
Abstract
Delayed cerebral vasospasm after subarachnoid hemorrhage is primarily due to sustained contraction of arterial smooth muscle cells. Its pathogenesis remains unclear. The degree of arterial constriction is regulated by membrane potential that in turn is determined predominately by K+ conductance (GK). Here, we identified the main voltage-gated K+ (Kv) channels contributing to outward delayed rectifier currents in dog basilar artery smooth muscle as Kv2 class through a combination of electrophysiological and pharmacological methods. Kv2 current density was nearly halved in vasospastic myocytes after subarachnoid hemorrhage (SAH) in dogs, and Kv2.1 and Kv2.2 were downregulated in vasospastic myocytes when examined by quantitative mRNA, Western blotting, and immunohistochemistry. Vasospastic myocytes were depolarized and had a smaller contribution of GK toward maintenance of their membrane potential. Pharmacological block of Kv current in control myocytes mimicked the depolarization observed in vasospastic arteries. The degree of membrane depolarization was found to be compatible with the amount of vasoconstriction observed after SAH. We conclude that Kv2 dysfunction after SAH contributes to the pathogenesis of delayed cerebral vasospasm. This may confer a novel target for treatment of delayed cerebral vasospasm.
Introduction
Cerebral vasospasm is a transient, self-limited narrowing of intradural subarachnoid arteries that occurs several days after subarachnoid hemorrhage (SAH) and can cause ischemia, stroke, and death. Vasospasm is chiefly due to sustained abnormal contraction of smooth muscle cells in the artery (Zhang and Macdonald, 2006). How and why this occurs is not well understood. Arterial diameter is regulated by smooth muscle membrane potential, which controls influx of external Ca2+ through voltage-gated Ca2+ channels (Wellman, 2006). Membrane potential in turn is largely determined by K+ conductance (GK), which is dominated by two K+ channels: voltage-gated K+ (Kv) and large-conductance Ca2+-activated K+ (BK) channels. BK and Kv channels open in response to depolarization, and the BK channel additionally can open in response to increased [Ca2+]i. Both channels thus act as a negative feedback to vasoconstriction by hyperpolarizing the membrane.
Kv channels are present in cerebral arteries of many species including cats, rabbits, rats, and dogs. Pharmacological blockade of Kv channels constricts cerebral arteries in vivo in rats (Quan and Sobey, 2000) and rabbits (Knot and Nelson, 1995). Evidence that K+ channels might be important in vasospasm after SAH was reported by Waters and Harder (1985), who found cat basilar artery myocytes to be contracted and depolarized by ∼15 mV after SAH. Vasospastic myocytes from dog basilar arteries also had a ∼17 mV depolarization under physiologic conditions in vitro, which was related to a decreased GK (Harder et al, 1987). This was consistent with results in other SAH models (Zuccarello et al, 1996; Quan and Sobey, 2000).
Despite reports suggesting a role of K+ channels in vasospasm after SAH, the functional and structural alterations in K+ channels that occur after SAH are not known. We tested the hypothesis that K+ channel dysfunction leads to membrane depolarization, arterial contraction, and vasospasm after SAH. Further, we used electrophysiological, isometric tension, molecular biological, and immunohistochemical methods to identify and characterize the type of K channel responsible for these changes in a dog model of SAH.
Materials and methods
Dog Model of Subarachnoid Hemorrhage and Isolation of Smooth Muscle Cells
Our methods for creation of SAH in dogs have been described (Macdonald, 2005; Xie et al, 2007). In brief, mongrel dogs (weighing 15 to 20 kg) were randomly divided into a control or an SAH group. Dogs were placed under general anesthesia and underwent baseline transfemoral cerebral angiography on day 0. Body temperature, arterial PCO2,and PO2, blood pressure, and heart rate were monitored during procedures and maintained within physiologic limits. Immediately after angiography, dogs were turned prone and the cisterna magna was punctured percutaneously with a spinal needle (Sherwood Medical, St Louis, MO, USA). A volume of 0.3 mL/kg CSF was allowed to drain spontaneously, after which 0.5 mL/kg of fresh, autologous, arterial, nonheparinized blood was withdrawn from the femoral artery and injected into the cisterna magna at a rate of 5 mL/min. Cisternal blood injection was repeated on day 2, resulting in consistently severe vasospasm 7 days later. Dogs were tilted 30° head down during cisternal blood injection and for 15 mins thereafter, after which they were turned supine, the femoral catheter was removed, and the femoral artery was ligated and the was wound closed. Control animals were killed after angiography on day 0, whereas all other animals underwent repeat angiography 7 days after SAH and were then killed. All procedures on animals were performed under general anesthesia and were approved by the Animal Care and Use Committee of The University of Chicago.
Smooth muscle cells were isolated from normal and vasospastic basilar arteries (Nikitina et al, 2007; Xie et al, 2007). Animals were euthanized under general anesthesia by exsanguination and perfusion with ice-cold phosphate-buffered saline (PBS, pH 7.4). The basilar artery was removed and segments were frozen in liquid N2 for study of mRNA, protein, and immunohistochemistry, placed in 4% paraformaldehyde for histology or used for electrophysiology. The pieces for molecular studies had the endothelium removed by passing a wire through the lumen multiple times and then flushing with PBS. For electrophysiology, artery segments were placed in dissection solution (in mmol/L: NaCl 130, KCl 5, MgCl2 1.3, 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) 10, glucose 5, with 100 U/mL penicillin and 0.1 mg/mL streptomycin). They were cut into small pieces and smooth muscle cells isolated by enzymatic digestion in a solution containing 500 U/mL collagenase type IV, 50 U/mL elastase, 100 U/mL DNase I, and 1 mg/mL trypsin inhibitor.
Isometric Tension
Our methods for isometric tension have been described previously (Macdonald et al, 2006; Young et al, 2007). Arteries were placed in Krebs—Henseleit buffer (in mmol/L: Na+ 139, K+ 5.9, Ca2+ 2.5, Mg2+ 1.2, Cl− 124, HCO3− 25, SO42− 1.2, H2PO4− 1.2, and glucose 11) bubbled with 95% O2 and 5% CO2 at 4°C. The endothelium was denuded by passing an angiography guide wire inside the arterial lumen. Rings of 3 mm long were cut and isometric tension was recorded using a custom-designed system (Young et al, 2007). Segments were stretched to a circumference equivalent to that seen angiographically before killing and allowed to equilibrate in normal [K+]o (5.9 mmol/L) for 60 mins. All contractions were measured relative to their preceding baseline and were expressed as a percentage of the maximal KCl-induced contraction.
Electrophysiology
Cells were plated in a low-profile, low-volume (180 μL) chamber (Warner Instruments, Hamden, CT, USA) mounted on a Diaphot 200 inverted microscope (Nikon Corp, Tokyo, Japan). Bath perfusion was by gravity, using a custom-built apparatus that permitted complete exchange of the extracellular medium within 5 secs.
Macroscopic currents were recorded using the whole-cell configurations of the patch-clamp method. Borosilicate pipettes were pulled on a Sutter P-97 programmable horizontal puller (Sutter Instrument, Novato, CA, USA) with resistances of 1.5 to 3 MΩ. Electrodes were heat-polished on an MF200 microforge (WPI, Sarasota, FL, USA). Currents were amplified using an Axopatch-1D amplifier (Axon Instruments, Union City, CA, USA) and digitized at 12-bits using a DigiData 1200 interface controlled by pClamp 6 (Axon Instruments). Series resistance in whole-cell recordings was usually <5 MΩ and was always compensated by ≥80%. Access resistance and cell capacitance were read from the amplifier compensation circuitry controls. All recordings were at room temperature (22°C). Grounding was established via a 3 mol/L KCl agar bridge and a standard Ag/AgCl pellet electrode. Macroscopic current analyses were performed using custom-written software in Igor (Wavemetrics, Lake Oswego, OR, USA). Leak subtraction was performed using P/4 protocols unless otherwise stated.
Macroscopic Kv Currents
Kv currents were isolated from BK currents by strong intracellular Ca2+ buffering and block of L-type Ca2+ channels by external Cd2+. The intracellular solution for recording Kv currents contained (in mmol/L): KCl 88.6, KOH 6.4, K2ATP 5, NaGTP 0.1, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA, tetrapotassium salt) 10, MgCl2 2.5, HEPES 10, pH 7.2 with KOH. Resulting currents were insensitive to paxilline (1 μmol/L) or glyburide (10 μmol/L), showing no contamination with BK current and no contribution of ATP-sensitive K+ (KATP) channels (Supplementary Figure 1). For lengthy protocols such as inactivation and recovery from inactivation, P/4 leak subtraction was omitted. Instead, brief hyperpolarizing pulses were included at the end of each episode to facilitate calculation and subtraction of linear leak.
Nystatin-Perforated Current and Voltage Clamp
Nystatin-perforated patch clamp was used to measure membrane potential under the current-clamp mode (I=0) of the Axopatch-1D. The pipette solution contained (in mmol/L): KCl 145, NaCl 5, MgCl2 1, HEPES 10, pH 7.2 with KOH. Final nystatin concentration was 200 μg/mL. In some experiments, Cl− was reduced to 30 mmol/L by replacement with aspartate. Bath solution excluded CdCl2 for membrane potential recordings, but included it along with 1 μmol/L paxilline in voltage-clamp Kv current recordings. Patch pipettes were dipped into internal solution and then backfilled with internal solution containing nystatin. Access resistance usually decreased to below 20 MΩ within 10 mins of cell contact and stabilized around 10 to 15 MΩ before experiments began. Membrane potential was stable 10 to 20 secs after switching from voltage to current clamp. Changes after drug applications were allowed to plateau, after which 30 secs of data were averaged as a representative membrane potential for that experiment.
Quantitative Real-Time mRNA Analysis and Western Blotting
Total RNA and protein were extracted with TRIzol (Gibco BRL, Life Technologies, Rockville, MD, USA) from individual basilar arteries denuded of endothelium as we described before (Aihara et al, 2004; Nikitina et al, 2007; Xie et al, 2007). RNase-free DNase (Promega, Madison, WI, USA) was used to eliminate contamination with genomic DNA. RNA reverse transcription was catalyzed using a SUPERSCRIPT First-Strand Synthesis System (Invitrogen Life Technologies, Carlsbad, CA, USA). RNA (5 μg) was used for reverse transcription in a final volume of 10 μL. The reaction mixture consisted of 50 ng of random hexamers per 1 μg of total RNA. To increase the sensitivity of PCR from first-strand cDNA, RNase H was used to remove the RNA template from cDNA after first-strand synthesis.
Primers and the probe for Kv2.1 were based on the sequenced dog product amplified by Epperson et al (1999) and verified using BLAST against human (GenBank accession number X68302) and pig (GenBank accession number AF026006) Kv2.1 sequences: TGAAGCCACCAGATTCTCCC (forward), GCTATGCTGCCCCCAGTCT (backward), CAGCCCTTTGGCATCACTTCCCAG (probe). Primers and the probe for Kv2.2 were based on the dog sequence of Kv2.2 (Schmalz et al, 1998) and were what we used before on dog basilar arteries with endothelium (Aihara et al, 2004). Primers, probes, and length of PCR products satisfied requirements for Primer Express software (version 1.5, Applied Biosystems, Foster City, CA, USA) for quantitative reverse transcription-PCR. Constitutively expressed 18S ribosomal RNA (18S rRNA; TaqMan Ribosomal RNA Control Reagent; Applied Biosystems) was used as an endogenous control. Quantitative PCR was then performed with real-time TaqMan technology using a Sequence Detection System model 7700 (Applied Biosystems) (Aihara et al, 2004). Expression levels of target genes were evaluated by the ratio of the threshold cycle (CT) number of target mRNA to 18S rRNA.
Protein was quantified by spectrophotometry (at 562 nm) with Micro BCA Protein Assay Reagents (Pierce Biotechnology, Rockford, IL, USA) and 10 mg per basilar artery was diluted in 1% sodium dodecyl sulfate (SDS) buffer (250 mmol/L Tris-HCl, pH 6.8, 10% sodium dodecyl sulfate, 10% glycerol) and separated on a 1.2% Tris-glycine gel (Invitrogen Life Technologies) in sodium dodecyl sulfate-polyacrylamide gel electrophoresis running buffer for 1 h at 160 V. Proteins were then transferred onto methanol-soaked Hybond P (polyvinylidene fluoride) membranes in buffer containing 3-[cyclohexylamino]-1-propanesulfonic acid and 10% methanol for 2 h at 60 V. For blotting, filters were first incubated with 5% skimmed milk in Tris-buffered saline with Tween 20 (20 mmol/L Tris-Cl, pH 7.6, 137 mmol/L sodium chloride, 0.1% Tween 20) at room temperature for 1 h to block nonspecific binding. Filters were incubated with mouse monoclonal Kv2.2 antibody (1:1,000 dilution; provided by Dr JS Trimmer, SUNY, Stony Brook, NY, USA) for 1 h or rabbit polyclonal Kv2.1 antibody (1:1,000 dilution; Alomone, Jerusalem, Israel). After washing, membranes were incubated for 1 h with horseradish peroxidase-conjugated anti-mouse (1:2,000; Santa Cruz Biotechnology, Santa Cruz, CA) or anti-rabbit IgG (1:500; Vector Laboratories, Burlingame, CA, USA). Detection was with the enhanced chemiluminescence Western blotting detection system (Amersham Health, Princeton, NJ, USA). Total protein loaded in each lane was visualized by staining the transferred polyvinylidene fluoride membrane with GelCode Blue Stain Reagent (Pierce Biotechnology). Densitometric analysis was carried out using a custom procedure written in Igor.
Immunohistochemistry
Tissues were sliced into 6-μm-thick sections on a cryostat at −20°C and were fixed on slides in 4% paraformaldehyde in PBS for 10 mins. After rinsing, the slides were incubated in 3% hydrogen peroxide for 5 mins and then 10% normal goat serum in PBS containing 0.025% Triton X-100. Kv2.2 (1:100 dilution) or Kv2.1 antibodies (20 mg/mL) were added overnight at 4°C. Negative controls replaced the primary antibody with mouse IgG2a (same isotype as primary antibody) at the same concentration. None showed specific staining. After washing with PBS, the slides were incubated with anti-mouse IgG-horseradish peroxidase (Envision kit; Dako, Carpinteria, CA, USA) or biotinylated anti-rabbit IgG (1:200, incubation solution; Vector Laboratories) for 30 mins. Antibody-antigen binding was detected using the diamino-benzidine (DAB) (Dako) or avidin-biotinylated enzyme complex (ABC)–horseradish peroxidase—DAB systems (PK-6100; Vector Laboratories). Slides were counterstained with hematoxylin and photographed under light microscopy.
Chemicals
All chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA), with the exception of BAPTA, which was obtained from Tef Labs (Austin, TX, USA). Hanatoxin was a gift from Dr Kenton Swartz (NINDS, Bethesda, MD, USA). Correolide was provided by Merck Company (Rahway, NJ, USA).
Statistical Analysis
An internal magnification standard was included in each angiogram. Vasospasm was assessed by comparing diameters of basilar arteries from day 0 to day 7 as measured at five predetermined points by two blinded observers using an optical micrometer. Computer simulations were run with SCoP version 3.52 (Simulation Resources Inc., Redlands, CA, USA). Data were compared using t-tests (Sigmaplot; SPSS, Chicago, IL, USA) or analysis of variance (Prism, GraphPad Software, San Diego, CA, USA). A P-value <0.05 was considered significant. Linear regression was performed using the least squares method and fits to curves were performed using the Levenburg—Marquardt algorithm in Sigmaplot or Igor. Goodness of fit was assessed by both visual inspection of fits and residuals and by the coefficient of determination (R2). All data were obtained from cells from at least five dogs and are presented as mean±s.e.m.
Results
Dog Model of SAH
Angiography showed SAH caused significant reduction in basilar artery diameter 7 days after the induction (1.52±0.03 mm for controls, 0.65±0.03 mm for SAH, P<0.001, n=28 SAH, n=21 control dogs). This corresponded to a 52%±5% decrease in diameter after SAH, P<0.001. We did not inject physiologic saline into the cisterna magna of control dogs but in subsequent experiments, this did not change basilar artery diameter or have any effect on expression or function of other ion channels (Xie et al, 2007). There were no significant differences in physiologic parameters between groups at day 0 or in SAH dogs between day 0 and day 7 (data not shown).
Kv Current in Control and SAH Smooth Muscle Cells: Voltage Dependence and Kinetics
The Kv current (IKv) was of the ‘delayed-rectifier’ type, activating and inactivating slowly, with no transient A-type rapidly inactivating component (Figure 1A). Steady-state availability (IKv inactivation) was determined using standard double-pulse protocols (Figure 1B), and steady-state conductance—voltage (GV) relationships for IKv were derived from normalized tail current amplitudes obtained at −20 mV (Figure 1A). An envelope of tails test (Supplementary Figure 2) was performed to test the validity of the latter method. The peak/tail current ratios were close to 0.3, as predicted by the Goldman—Hodgkin—Katz current equation for an otherwise nonrectifying channel with a K+ gradient of 5/145 mmol/L. Tail current ratios remained constant over time, suggesting that the initial amplitude of tails can be used to provide a quantitative measure of IKv. This also suggests that IKv is dominated by a single component or at least by components that have similar IV relationships, kinetics, and rectification (Sanguinetti and Jurkiewicz, 1990; Carl et al, 1996).
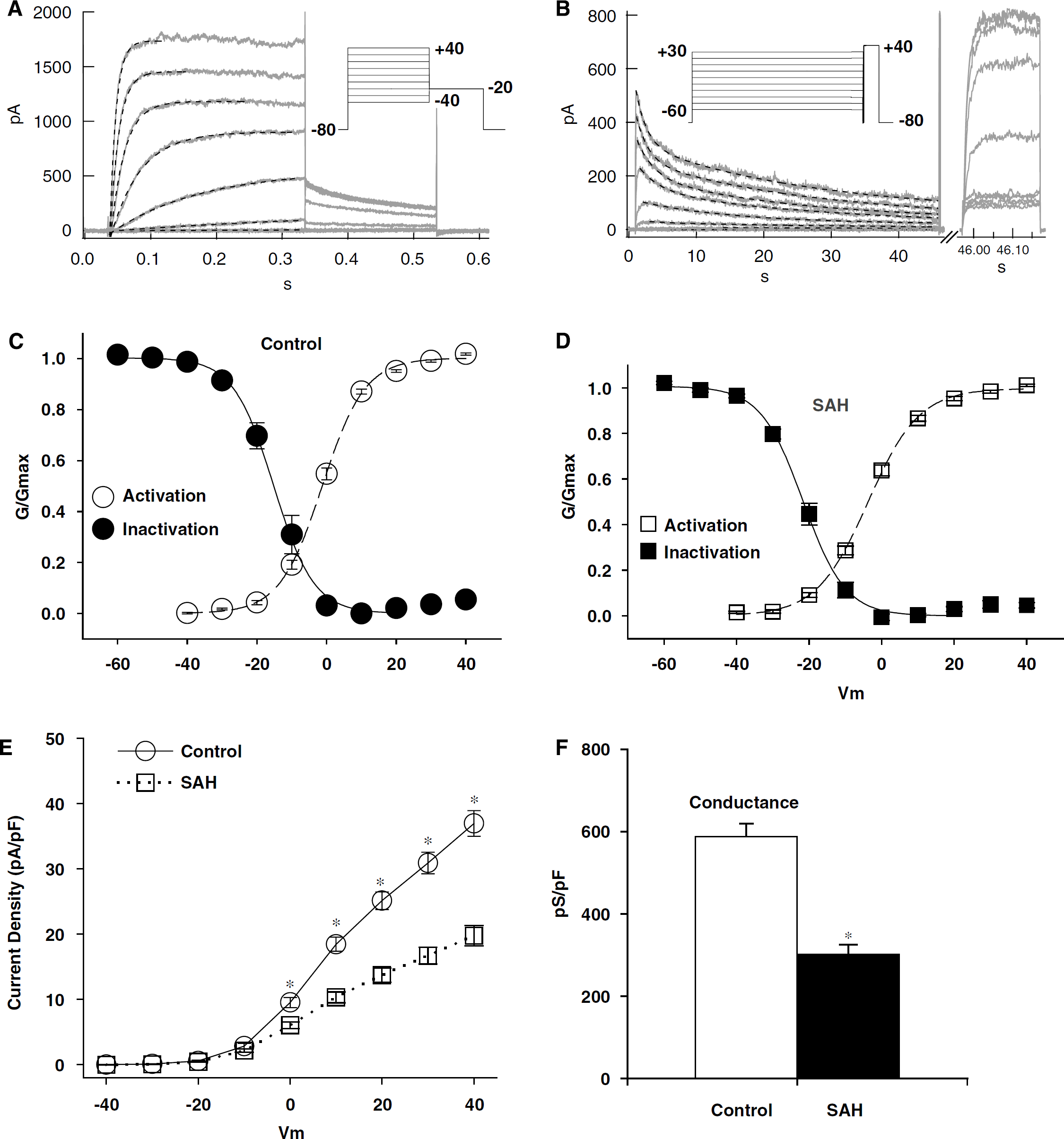
Kv currents in control and vasospastic myocytes. Typical currents recorded during activation (
Summary of Kv current characteristics and pharmacology a
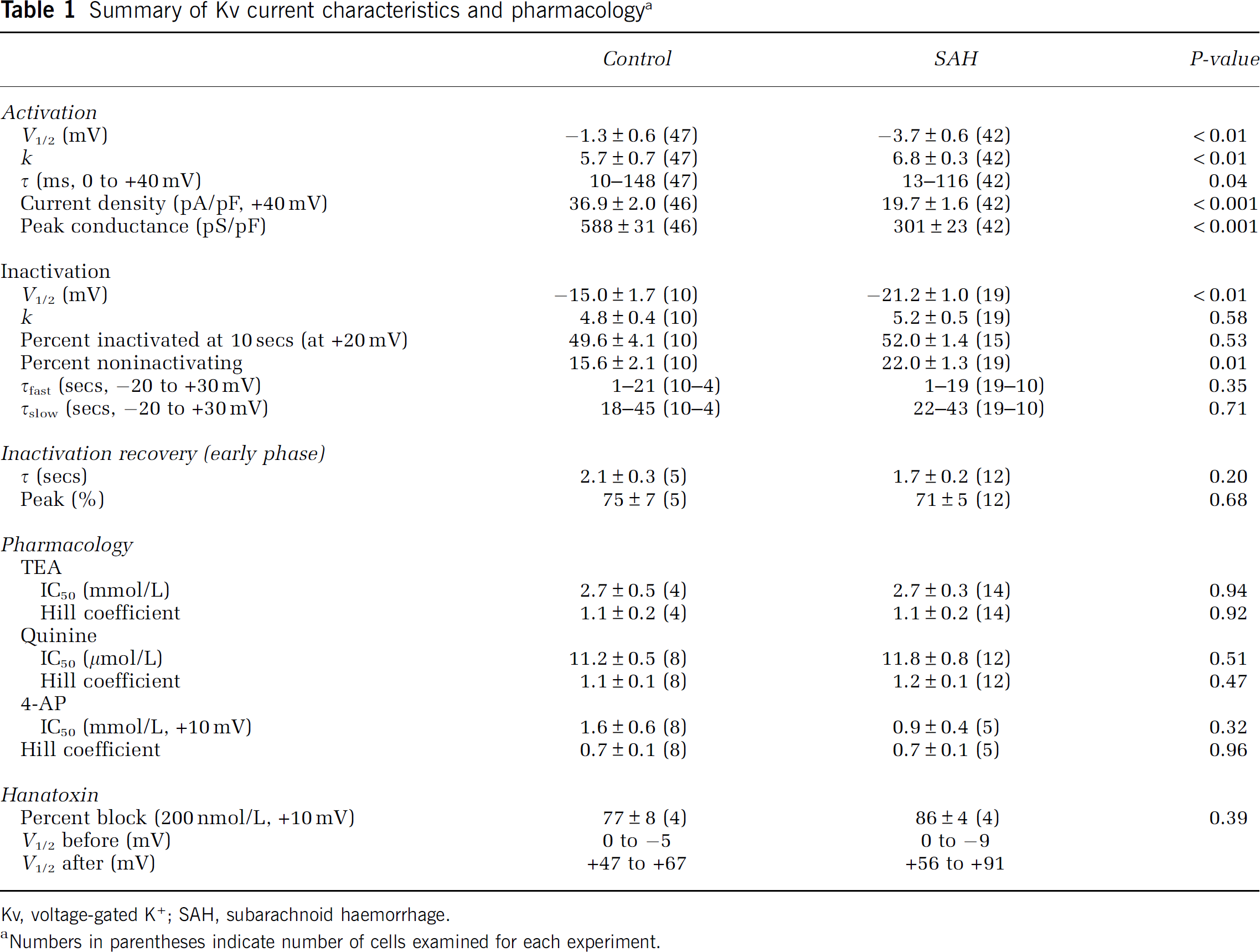
Kv, voltage-gated K+; SAH, subarachnoid haemorrhage.
Numbers in parentheses indicate number of cells examined for each experiment.
Although the voltage dependence of activation and inactivation appeared broadly similar between control and vasospastic myocytes (Figures 1C and 1D), the midpotential for activation was somewhat left shifted (2.4 mV) and the midpotential for inactivation even more so (6.2 mV) in SAH (Table 1). Kv currents from vasospastic cells were significantly smaller (Figure 1E, P<0.001 at Vm from 0 to +40). To ensure that this was not simply due to a shift in the GV relationship after SAH (unlikely, since V1/2 for activation was more hyperpolarized in SAH), maximum control and vasospastic slope conductance (Smirnov et al, 1994) were compared between +20 and +40 mV (where channel activation was near complete). The resulting conductance density of IKv (pS/pF) was nearly halved in vasospastic myocytes, which is significantly smaller than that in normal control cells (P<0.001, Figure 1F and Table 1).
The time course of IKv activation was well fitted by single exponentials at depolarized potentials ≥0 mV in both control and vasospastic myocytes (Supplementary Figure 3). The voltage dependence of the resulting time constants was shifted significantly negatively (two-way analysis of variance, P=0.04, Table 1), with slightly faster rise times at less depolarized potentials, and vice versa in the SAH group. IKv inactivation was very slow and complex, and was best fit by the sum of two exponentials (τfast and τslow), with less apparent voltage dependence for the slower component in both groups (Table 1, Supplementary Figure 3). Inactivation was incomplete, with a smaller inactivation-resistant fraction in control than in SAH (17% versus 22%, P=0.01, Table 1). Recovery from inactivation was similarly complex in both groups and appeared to take place in at least two phases, with an early phase (t=1.7–2.1 secs) contributing to 70% to 80% of recovery (Table 1, Supplementary Figure 3). Full recovery often took over a minute.
Kv Current in Control and SAH: Pharmacology
The preceding results showing similar Kv current kinetics in control and vasospastic myocytes suggest that one type of Kv channel predominantly contributes to Kv current in both groups. Pharmacological investigations were undertaken to identify this Kv channel. Tetraethyl ammonium (TEA) and quinine blocked IKv in both control and vasospastic myocytes equally well (IC50=2.7 versus 2.7 mmol/L and 11.2 versus 11.8 μmol/L for TEA and quinine, respectively, P>0.5, Table 1 and Supplementary Figure 4). Block by 4-aminopyridine (4-AP) was voltage dependent, with less block at more depolarized potentials (Figures 2A and 2B, Table 1). There was no difference between control and SAH animals in the 4-AP blockade efficiency (Figures 2C and 2D). For example, IC50 value was 1.6 versus 0.9 mmol/L at +10 mV in control and SAH, respectively (Figure 2D, Table 1, P>0.3). This pharmacological profile is nearly identical to that of Kv2.2 in dog gastrointestinal smooth muscle (Schmalz et al, 1998) and, together with the preceding kinetic experiments, strongly indicates Kv2.2 to be the predominant Kv channel in dog basilar artery. Subsequent investigations were thus designed to prove this.
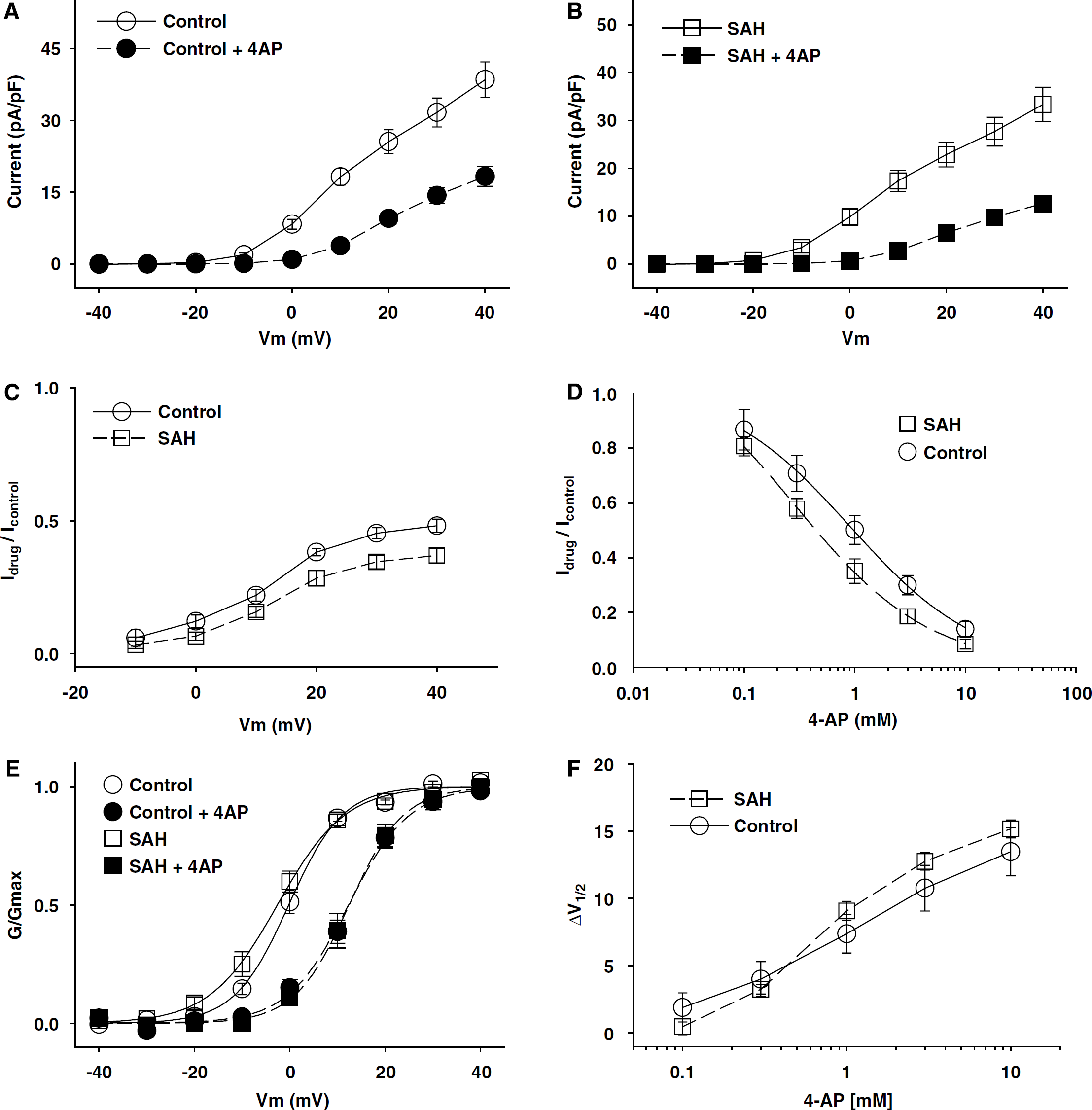
Kv current block by 4-AP. Kv current in control (
Evaluation of IV curves before and after 4-AP (Figure 2) suggested a right shift in current activation, which has been described for some Kv channels after 4-AP application, including Kv2 channels (Kirsch and Drewe, 1993; Kerr et al, 2001). This was confirmed and found to be dose-dependent after examining tail currents before and after 4-AP (Figure 2E). Such voltage-dependent behavior of 4-AP can be explained using a model proposed by Armstrong and Loboda (2001). In this model, the channel must open for 4-AP to gain access to its blocking site (use dependence); once there, it promotes channel closure, thus trapping itself within the channel. This can be tested by allowing 4-AP to reach steady-state inhibitory concentrations while keeping the channel hyperpolarized and closed. The first depolarizing pulse under these conditions should initially show little/no sign of block but subsequently show decreased current (block onset) as 4-AP gains access to open channels. Subsequent pulses should be fully blocked. Similarly, allowing blockade to be washed off while keeping the channel hyperpolarized and closed should keep 4-AP trapped in the channel. In this test, the first depolarizing pulse should show gradual block offset as 4-AP escapes from newly opened channels, subsequent pulses will not show further recovery. Both predictions were confirmed experimentally (Supplementary Figure 5) and have been observed in Kv2 channels by others (Kirsch and Drewe, 1993).
The kinetics and pharmacology of IKv in control and vasospastic myocytes in this study were closest to that of Kv2 channels reported previously (Coetzee et al, 1999). To confirm this, currents were recorded before, during, and after application of 200 nmol/L hanatoxin (Figures 3A to 3C), which specifically decreases current through Kv2 channels by acting as a gating modifier rather than a pore blocker (Swartz and MacKinnon, 1997). Hanatoxin reversibly blocked 77% to 86% of IKv in control and vasospastic cells at +10 mV (Table 1 and Figure 3D). A clear shift in the GV curve could be shown for all cells tested at potentials depolarized enough to elicit suitable tails for GV analysis (Figures 4E and 4F). Electrophysiological evidence against the presence of Kv1 class channels was provided by application of correolide (a selective Kv1 antagonist; Albarwani et al, 2003), 1 μmol/L, to control cells, which had no effect (Table 1 and Figure 4D). These results suggest that the dominant component of IKv in both control and SAH might be Kv2 channels.
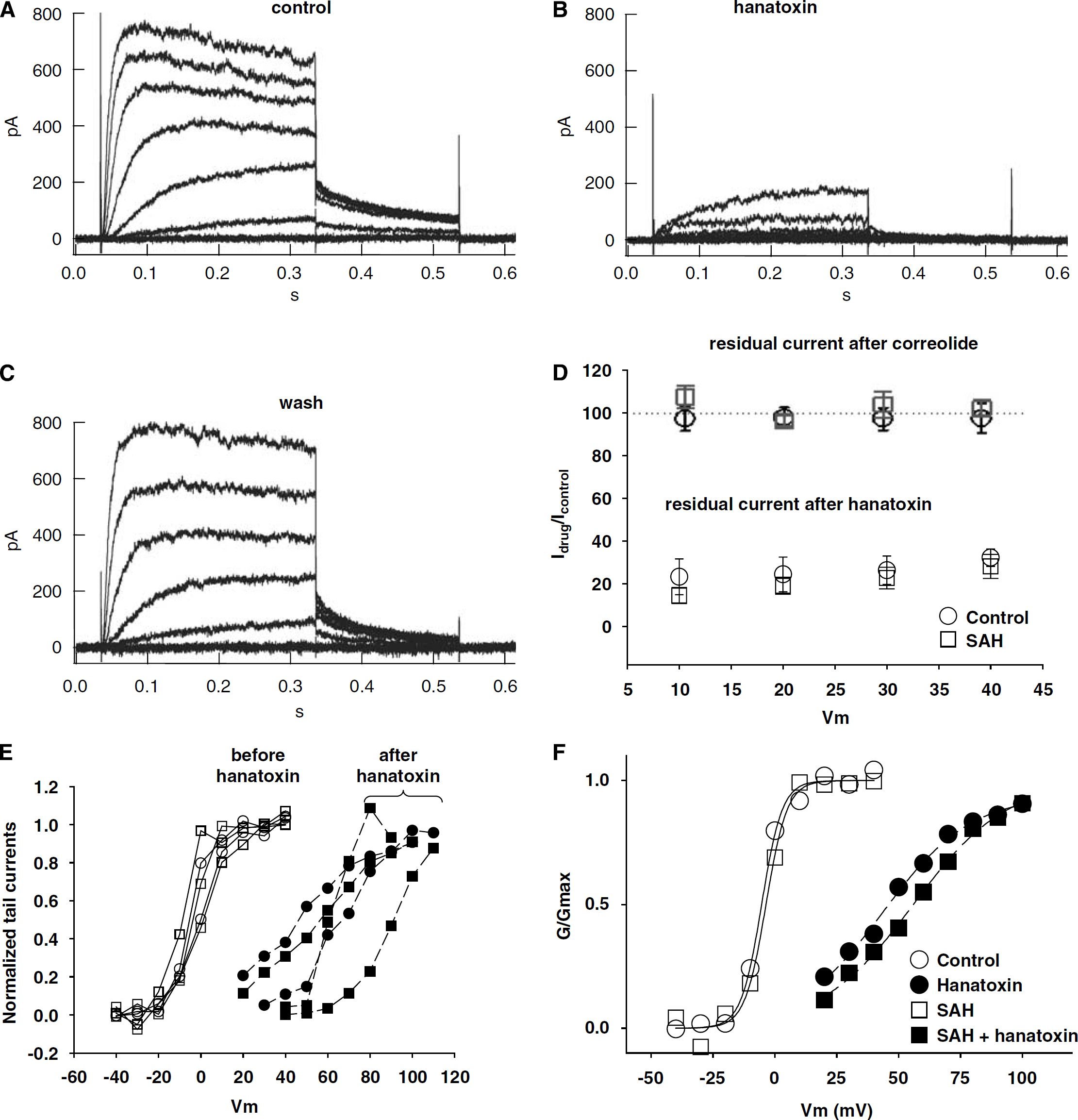
Kv current block by hanatoxin but not correolide. A control myocyte before (
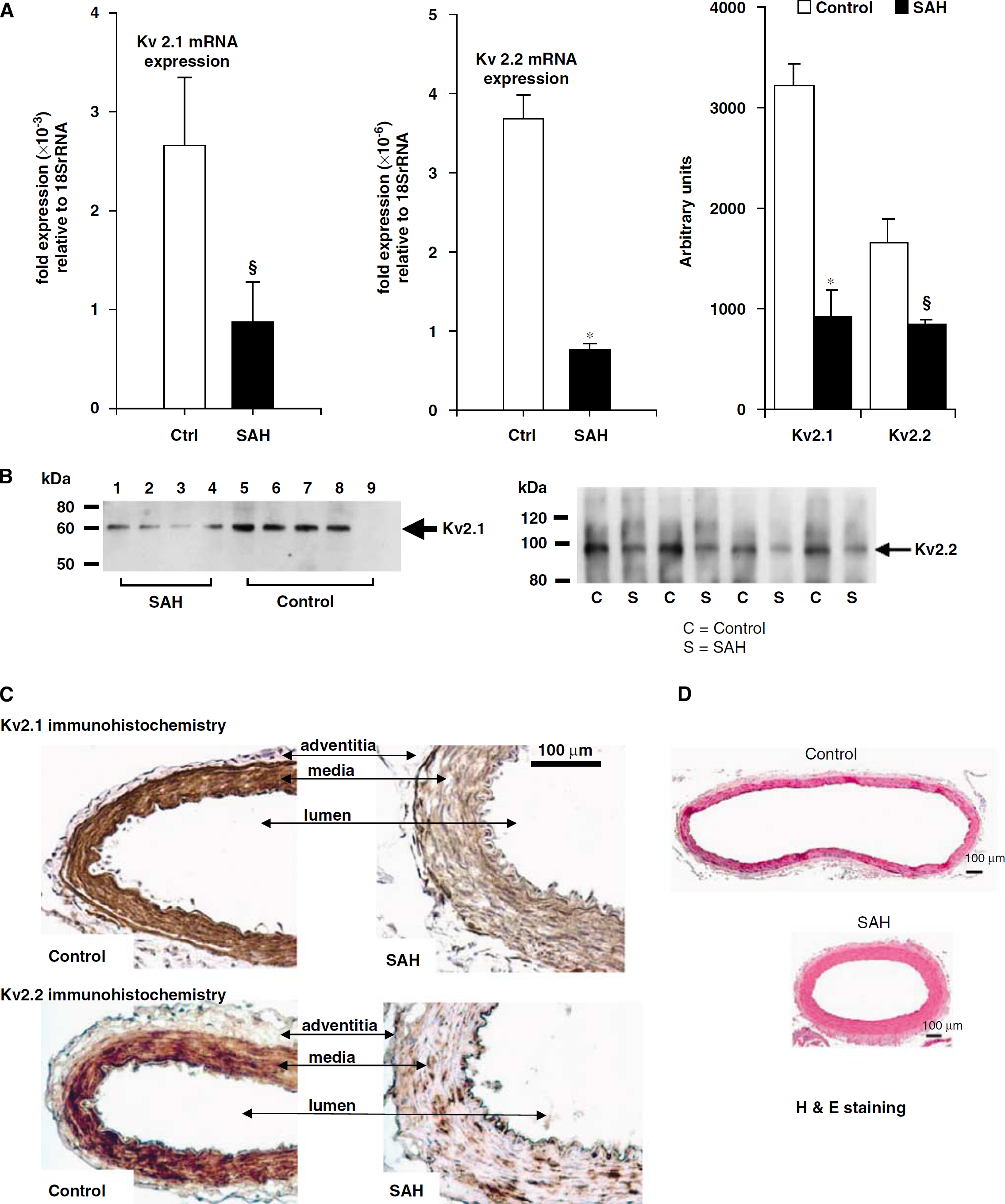
Kv2.1 and Kv2.2 mRNA, protein, and immunostaining. Decreased Kv2.1 and 2.2 mRNA (
Molecular Biology and Immunohistology of Kv2.2 Channels in Control and SAH
The molecular identity of the Kv channels responsible for the observed electrophysiological alterations was identified by quantitative real-time PCR, Western blotting, and immunohistochemistry. There was an over three-fold reduction in Kv2.1 and Kv2.2 mRNA in SAH (Figure 4A, Kv2.1 mRNA: 2.66±0.69 for control, 0.87±0.41 for SAH, P<0.05, n=8 for control, 9 for SAH; Kv2.2 mRNA: 3.68±0.3 for control, 0.76±0.08 for SAH, P<0.001, n=7 dogs for both groups). The decrease in mRNA was also reflected in a decrease in both proteins (Figures 4A and 4B), where Western blotting showed decreased protein for Kv2.1 and 2.2 (Figure 4A, Kv2.1: 3,211±233 for control, 918±265 for SAH, P<0.001 n=4 for both groups; Kv2.2: 1,663±240 for control, 849±48 for SAH, P<0.05, n=4 dogs for both groups). Similarly, immunohistochemistry for Kv2.1 and 2.2 showed less staining in basilar artery sections from SAH versus control animals (Figure 4C, n=4 animals for each group).
Contribution of Kv Channels to Basilar Artery Membrane Potential and Tone
To investigate whether reduced Kv currents could cause vasospasm, we used the nystatin-perforated patch-clamp method to record membrane potential of nondialyzed normal and vasospastic myocytes. Resting membrane potential of vasospastic myocytes (−27.2±1.4 mV, n=40) in physiologic [K+]o was significantly depolarized (P<0.001, Figure 5A) by ∼9.5 mV when compared to control myocytes (−36.7±1.0, n=57).
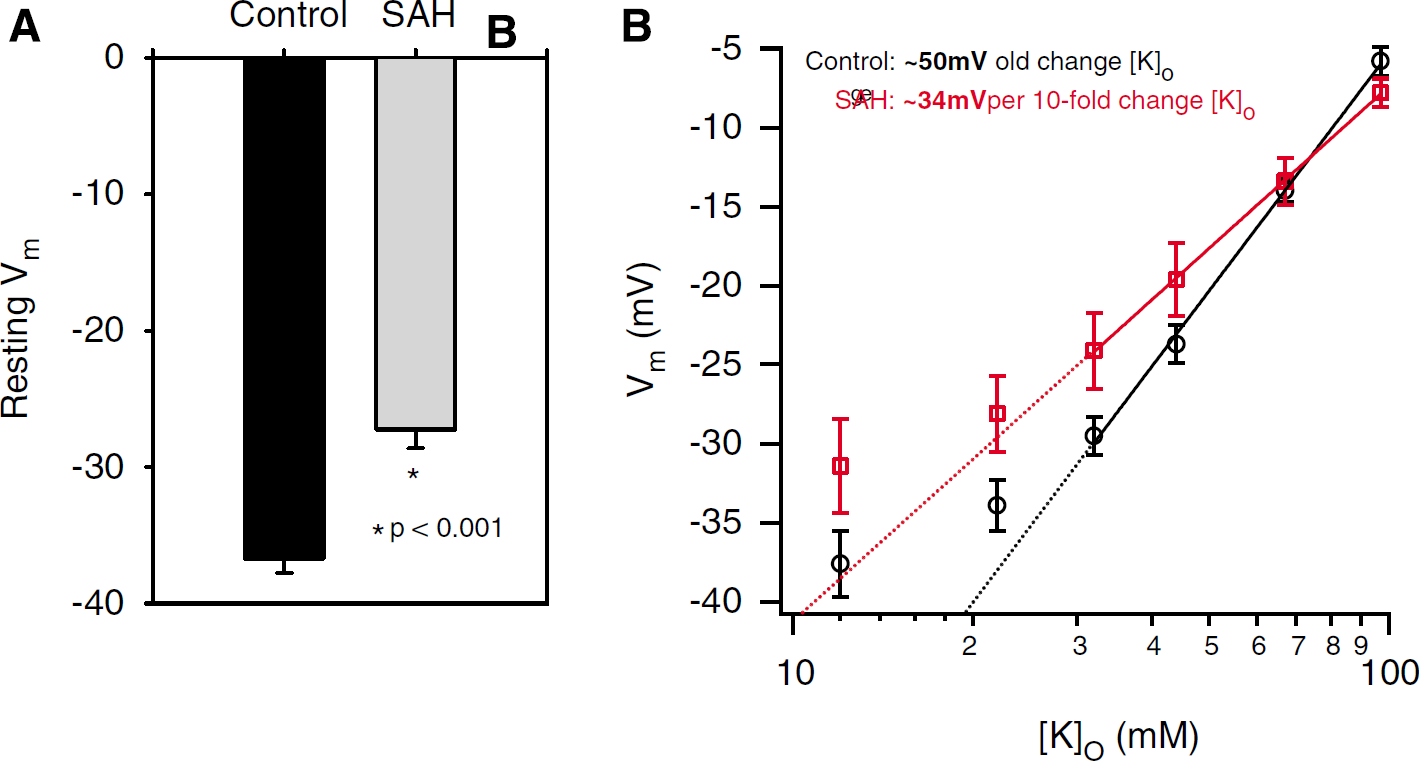
Membrane potential and modulation by EK in control and SAH. (
The relative contribution of GK to the resting potential of a cell can be estimated by measuring membrane potential (Em) at different concentrations of external K+ ([K+]o). The slope of Em versus log([K+]o) should be close to that predicted by the Nernst equation (∼58.6 mV per decade change in [K+]o at 22°C) for membranes where GK is dominant (Harder et al, 1987; Bratz et al, 2002). We used this method to measure the membrane potential by nystatin-perforated patch methods of control and vasospastic myocytes at differing [K+]o (Figure 5B). K+ concentrations were chosen for fitting the data based on Knot and Nelson (1998), who reported that the membrane potential of cerebral arteries was close to the theoretical K+ equilibrium potential (EK) when [K+]o>16 mmol/L. Similarly, Fujiwara et al (1982) found Em versus log([K+]o) was linear when [K+]o>20 mmol/L in the dog basilar artery. Accordingly, data for [K+]o>22 mmol/L were fit to the Nernst equation, resulting in significantly different slopes for control (49.8±1.6 mV per 10-unit change in [K+]o) versus vasospastic myocytes (33.9±0.3 mV per 10-unit change in [K+]o, P<0.001, Figure 5B).
The depolarization and decreased slope of Em versus log([K+]o) suggests diminished outward K+ current in vasospastic cells. To examine the relative contributions of Kv and BK currents to the maintenance of membrane potential and to the reduction in GK, effects of paxilline and 4-AP on membrane potential and whole-cell outward currents were investigated using a nystatin-perforated patch-clamp in control and vasospastic myocytes. Figure 6A shows a typical voltage-ramp experiment illustrating two additive components (separated by a consistent ‘hump’) representing BK and Kv currents. Similar results were observed when Ca2+ was removed from the extracellular solution. BK current was rarely observed at depolarizations below +20 mV, whereas visible Kv current activation could usually be seen by depolarization to ≥−30 mV. These data suggest that, in dog basilar artery myocytes at rest, Kv current is predominant over BK current within the physiologic range of vascular smooth muscle membrane potentials. If so, resting membrane potential should be more affected by Kv rather than by BK block. This was tested by examining the effects of paxilline and 4-AP on the resting membrane potential of basilar artery myocytes. Figure 6B shows this experiment, where paxilline had little effect on membrane potential, whereas 4-AP caused ∼12 mV depolarization. Overall, paxilline depolarized myocytes by 1.7±0.6 (n=7) versus 1.3±0.2 mV (n=3) in control and SAH cells, respectively (Figure 6C, P>0.05), whereas 4-AP depolarized myocytes by 10.5±0.9 (n=5) versus 7.2±1.1 mV (n=7) in control and SAH, respectively (Figure 6C, P<0.05). 4-AP caused significantly greater depolarizations than paxilline in both groups of myocytes (Figure 6C, P<0.05, two-way analysis of variance). Similarly, 4-AP evoked significantly larger contractions than paxilline in control dog basilar artery rings without endothelium under isometric tension (64.3%±10.3% versus 16.9%±5.3%, n=22 and 9, respectively, P<0.01, Figure 6D).
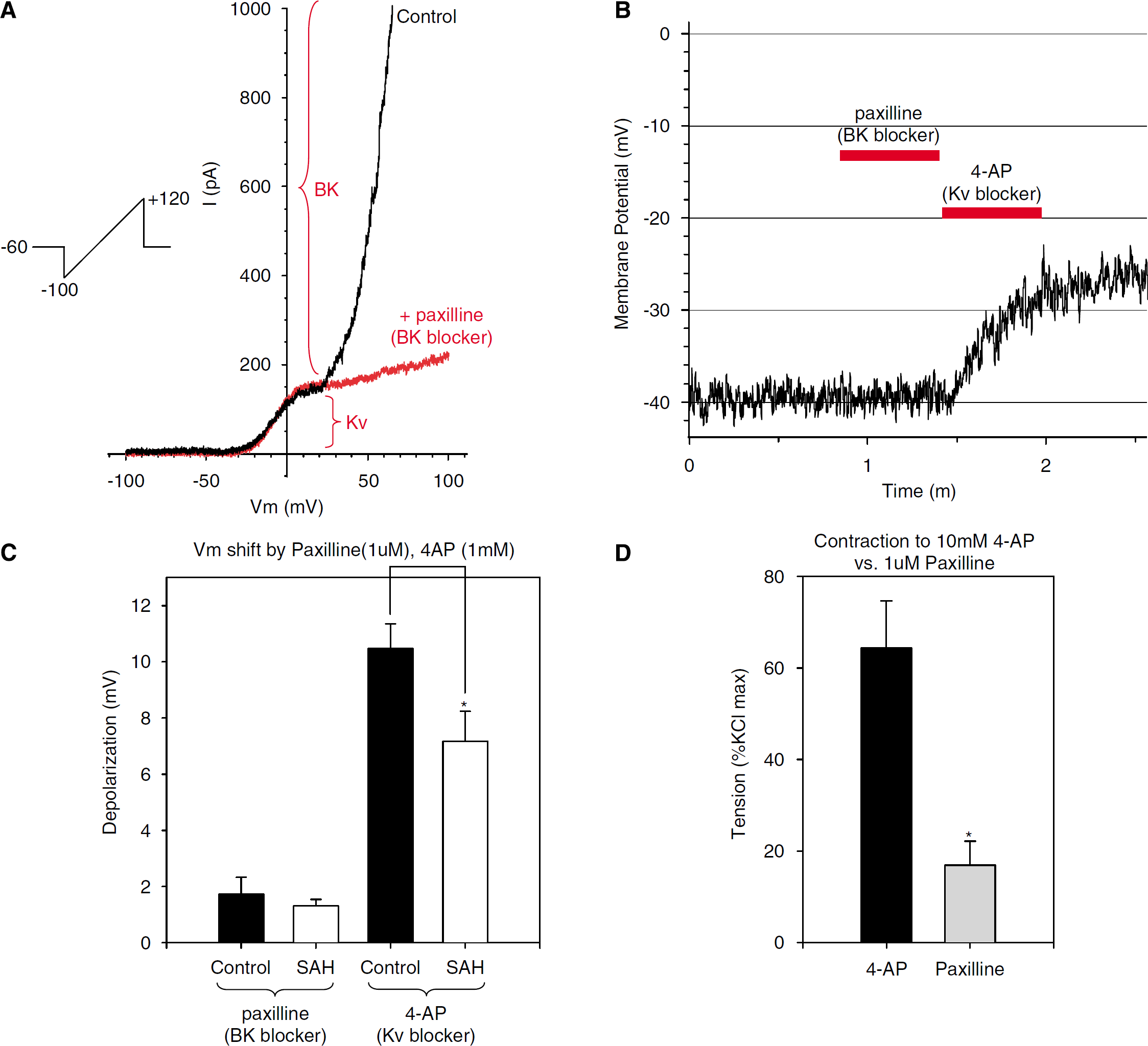
Comparison of contribution of Kv and BK currents to resting membrane potential and tone. (
There is a steep relation between cerebral artery diameter and resting membrane potential as determined by [K+]o (Fujiwara et al, 1982; Knot and Nelson, 1998). Accordingly, we estimated how the degree of depolarization observed in vasospastic myocytes might affect the tone of vasospastic arteries using different [K+]o. We found that KCl-induced changes in tone in control basilar artery rings without endothelium under isometric tension were correlated with KCl-induced depolarization in current-clamped myocytes (Supplementary Figure 6). Control arteries were used, as the purpose was to model the relationship between tone and membrane potential. The degree of membrane depolarization in vasospastic myocytes is compatible with >40% increase in arterial wall tension under these experimental conditions (Supplementary Figure 6).
Discussion
Novel Findings and Central Hypothesis
We report several novel findings. First, there was a significant decrease of Kv current in vasospastic myocytes as compared to normal controls (Figure 1). Second, this decreased Kv current is compatible with the degree of depolarization observed in these cells (Figure 5), which is consistent with the vasoconstriction observed after SAH (Supplementary Figure 6). Third, electrophysiological and molecular data suggest that the channel affected is of the Kv2 class (Figure 4).
Membrane depolarization due to decreased Kv current will lead to contraction mainly by increasing the open probability of voltage-gated Ca2+ channels and increasing Ca2+ influx. Depolarization of vasospastic basilar artery myocytes brings Em closer to the physiologic activation range of voltage-gated L-type Ca2+ channels. Fleischmann et al (1994) reported that window current for these channels occurs between −40 and −20 mV, peaking at −30 mV, with [Ca2+]i falling off steeply in either direction. In support of this, there may be increased intracellular Ca2+ in vasospastic smooth muscle (Kim et al, 1996; Butler et al, 1996). That L-type voltage-gated Ca2+ channels are available during vasospasm is supported by experimental and clinical results showing that local application of high doses of nicardipine to cerebral arteries after SAH prevents vasospasm (Kawashima et al, 2000; Kasuya et al, 2005; Barth et al, 2007).
Identity of Kv Channels in Dog Basilar Artery
Our data suggest that IKv in dog basilar myocytes is carried primarily through one type of Kv channel. The envelope of tails test was unable to separate IKv into kinetically discernable components (Supplementary Figure 2) and dose—response curves for TEA, 4-AP, and quinine were well fit by single isotherms (Figure 2 and Supplementary Figure 4). This argues against more than one pharmacologically distinguishable binding site. Excluding 4-AP, which shows voltage-dependent block, the near-complete block affected by both TEA and quinine is consistent with a single channel dominating IKv in these cells (Figure 2 and Supplementary Figure 5). This view is reinforced by the extent of current inhibition (∼80%) and GV shift (∼50–90 mV) by hanatoxin (200 nmol/L), which is more selective than TEA or quinine and by the lack of effect of correolide (Figure 3).
Electrophysiological experiments were used to further characterize IKv. The extremely slow inactivation kinetics rule out rapidly or moderately rapidly inactivating A-type Kv channels (Kv1.3, Kv1.4, Kv1.7, Kv3.3, Kv3.4, Kv4) (Coetzee et al, 1999). The slow activation kinetics suggest that these currents are not carried by a Kv1 family delayed rectifier such as Kv1.1, Kv1.5, or Kv1.6 (Coetzee et al, 1999). Only two members of the Kv3 family (Kv3.1 and Kv3.2) show comparable slowly activating, slowly inactivating currents, but both are exquisitely sensitive to submillimolar TEA (IC50=0.15 to 0.2 mmol/L) (Coetzee et al, 1999). Similarly, the pharmacological profile of Kv1.5 cloned from dog vascular smooth muscle argues against its presence in dog basilar artery (IC50=200 and 365 μmol/L for 4-AP and quinine, respectively, Kv1.5 not blocked by TEA, antagonized by correolide, see Table 1, Figure 2, Figure 3 for comparison) (Overturf et al, 1994; Albarwani et al, 2003).
The remaining candidate is Kv2. The pharmacological profile of IKv in control and vasospastic myocytes was nearly identical to that of cloned dog Kv2.2 (IC50=2.6 mmol/L for TEA, 1.5 mmol/L at +10 mV for 4-AP, and 13.7 μmol/L for quinine) (Schmalz et al, 1998), suggesting that this channel underlies IKv in dog basilar artery. This was confirmed using hanatoxin, which blocks Kv2 channels with high affinity (Kd=42 nmol/L). Hanatoxin block of Kv2 current is primarily accomplished by shifting the GV curve of the channel to more depolarized potentials rather than actual pore blockade (Swartz and MacKinnon, 1997), which was also observed in dog basilar IKv (approximately 50–90 mV shift, Figures 3E to 3F).
The Kv2 family has two members, Kv2.1 and Kv2.2, with high homology (Hwang et al, 1992). Kinetically, Kv2.1 and Kv2.2 are essentially identical when expressed in oocytes and Kv2.1/2.2 heterotetramers are not readily distinguished from homomultimeric Kv2.1 or Kv2.2 channels (Burger and Ribera, 1996; Blaine and Ribera, 1998). Although Kv2.1 and 2.2 can heteromultimerize when heterologously expressed, they have thus far not been detected together in nature (Blaine and Ribera, 1998). Our molecular biologic data showed that both Kv2.1 and 2.2 transcripts and protein presented in dog basilar artery, which suggest that the molecular identity of the Kv channel mediating delayed-rectifier currents in dog basilar artery is Kv2, likely including Kv2.1 and 2.2.
These findings are different from other reports of Kv channel subtypes in cerebral arteries that describe Kv1 class channels in rodent and human cerebral arterioles (Cheong et al, 2001a, 2001b; Albarwani et al, 2003). It is speculation as to whether this is due to differences in sizes of the arteries studied, species, or to existence of differences between arteries of the anterior and posterior circle of Willis.
Comparison of Kv Currents in Control and SAH
Kv currents were found significantly smaller in vasospastic myocytes after SAH as compared to control myocytes (Figure 1). However, overall kinetic and pharmacological features remained similar (Table 1). This suggests that the reason for diminished IKv after SAH is functional downregulation rather than a change in Kv channel kinetics or subtype. This is reinforced by the observed decrease in Kv2.1 and 2.2 mRNA, protein in Western blotting, and immunohistological staining in dog basilar arteries after SAH (Figure 4). This work thus identifies at a functional and molecular level a target mechanism involving a specific K+ channel dysfunction, which may underlie the smooth muscle cell depolarization and contraction observed during vasospasm after SAH in dogs (Harder et al, 1987), cats (Waters and Harder, 1985), rabbits (Zuccarello et al, 1996), and rats (Quan and Sobey, 2000).
We did not determine how SAH decreases Kv2 expression. One possibility could be enhanced oxidative stress, as it has been shown to decrease Kv function and is hypothesized to play a role in loss of vasodilatory mechanisms seen in hyperglycemic states (Liu et al, 2001). Furthermore, free radical-mediated injury catalyzed by iron within or released from hemoglobin in the antioxidant-poor subarachnoid space has been implicated in the pathogenesis of cerebral vasospasm (Macdonald and Weir, 1994).
Regulation of Cerebrovascular Tone and Membrane Potential by K+ Channels in Normal and Vasospastic Arteries
Membrane potentials recorded from cerebral arteries under physiologic conditions in vitro range between −36 and −46 mV (Faraci and Sobey, 1998). The membrane potentials recorded for control basilar artery myocytes in this study are within this range and are close to those recorded by Nagao and Vanhoutte (1993) using sharp microelectrodes from dog basilar artery rings under isometric tension (−43 mV). In contrast, the membrane potential of vasospastic myocytes was significantly depolarized by ∼9.5 mV, which is consistent with studies in dogs (17 mV; Harder et al, 1987), cats (15 mV; Waters and Harder, 1985), and rabbits (10 mV; Zuccarello et al, 1996, Figure 5A).
The slope of Em versus log([K+]o) was 50 mV per 10-unit change in [K+]o in control basilar artery myocytes (close to that predicted by the Nernst equation, suggesting that EK regulates Em under these circumstances). This slope was reduced to 34 mV per 10-fold change in [K+]o in vasospastic myocytes, suggesting decreased contribution of GK to the resting membrane potential. This decrease in slope during vasospasm agrees with measurements of membrane potential in pressurized vasospastic dog basilar arteries, where the slope was 30 mV per decade change in [K+]o (Harder et al, 1987).
The ∼9.5 mV depolarization observed in vasospastic myocytes correlated with >40% contraction under isometric tension (Supplementary Figure 6). This increased wall tension is qualitatively similar to the ∼53% vasoconstriction observed angiographically 7 days after SAH in the dogs. The findings also are compatible with our measurements of membrane potential in pressurized vasospastic dog basilar arteries that showed an ∼15 mV depolarization of these arteries compared to control (Weyer et al, 2006).
Conclusions
We used electrophysiological, molecular biologic, immunohistological, and isometric tension measuring techniques to show that Kv2 class channels underlie the depolarization and likely contractile vasospasm that occurs after SAH in dogs. This mechanism suggests potential pharmacological targets for vasospasm after SAH. Given the fact that there are limited therapeutic manipulations available clinically for SAH and its subsequent vasospasm, our study may provide a support for a new direction in development of novel antivasospasm drugs.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.