
Abstract
Cardiac arrest (CA) causes hippocampal neuronal death that frequently leads to severe loss of memory function in survivors. No specific treatment is available to reduce neuronal death and improve functional outcome. The brain's inflammatory response to ischemia can exacerbate injury and provides a potential treatment target. We hypothesized that microglia are activated by CA and contribute to neuronal loss. We used a mouse model to determine whether pharmacologic inhibition of the proinflammatory microglial enzyme soluble epoxide hydrolase (sEH) after CA alters microglial activation and neuronal death. The sEH inhibitor 4-phenylchalcone oxide (4-PCO) was administered after successful cardiopulmonary resuscitation (CPR). The 4-PCO treatment significantly reduced neuronal death and improved memory function after CA/CPR. We found early activation of microglia and increased expression of inflammatory tumor necrosis factor (TNF)-α and interleukin (IL)-1β in the hippocampus after CA/CPR, which was unchanged after 4-PCO treatment, while expression of antiinflammatory IL-10 increased significantly. We conclude that sEH inhibition after CA/CPR can alter the transcription profile in activated microglia to selectively induce antiinflammatory and neuroprotective IL-10 and reduce subsequent neuronal death. Switching microglial gene expression toward a neuroprotective phenotype is a promising new therapeutic approach for ischemic brain injury.
INTRODUCTION
Cardiac arrest (CA) is a major cause of death and disability in the developed world. In all, 1 out of 1,000 adults in the United States experiences CA every year. 1 Thanks to improved cardiopulmonary resuscitation (CPR) techniques and the widespread adoption of therapeutic hypothermia as a treatment option in recent years, survival rates after CA/CPR have improved. Overall survival is now above 20% in specialized centers,1,2 which puts new emphasis on the significant cognitive problems many patients face after CA. The majority of survivors show signs of decreased memory or executive cognitive function, which is a cause of significant and sustained disability for many.2,3 The memory impairment is believed to be caused by neuronal death and atrophy of the ischemia-sensitive hippocampus, which has a central role in acquisition of new memories. 4 Even relatively brief periods of global ischemia cause a distinct pattern of brain injury in surviving patients, consisting of selective and delayed death of neurons within ischemia-sensitive areas, mostly the hippocampus and basal ganglia. 5 Neuronal death is delayed by several days after CA/CPR, 6 which offers a unique window of opportunity for therapeutic interventions. At the same time, drug application is possible very early after resuscitation from CA, as trained medical personal is at the patient's side during resuscitation. Unfortunately, however, efforts to capitalize on the potential for preventing delayed neuronal death after CA/CPR are hampered by a lack of understanding of underlying mechanisms. No specific treatment options are therefore available for survivors of CA/CPR beyond hypothermia.
Brain inflammation is increasingly recognized as a critical component of brain injury. Microglia, the brain resident immune cells, are activated in response to injury and orchestrate the brain's inflammatory response. Microglia are activated after CA/CPR and migrate to injured brain regions where they remain activated for many weeks 7 and may contribute to delayed neuronal death. Accordingly, studies using the antibiotic minocycline, a nonspecific inhibitor of inflammation, suggest, albeit inconsistently, that antiinflammatory strategies may improve neuronal survival after global ischemia.8,9 However, few investigations have carefully evaluated the role of microglia in delayed neuronal death after CA/CPR.
We set out to investigate whether inhibition of a proinflammatory microglial enzyme, soluble epoxide hydrolase (sEH), after CA/CPR can reduce activation of microglia and subsequently reduce neuronal death in the hippocampus. Soluble epoxide hydrolase metabolizes eicosatrienoic acids, cytochrome P450 epoxygenase produced metabolites of arachidonic acid, which have antiinflammatory and neuroprotective properties. 10 The sEH activity contributes to brain inflammation after stroke 11 and pharmacologic inhibition or genetic deletion of sEH consequently reduce infarct size after experimental stroke.12,13
It remains unclear, however, if sEH also contributes to activation of neurotoxic microglia and subsequent neuronal death after CA/CPR and whether inhibition of sEH might thus be beneficial. In this study, we therefore examined whether the clinically relevant application of an sEH inhibitor after successful resuscitation from experimental CA can suppress inflammation and activation of microglia in the ischemia-susceptible hippocampus as well as improve neuronal survival and subsequently enhance memory function.
MATERIALS AND METHODS
Experimental Animals
All experimental protocols were performed in accordance with the National Institutes of Health guidelines for the care and use of animals in research and approved by the Institutional Animal Care and Use Committee at Oregon Health and Science University. Experimental animals were randomized to treatment groups. The experimenters were blinded to assigned treatment throughout all experiments and analyses.
Cardiac Arrest and Cardiopulmonary Resuscitation
Adult (20 to 25 g body weight) male C57BL/6 mice (Charles River Laboratories International, Wilmington, MA, USA) were subjected to CA/CPR as previously described. 14 Briefly, mice were anesthetized with isoflurane (3% for induction, 1.5% to 2% for maintenance) in oxygen-enriched air and orally intubated with a 22-G catheter. Mechanical ventilation was maintained at a rate of 150 breaths/min using a mouse ventilator (Minivent; Hugo Sachs Elektronik, March-Hugstetten, Germany). Core body temperature and head temperature were monitored throughout the experiment using temperature probes inserted into the rectum and left temporalis muscle, respectively, and controlled with a heating lamp and heating pad. Electrocardiogram (EKG) was monitored continuously with subcutaneous needle electrodes. Cardiac arrest was induced by injecting 50 μL ice-cold 0.5 mol/L potassium chloride into a PE-10 catheter inserted into the right internal jugular vein and confirmed by EKG asystole. During CA, head temperature was maintained at 38.9 ± 0.02 °C while body temperature was allowed to decrease to 29.9 ± 0.03 °C. The CPR was initiated after 8 minutes of CA by injection of 0.5 to 1 mL of epinephrine solution (16 μg epinephrine/mL) and chest compressions at a rate of 300/min. Ventilation was resumed with 100% oxygen at a rate of 200/min. Return of spontaneous circulation (ROSC) was assessed by reappearance of electrical activity on the EKG monitor and observation of visible cardiac contractions. Animals were excluded from the study if ROSC was not achieved within 2 minutes.
In all, 5 mg/kg 4-phenylchalcone oxide (4-PCO, Enzo Life Sciences, Farmingdale, NY, USA) or vehicle (dimethylsulfoxide, DMSO) was diluted in 500 μL sterile 0.9% saline and administered by intraperitoneal injection 4 minutes after ROSC. Injections were repeated 24 hours later in animals scheduled for 3-day survival after CA/CPR.
Additional animals underwent sham surgery. These animals were instrumented and monitored as above, but no potassium chloride or epinephrine was injected.
A separate group of mice (
Histologic Analysis
One (
Fluorojade B Staining
We additionally used Fluorojade B staining to identify dead and dying neurons in hippocampal CA1 3 days after CA/CPR. Deparaffinized sections adjacent to the ones used for H&E were stained for 20 minutes in 0.0004% Fluorojade B solution (EMD Millipore, Billerica, MA, USA) after 10 minutes incubation with 0.06% potassium permanganate. The density of Fluorojade B-positive dead/dying neurons in the CA1 regions of both hemispheres was assessed at × 40 magnification using newCAST software (Visiopharm, Hørsholm, Denmark). Cells were counted in an average of eight counting frames (50 × 50 μm) per CA1 region, which were selected by systematic uniform random sampling.
Immunohistochemistry
Adjacent hippocampal sections were stained for activated microglia using a Mac-2 antibody (Cedarlane, Burlington, NC, USA) that recognizes galectin-3, which is expressed by activated microglia. 15 Sections were deparaffinized and stained overnight with Mac-2 antibody (1:200), followed by incubation with a biotin-labeled goat-anti-rat secondary antibody (1:150) and Cy-3 linked streptavidin (1:700; both from GE Healthcare, Piscataway, NJ, USA). These conditions allow selective staining of activated microglia only, with no Mac-2-positive cells apparent in naïve or sham-operated animals. Photomicrographs of Mac-2-stained sections were obtained at low magnification (x 5). For analysis of microglial activation, ImageJ software (National Institutes of Health) was used to quantify the percentage of pixels above background staining (thresholded area) in a region of interest manually drawn around the hippocampus (see Figure 1) on three coronal sections adjacent to the sections used for cell death analysis.
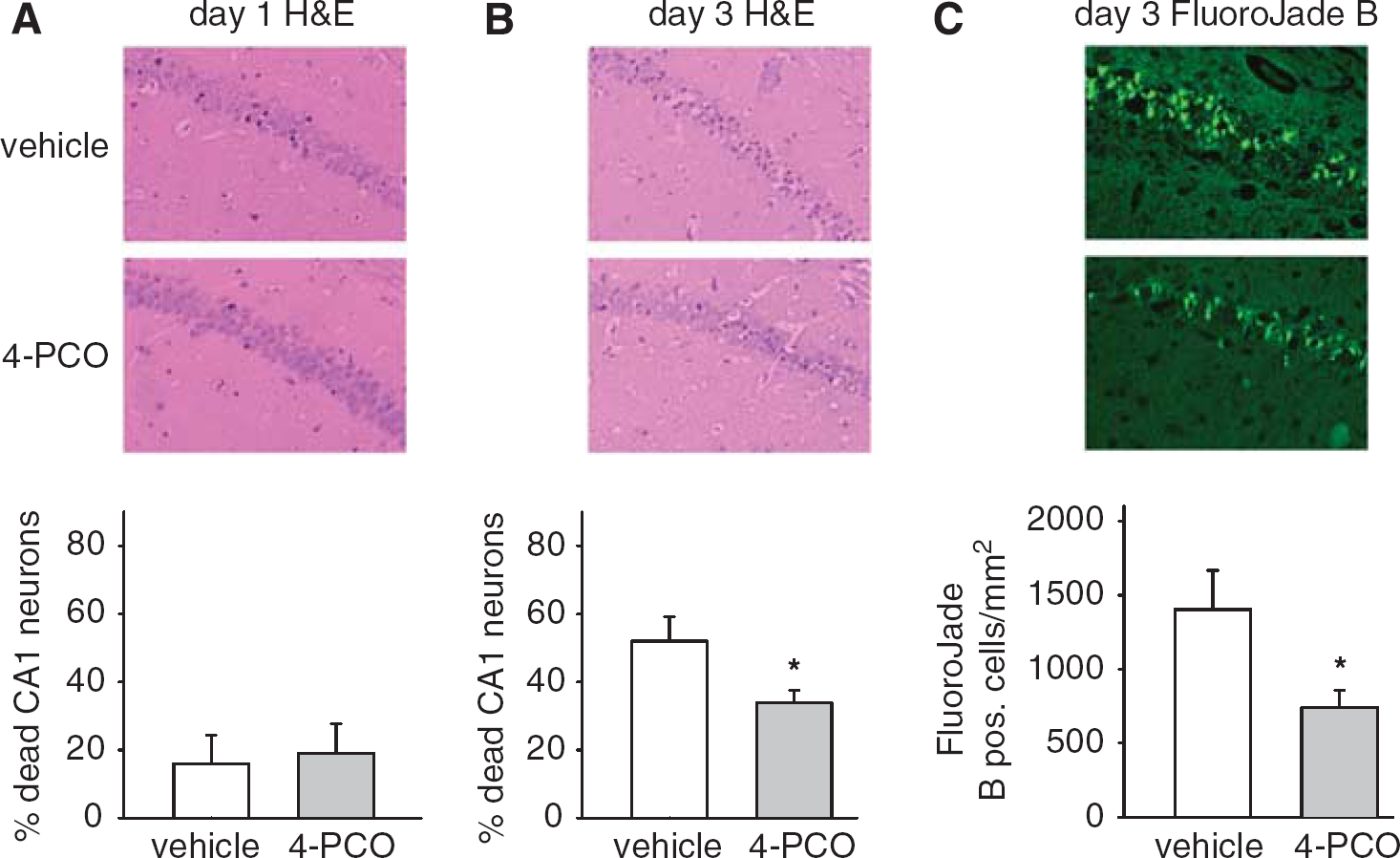
Inhibition of soluble epoxide hydrolase reduces delayed death of CA1 neurons after cardiac arrest. Few CA1 neurons are dead or dying on the first day after CA/CPR independent of treatment (
Quantitative RT-PCR
Mice were deeply anesthetized and transcardially perfused with 0.9% saline 1 day after CA/CPR (
Analysis of Nuclear Factor-κB Activity
Mice were deeply anesthetized and transcardially perfused with 0.9% saline 24 hours after CA/CPR (
Isolation of Microglia from Adult Mouse Brain After Cardiac Arrest/Cardiopulmonary Resuscitation by Magnetic Bead-Assisted Cell Sorting
Naïve mice (
Fear Conditioning
We used a fear conditioning paradigm to assess long-term deficits in memory and learning after CA/CPR. An additional cohort of mice was followed for 10 days after CA/CPR. During this time, mice received daily intraperitoneal injections of vehicle (
Data Analysis and Statistics
SigmaStat software (Systat Software Inc., Chicago, IL, USA) was used for statistical analysis. Groups were compared by two-tailed unpaired Student's
RESULTS
Inhibition of Soluble Epoxide Hydrolase Reduces Delayed Neuronal Death After Cardiac Arrest
Neuronal death was delayed after CA/CPR. Few CA1 neurons showed signs of ischemic injury and death (eosinophilic cytoplasm and pyknotic nucleus) 1 day after CA/CPR, independent of treatment (Figure 1A). Three days after CA/CPR, neuronal death was widespread, with 52±7% of CA1 neurons dead or dying in vehicle-treated mice (Figure 1B). Mice treated with 5 mg/kg intraperitoneal of sEH inhibitor 4-PCO after resuscitation experi-enced significant protection against ischemic cell death, exhibiting only 34±4% of dead or dying CA1 neurons on day 3 (Figure 1B;
4-Phenylcalcone Oxide Treatment Prevents Hippocampal Memory Deficit After Cardiac Arrest/Cardiopulmonary Resuscitation
To determine whether the CA1 injury sustained after CA/CPR is functionally relevant, we used a fear conditioning paradigm to test learning and memory. We compared contextual and cued conditioning to assess whether mice were able to acquire new memory in a hippocampus-dependent (contextual) versus hippocampus-independent (cued) task 8 to 9 days after CA/CPR. Two-way repeated measures ANOVA (drug treatment; contextual or cued test) revealed a significant test × drug interaction (
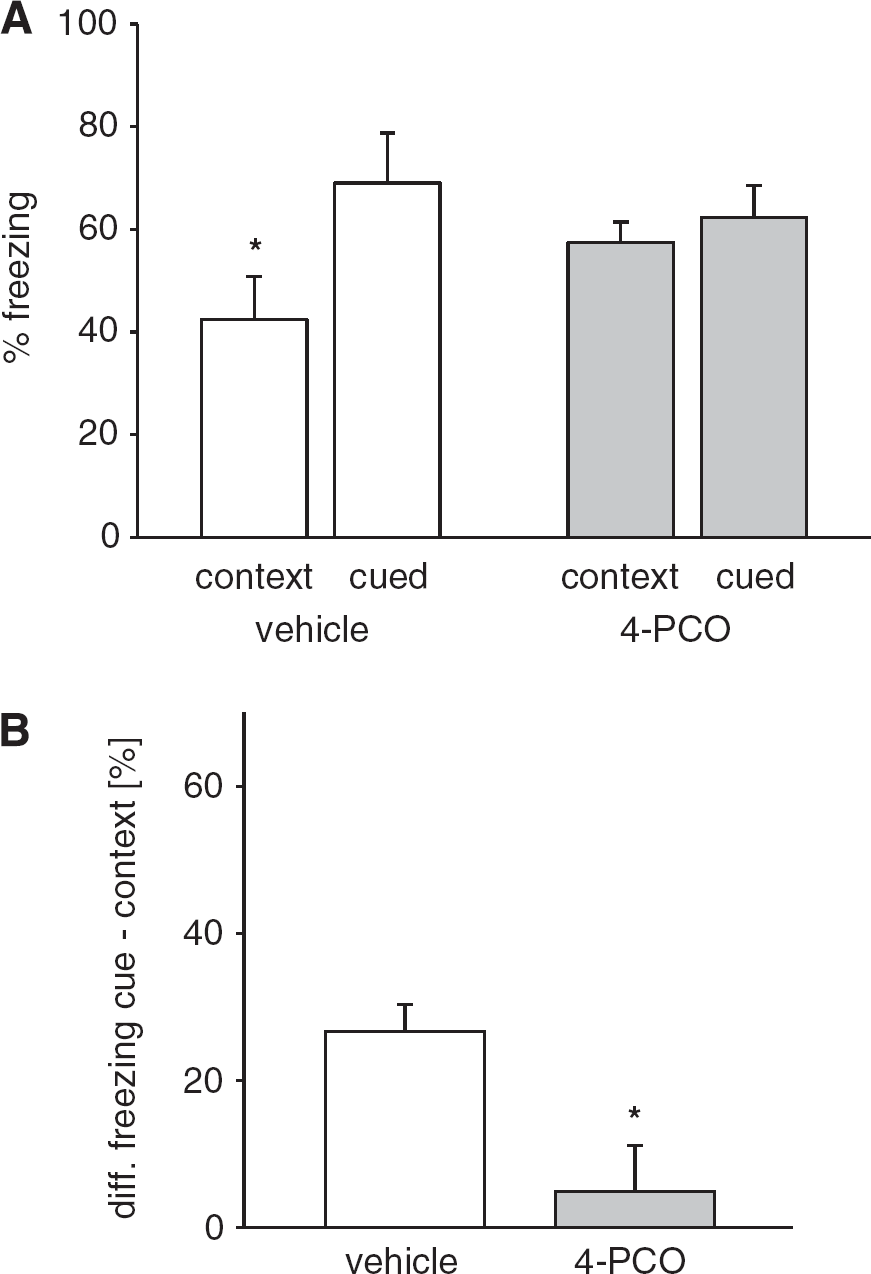
Inhibition of soluble epoxide hydrolase protects hippocampus-dependent memory after CA/CPR. Mice were assessed by fear conditioning 9 to 10 days after CA/CPR. (
Baseline freezing behavior in a novel environment was not different between treatment groups. To more closely examine memory function on the level of the individual mouse, we additionally compared the difference between freezing response to cue versus context in each individual. We found that while vehicle-treated mice consistently exhibited a deficit in contextual compared with cued freezing (27±4% difference), this deficit was absent in 4-PCO-treated mice (5±6% difference,
Microglial Activation is Increased Relative to Neuronal Death After 4-Phenylchalcone Oxide Treatment
We used immunolabeling for galectin-3/Mac-2, a marker of activated microglia, to assess the microglial response to CA/CPR. No Mac-2-positive activated microglia were present in hippocampus of mice after sham surgery. One day after CA/CPR, Mac-2 positive, phenotypically activated microglia with plumb cell bodies and short processes began to appear in the injured hippocampus, most pronounced in the area of the dentate gyrus. Microglial activation increased over time and was much more pronounced 3 days after CA/CPR, when Mac-2-positive microglia were present throughout the CA1 area and the dentate gyrus (Figure 3B). Activated microglia are thought to contribute to delayed neuronal death after CA/CPR by releasing neurotoxic mediators including pro-inflammatory cytokines. Accordingly, we expected that the number of dead CA1 neurons on day 3 would increase as more Mac-2-positive activated microglia appear in the hippocampus. We found that CA1 death and microglial activation (Mac-2-positive area) were indeed correlated on day 3 in both treatment groups with
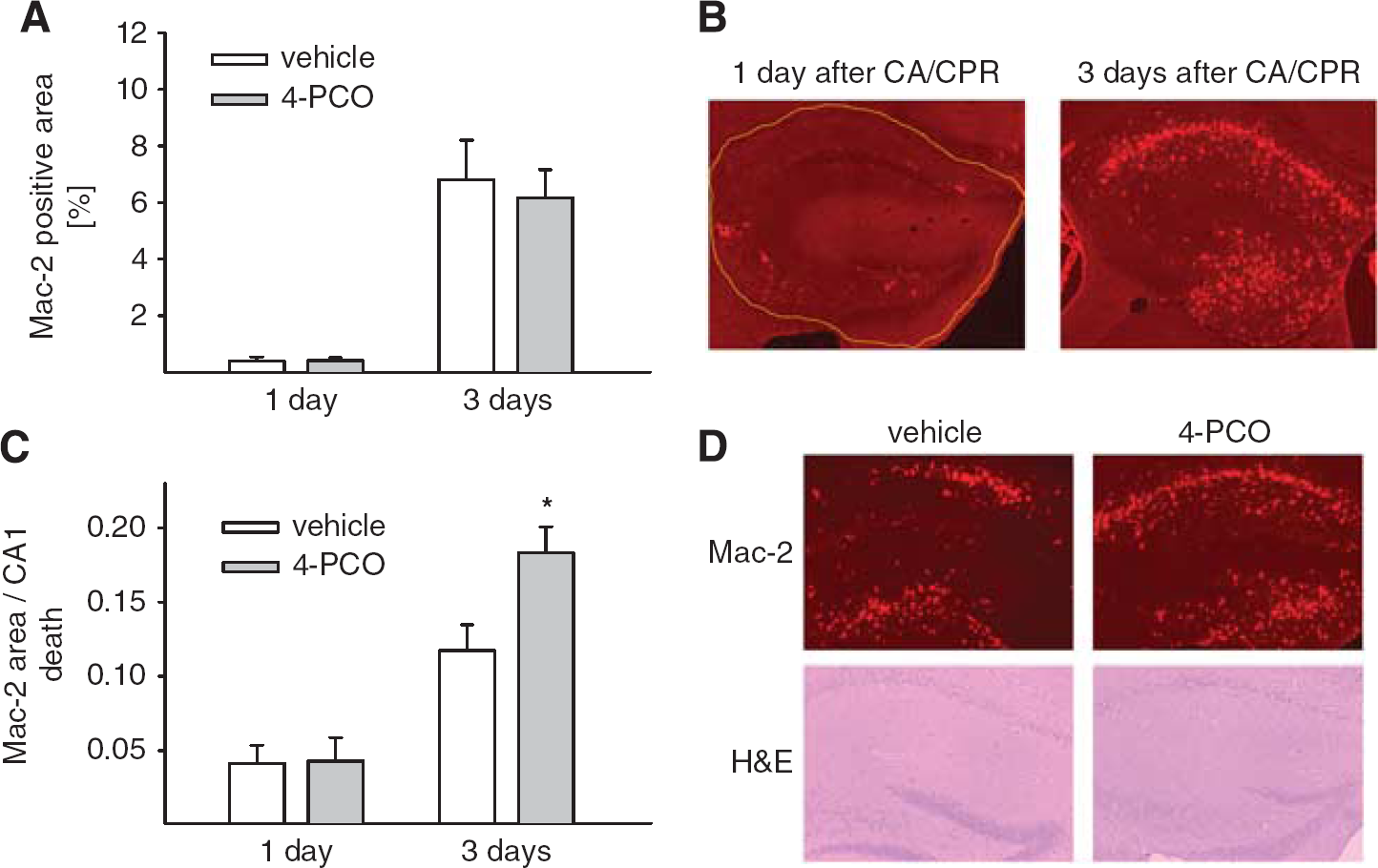
Inhibition of soluble epoxide hydrolase increases microglial activation relative to neuronal death after CA/CPR. (
4-Phenylchalcone Oxide Induces an Anti-Inflammatory Phenotype in Hippocampal Microglia After Cardiac Arrest
To address whether 4-PCO changed the inflammatory milieu created by activated microglia after CA/CPR, we next compared activation of proinflammatory transcription factor NF-κB and expression of inflammatory cytokines between vehicle and 4-PCO-treated mice. The NFκB p65 DNA binding activity was more than fourfold increased in hippocampal nuclear extracts from vehicle-treated mice 1 day after CA, compared with sham surgery (
DISCUSSION
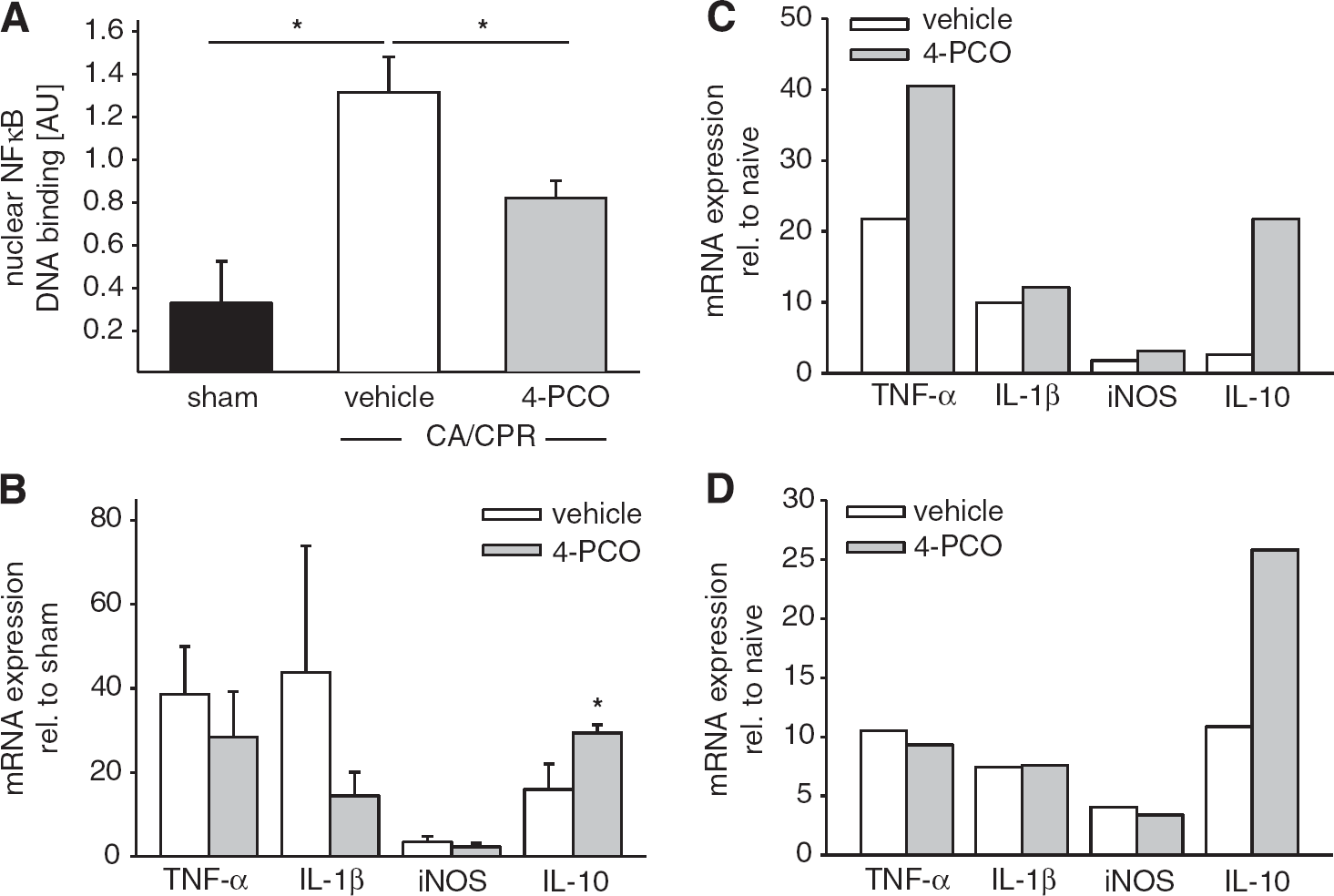
Inhibition of soluble epoxide hydrolase increases antiinflammatory cytokine expression in hippocampal microglia after CA/CPR. (
Our study has three main findings. First, CA/CPR in our mouse model causes early hippocampal inflammation and activates microglia, followed by delayed neuronal death in the CA1 region 3 days after the insult. Second, this delayed neuronal death can be significantly reduced, and hippocampus-dependent memory function protected, by an inhibitor of sEH administered after successful resuscitation, a clinically relevant treatment regimen.
Third, sEH inhibition induces expression of IL-10 in the hippocampus after CA/CPR, which may reduce microglial toxicity and contribute to improved neuronal survival.
The pronounced increase in the number of Mac-2 expressing activated microglia that we saw in the hippocampus over the first days after CA/CPR is in line with other studies using models of global ischemia and reperfusion that find a similarly brisk response from microglia with significant proliferation in ischemia-sensitive areas7,15 and activation that is sustained for many weeks after the insult. 17 Resting microglia constantly survey their environment with their highly mobile processes, sensing input from neurons under their guard. 18 Ischemia/reperfusion injury causes the release of danger-associated molecules such as heat-shock proteins from injured neurons, which are recognized by toll-like receptors on microglia and classically induce an NFκB-dependent increase in transcription of IL-1β, TNF-α, and related cytokines. 19 The NFκB activation and increased hippocampal cytokine transcription in our study support that this pathway is relevant after CA/CPR. Traditionally, microglial activation is seen as part of a death pathway after ischemia/reperfusion as unopposed microglial activation in response to sterile injury exacerbates damage and causes bystander death of healthy or mildly injured neurons. 20 This notion is further supported by the fact that blocking inflammatory cytokines typically produced by activated microglia, such as TNF-α 21 and IL-1β 22 reduces stroke injury. In contrast, however, more recent work points at a significantly more complex role of microglia, which can act as both detrimental and supportive players after ischemia: selective ablation of proliferating microglia, for example, exacerbated stroke injury. 23 Similarly, suppression of microglial activation after CA/CPR has lead to mixed results, with some studies finding improved neuronal survival when antiinflammatory agents are provided, 24 whereas there is no effect in other studies. 25 Inhibition of sEH in our study reduced neuronal death without reducing the number of activated microglia, suggesting that neuronal death is not always a necessary consequence of microglial activation. Rather, there may be less toxic—or even supportive—types of activated microglia that are preferentially induced by sEH inhibition after CA/CPR.
It is increasingly recognized that myeloid cells outside the brain, which are related to microglia, such as blood-borne monocytes, can assume different activated phenotypes depending on the activating stimulus. Some of the phenotypes assumed in response to ‘alternative’ activation have enhanced capacity for phagocytosis, which is beneficial during clean-up, or even produce antiinflammatory mediators, which may help terminate the inflammatory response. 26 Early evidence supports that microglia as well can assume at least two different phenotypes, the classically activated microglia that produced toxic TNF-α, IL-1β, and nitric oxide as well as an alternatively activated microglial phenotype that produces IL-10. 27 The selective increase of IL-10 in response to sEH inhibition in our study may be evidence of such alternative activation of microglia, which adds to the emerging notion that microglia are not always and exclusively toxic, but can be induced to assume a more nurturing, neuroprotective phenotype. Further study is needed to more fully characterize this potentially beneficial microglial phenotype and define how it can be induced by sEH inhibition.
Soluble epoxide hydrolase is a proinflammatory enzyme widely expressed in the brain.
12
Inhibition of sEH reduces systemic inflammation and blocks activation of NFκB.
28
Similarly, inhibition of sEH reduces infarct size after ischemic stroke.
12
The reduction in ischemic injury by sEH inhibition may partly be a direct cytoprotective effect, as neurons themselves express sEH and sEH inhibitors reduce death in neuronal cultures after
Interleukin-10 is a neuroprotective, antiinflammatory cytokine that is expressed by most immune cells, including myeloid cells such as microglia.
33
It is traditionally seen as an antiinflammatory cytokine that can inhibit proliferation of T cells and production of proinflammatory cytokines. It also inhibits NFκB activation,
34
which may have contributed in a negative feedback loop to the reduced levels of nuclear NFκB p65 we observed in 4-PCO-treated mice. Production of IL-10 protects from fatal systemic inflammation in sepsis,
34
similar to the protection that is afforded by sEH inhibition in septic shock,
35
and IL-10-deficient mice have increased mortality from endotoxemia. As expression of proinflammatory cytokines was unchanged in 4-PCO-treated mice, it is less likely that the antiinflammatory action of IL-10 is critical for neuroprotection and improved functional outcome after inhibition of sEH. In addition to its antiinflammatory capacity, IL-10 is also directly neuroprotective, increasing AKT phosphorylation and reducing oxygen-glucose deprivation induced death in cultured neurons
36
as well as protecting neurons against glutamate toxicity.
37
This direct cytoprotection involves IL-10 receptor-dependent activation of the Jak/STAT and AKT pathways leading to increased expression of antiapoptotic Bcl-2 family members.36,37 Inhibition of sEH increases Bcl-2 expression in diabetic nephropathy,
38
suggesting that IL-10-dependent upregulation of antiapoptotic mediators may be one of the mechanisms by which inhibitors of sEH are protective. Both the antiinflammatory and neuroprotective effect may contribute to the benefits afforded by IL-10 after ischemia
Transcriptional regulation of IL-10 is complex. The most important stimulus for IL-10 production in immune cells is ligation of toll-like receptors, which induces IL-10 transcription via activation of NFκB. However, this can be enhanced by additional transcription factors, most importantly cyclic adenosine monophosphate (cAMP)-response element binding protein.
33
Ischemia/reperfusion causes massive norepinephrine release in the hippocampus,
46
which increases cAMP levels via activation of β2-adrenergic receptors. Enhanced cAMP-response element binding protein-mediated transcription in response to cAMP increase may therefore contribute to the induction of hippocampal IL-10 expression that is seen in response to norepinephrine.
47
Inhibition of sEH may be able to further augment this response as sEH inhibitors stabilize cAMP levels
A question of great clinical interest remains whether inhibition of sEH can provide additional protection when combined with clinically used hypothermia. Interestingly, exogenously applied IL-10 improves long-term survival of CA1 neurons after global ischemia when combined with mild hypothermia, while neither hypothermia nor IL-10 alone was able to achieve similar protection. 49 Future studies are needed to investigate whether enhancing endogenous IL-10 levels by sEH inhibition will further augment protection achieved by mild hypothermia after CA/CPR. Previous clinical trials have already established that preclinical administration of potentially neuroprotective drugs immediately after resuscitation is feasible and safe. 50
In summary, our study adds to the growing understanding of the multifaceted role of microglia after brain injury and introduces a promising new treatment regimen after CA/CPR. We found that microglia can be induced to produce beneficial cytokines, namely IL-10, after CA/CPR. Induction of IL-10 may be responsible for the improved survival of hippocampal neurons and the protection of hippocampal memory function achieved by inhibition of sEH after CA/CPR. We conclude that application of an sEH inhibitor after successful resuscitation from experimental CA can improve neuronal survival in the ischemia-susceptible hippocampus and protect memory function, possibly by changing the expression pattern of microglia toward an antiinflammatory, neuroprotective phenotype. Interventions that alter microglial phenotype rather than indiscriminately suppressing microglial activation hold promise for future therapies to reduce brain damage and to improve functional outcome in survivors of CA.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.