel opener BMS-191095 dose-dependently depolarized isolated rat brain mitochondria without an increase in ROS generation; (2) long-term BMS treatment induced prolonged protection along with sustained mitochondrial depolarization, reduced basal ROS generation, and improved ATP levels in cultured neurons; and (3) BMS activated the PI3K signal-transduction pathway that led to the phosphorylation of Akt and Gsk3β, increased expression and activity of catalase, and ultimately elicited neuroprotection.
Since the first report on the presence of a small conductance ATP-sensitive potassium channel in the inner mitochondrial membrane by Inoue et al (1991), the selective pharmacological activation of mitoKATP channels became a major target of both immediate and delayed PC studies. However, the exact steps of PC after mitoKATP activation are still unclear. The role of mitoKATP channels in ROS generation is controversial. ROS released by mitochondria and acting as second messengers could provide a link between mitoKATP opening and the activation of cytoprotective pathways. However, most of the pharmacological studies on the mitoKATP channel showing ROS generation were performed using DZ, which was shown to inhibit the mitochondrial electron transport chain at Complex II (succinate dehydrogenase) resulting in excessive ROS release (Kis et al, 2003; Minners et al, 2007). Others have shown that DZ may also increase mitochondrial ROS generation by inhibiting Complex I (Andrukhiv et al, 2006). Indeed, ROS seem to play a pivotal role in the tolerance-inducing effect of DZ, as the neuroprotection could be completely blocked with an SOD mimetic if administered during the PC phase (Kis et al, 2003). Conversely, Facundo et al (2006) tested the redox effects of mitoKATP activation and found no evidence of increased mitochondrial ROS generation by opening of the mitoKATP channels. Furthermore, Ferranti et al (2003) showed that mitochondria depolarized by K+ entry generate less ROS. To clarify this issue, studies using more selective activators of the mitoKATP channels such as BMS are needed. In our experiments, comparable to our previous studies on isolated piglet brain mitochondria (Busija et al, 2005) and on cultured neurons (Mayanagi et al, 2007), BMS induced dose-dependent depolarization of the inner membrane of isolated rat brain mitochondria without ROS generation. Hence, we provide strong evidence that mitochondrial ROS generation is independent from the activation of the mitoKATP channels. Consequently, it is likely that ROS generation is a nonspecific side effect of some less selective mitoKATP openers. It is worth mentioning, however, that ROS produced during PC is known to activate cytoprotective pathways, which may act synergistically with mitoKATP activation induced by nonselective openers. Additionally, ROS may directly or indirectly modulate the activation of mitoKATP channels.
We have also shown that long-term treatment of cultured neurons with BMS leads to sustained mitochondrial depolarization and a parallel reduction in basal ROS production. Furthermore, this effect was reversed by the putatively selective mitoKATP blocker 5HD. These results are in agreement with previous studies from our laboratory in which we showed that at least 5 mmol/L 5HD is needed to sufficiently antagonize the effects of BMS and that doses of 1 mmol/L or less are ineffective (Kis et al, 2004; Mayanagi et al, 2007). The exact mechanism of sustained depolarization is unknown. Nevertheless, in a previous study, we found a similar phenomenon after a different PC stimulus, namely, transient energy deprivation (Gáspár et al, 2006). Therefore, it seems that sustained depolarization of mitochondria may play a crucial role in delayed PC induced by various stimuli.
K+ influx through mitoKATP channels might preserve mitochondrial integrity by maintaining matrix volume and reducing Ca2+ accumulation (Busija et al, 2004; Domoki et al, 2004). This is particularly important during an ischemic or chemical challenge and allows the rapid restoration of normal ATP production by maintaining the optimal configuration of respiratory complexes. Our results appear to support this hypothesis, as we found higher basal levels of ATP after long-term BMS treatment despite sustained mitochondrial depolarization. This may seem controversial; nevertheless, a similar trend in Δψm and ATP content was observed in neuronal cultures after delayed PC with transient energy deprivation (Gáspár et al, 2006). Moreover, BMS-preconditioned neurons better preserved ATP during a glutamate challenge and, consequently had a better survival rate. In our experiments, neuronal viability was assessed with a probe that measures the reducing capacity of cells that is primarily dependent on mitochondrial NADH generation. Thus, increased ATP content and preserved viability in BMS-treated neurons also indicate preserved mitochondrial integrity.
To further explore the immediate effects of BMS, we performed Western blot analysis of two key components of the PI3K signaling pathway, namely, Akt (also known as protein kinase B) and Gsk3β, and found that both enzymes were phosphorylated within 15 mins of the initiation of BMS treatment. However, whereas activity of Akt returned to baseline within 3 h, Gsk3β remained highly phosphorylated until the end of the measurement (3 h). Akt is a serine/threonine protein kinase with high sequence homology to both protein kinase A and PKC; its role in neuronal survival was recently reviewed by Brunet et al (2001). Among many others, active Akt phosphorylates Bad (Kamada et al, 2007) and Gsk3β (Grimes and Jope, 2001) and thereby induces antiapoptotic effects. Intracerebroventricular infusion of the PI3K inhibitor Wrt before ischemic PC was shown to inhibit both Akt phosphorylation and protection against bilateral common carotid artery occlusion in the Mongolian gerbil model (Yano et al, 2001). Glycogen synthase kinase 3β is a critical component of a vast number of cellular signaling pathways (for a detailed review see Grimes and Jope, 2001). As one of the central targets of the PI3K—Akt pathway, Gsk3β is also a key modulator of apoptosis. The enzyme is constitutively active and is primarily regulated through inhibition by phosphorylation. Liang and Chuang (2007) showed that inhibition of either isoforms of Gsk3 completely blocks glutamate excitotoxicity and protects rat cortical neuronal cultures. Furthermore, Kajta et al (2007) reported that pharmacological inhibition of Gsk3β enhanced the neuroprotective effect of the selective estrogen receptor modulator genistein against glutamate challenge. These results support our findings with BMS concerning inhibition of Gsk3β and almost complete protection against glutamate excitotoxicity. However, it seems that Gsk3β is not the ultimate effector of long-term BMS treatment but rather an important component of the cytoprotective cascade, as its phosphorylation level did not differ from the control at the time of glutamate exposure.
The activation of the PI3K pathway is usually linked to cell surface receptors such as receptor tyrosine kinases and G-protein-coupled receptors (Brunet et al, 2001; Oudit et al, 2004). This might suggest that the effects of BMS are also mediated by binding to such a receptor. We are unaware, however, of any targets other than the mitoKATP channel. Moreover, Wrt was unable to block the mitochondrial depolarizing effect of BMS, suggesting that the opening of mitoKATP channels is upstream to PI3K activation. The proposed mechanism of mitoKATP activation is depicted in Figure 7. A potential retrograde signaling link between the activation of mitoKATP channels and the PI3K pathway could be the production of ROS by mitochondria. However, our results clearly show that BMS opens mitoKATP channels without ROS generation. Calcium is one of the possible links between mitochondria and cytosolic signaling pathways, and it was shown to be related to the PI3K cascade by several mechanisms. First, the small GTP-binding protein p21ras that is upstream to PI3K is activated by Ca2+ (Finkbeiner and Greenberg, 1996). Second, the Ca2+/calmodulin-dependent protein kinase (CaMK) II may activate PI3K, and the calmodulin-binding sequence in PI3K may facilitate the association of calmodulin with PI3K (Wang et al, 2007a). Third, Akt was shown to be phosphorylated by CaMK kinase (Yano et al, 2005). Finally, Bickler and Fahlman (2004) showed that moderate increases in [Ca2+]c induced protection against a subsequent lethal stress in hippocampal slice cultures by the activation of cytoprotective kinases, including Akt. An elevation of [Ca2+]c was also observed after the dissipation of Δψm, and this led to the activation of Ca2+-sensitive signaling pathways (Biswas et al, 2005). Consistent with these observations, we have also found that the application of BMS resulted in a mild and transient increase in [Ca2+]c and a subsequent phosphorylation of Akt and Gsk3β. Another possible ROS-independent mitochondrial mechanism of Akt activation could be the inhibition by increased NADH/NADPH ratio of phosphatase and tensin homologue deleted on chromosome 10 (PTEN) that dephosphorylates Akt (Pelicano et al, 2006). Finally, Shang et al (2005) found that mitochondrial depolarization, independently of ROS formation, led to the dephosphorylation of death-associated protein kinase through the activation of a class III PI3K in a neuroblastoma cell line. The authors proposed the role of lipid second messengers in the activation of PI3K released from the mitochondrial membrane during depolarization.
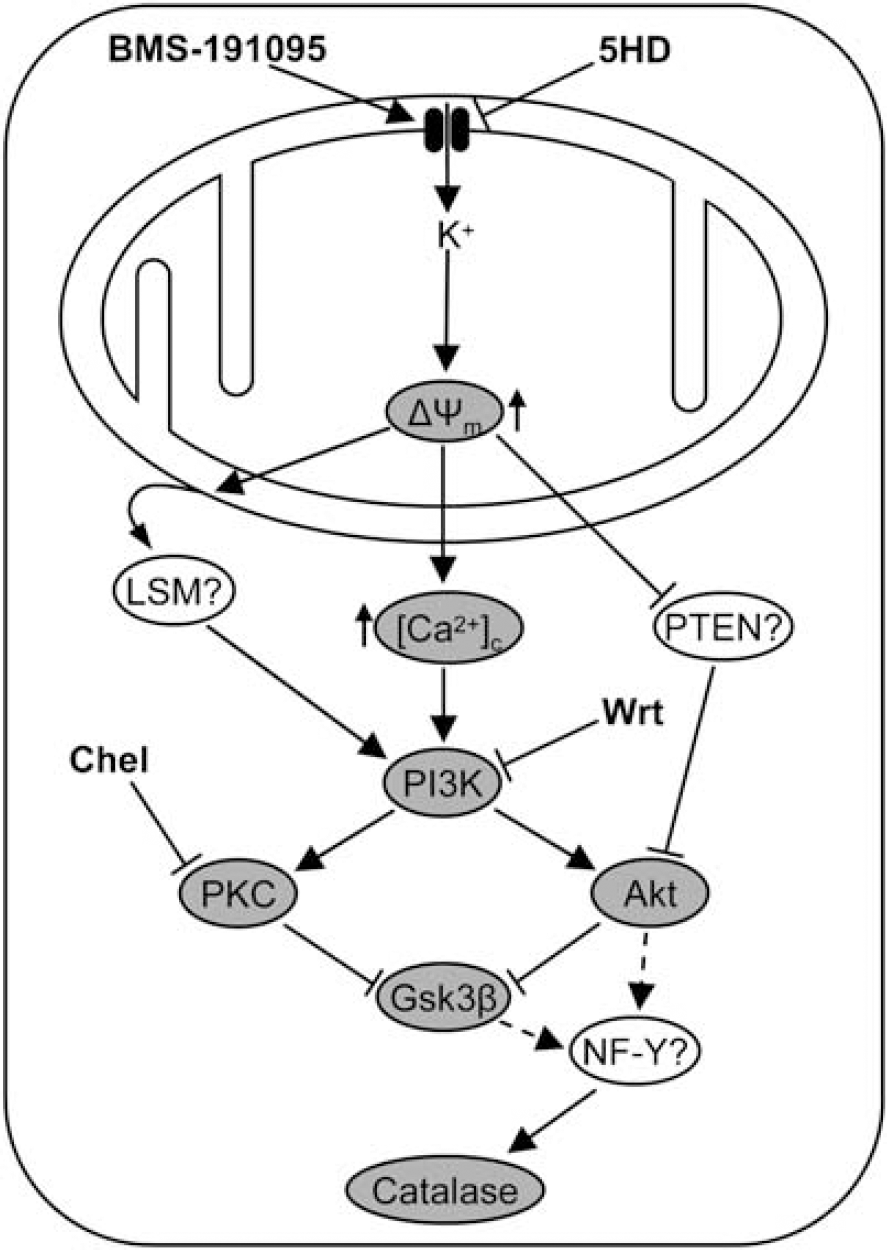
A schematic diagram illustrating the activation of signaling pathways of preconditioning after mitochondrial ATP-sensitive potassium (mitoKATP) channel opening. Activation of mitoKATP channels by BMS-191095 induces the dissipation of the mitochondrial membrane potential (Δψm) without an elevation in reactive oxygen species generation. Mitochondrial depolarization causes an increase in cytosolic free calcium ([Ca2+]c) levels, inhibits phosphatase and tensin homologue deleted on chromosome 10 (PTEN), and may induce the release of lipid second messengers (LSM) from the mitochondrial membrane. These effects give rise to the activation of phosphoinositide 3-kinase (PI3K), Akt, protein kinase C (PKC), inhibition of glycogen synthase kinase 3β (Gsk3β), and to the activation of NF-Y that is involved in the transcription of catalase. Solid arrows indicate activation and broken arrows show putative activation; whiskered lines indicate inhibition; gray ellipses indicate components of the signaling pathway that were discussed in this study. Although catalase upregulation provides the bulk of the neuroprotection, it seems likely that the activation of these pathways also elicit other beneficial cellular changes. 5HD, 5-hydroxydecanoic acid; Chel, chelerythrine; PI3K, phosphoinositide 3-kinase; Wrt, wortmannin.
Next, we tested the delayed effects of BMS PC and found that Wrt dose-dependently and completely antagonized the neuroprotective effect of BMS, providing evidence on the crucial role of the PI3K signaling pathway in the initiation and/or mediation of PC. Conversely, the SOD mimetic M40401 had no effect on the protection, showing that indeed ROS did not have a role in the initiation of PC by BMS. The KCa channel inhibitor Pax was also ineffective, proving again the selectivity of BMS. Interestingly, subtoxic doses of the PKC inhibitor Chel by itself induced protection against glutamate challenge. Although the beneficial role of PKC activation (especially that of its ε isoform) in PC was shown in various models, our results are in agreement with previous reports showing that in neurons the inhibition or downregulation of PKC confers protection against ischemia and other insults (Favaron et al, 1990). Additionally, Reshef et al (1997) reported that both activation and inhibition of PKC elicit neuroprotection. These authors concluded that differential activation (or inhibition) of PKC isoforms might be responsible for the protection. Surprisingly, cotreatment with Chel did not significantly change the viability of BMS-preconditioned neurons exposed to glutamate. Nevertheless, this finding may be explained as the parallel actions of Chel: PC by PKC inhibition, and antagonizing the effects of BMS. Additionally, the activation of PKC was shown to be secondary to that of PI3K (Tong et al, 2000); this may explain the complete inhibition of PC by Wrt. Finally, several isoforms of PKC were reported to inactivate Gsk3β (Grimes and Jope, 2001).
We also examined the effects of long-term BMS treatment on the expression of several cytoprotective proteins and found that the expression of catalase increased at both transcriptional and translational levels. A similar, although not entirely parallel, increase in its enzymatic activity was also found. Furthermore, Wrt abolished catalase overexpression showing that catalase was activated by the PI3K pathway. Finally, to examine whether elevated catalase levels play any role in the neuroprotective effect of BMS, we performed viability experiments in which the catalase inhibitor 3AT was used to antagonize the protective effect of BMS against glutamate challenge and found a partial and dose-dependent inhibition of the protection. The expression of catalase is regulated both transcriptionally and posttranscriptionally. Our results indicate that BMS-induced catalase overexpression was, at least in part, due to transcriptional activation. Luo and Rando (2003) reported that in mice the catalase expression is regulated by the CCAAT-binding factor NF-Y. In addition, Lee et al (2005) showed that Akt upregulates the DNA-binding activity of NF-Y. Furthermore, the presence of NF-Y was reported in several neural cell lines. Thus, NF-Y may provide a link between Akt activation and catalase overexpression. Although increased levels of catalase provided much of the protection against neuronal cell death during glutamate exposure, it is likely that other factors also participate in the mediation of BMS-induced PC.
In conclusion, our results provide novel components to the mechanism of action of delayed PC induced by long-term BMS treatment showing that activation of mitoKATP channels is independent of ROS generation. Long-term treatment with BMS results in sustained mitochondrial depolarization, reduced basal ROS formation, and improved ATP homeostasis. Together with the activation of the PI3K—Akt—Gsk3β axis and upregulation of catalase, these effects play important roles in BMS-induced neuroprotection.
Footnotes
Acknowledgements
We gratefully thank Nancy Busija, MA, for critical reading of the paper. BMS-191095 was a generous gift from Bristol-Myers Squibb (Princeton, NJ, USA).