
Abstract
As clinical trials of pharmacological neuroprotective strategies in stroke have been disappointing, attention has turned to the brain's own endogenous strategies for neuroprotection. Recently, a hypothesis has been offered that modified reperfusion subsequent to a prolonged ischemic episode may also confer ischemic neuroprotection, a phenomenon termed ‘postconditioning’. Here we characterize both in vivo and in vitro models of postconditioning in the brain and offer data suggesting a biological mechanism for protection. Postconditioning treatment reduced infarct volume by up to 50% in vivo and by ∼30% in vitro. A duration of 10 mins of postconditioning ischemia after 10 mins of reperfusion produced the most effective postconditioning condition both in vivo and in vitro. The degree of neuroprotection after postconditioning was equivalent to that observed in models of ischemic preconditioning. However, subjecting the brain to both preconditioning as well as postconditioning did not cause greater protection than each treatment alone. The prosurvival protein kinases extracellular signal-regulated kinase (ERK), p38 mitogen-activated protein kinase (MAPK), and Akt show prolonged phosphorylation in the cortex of postconditioned rats. Neuroprotection after postconditioning was inhibited only in the presence of LY294002, which blocks Akt activation, but not U0126 or SB203580, which block ERK and P38 MAP kinase activity. In contrast, preconditioning-induced protection was blocked by LY294002, U0126, and SB203580. Our data suggest that postconditioning may represent a novel neuroprotective approach for focal ischemia/reperfusion, and one that is mediated, at least in part, by the activation of the protein kinase Akt.
Introduction
The neuroprotective concept of preconditioning is based on the observation that a brief noninjurious episode of ischemia is able to protect the brain from a subsequent longer ischemic insult (Dirnagl et al, 2003). Such preconditioning as a strategy to attenuate the pathophysiological consequences of ischemia—reperfusion injury would be focused on pretreatment situations, such as protection before cardiac bypass surgery. A nonpharmacological neuroprotective strategy for administration after ischemia onset, however, remains illusive. Based on recent studies on the heart (Darling et al, 2005; Hausenloy et al, 2005; Kin et al, 2005; Yellon and Hausenloy, 2005) and proof of principle experiments in the brain (Burda et al, 2006; Danielisova et al, 2006; Zhao et al, 2006), we addressed the hypothesis that modified reperfusion in the same artery occluded to produce a prolonged episode of focal cerebral ischemia, may confer postischemic neuroprotection, a phenomenon termed ‘postconditioning.’
In the published studies in myocardium describing postconditioning, a number of issues are presented. Yellon has suggested that pre- and postconditioning are similar phenomena with the effectors being a similar group of downstream signaling cascades (Yellon and Hausenloy, 2005). Alternately, the mechanisms regulating postconditioning might be entirely different than preconditioning, as the rapidity of onset of postconditioning-induced neuroprotection contrasts with a significant temporal delay for (protein synthesis dependent) preconditioning-induced neuroprotection. Further, postconditioning may not involve the activation of endogenous neuroprotection, but rather merely attenuate the burst of free radicals occurring with reperfusion. Indeed, it has been suggested that the protection of postconditioning could be accomplished by a gradual increase in the reperfusion rate (Burda et al, 1991, 1995). Other hypotheses have been offered as well, such as postconditioning resulting from intravascular adenosine wash out (Kin et al, 2005) or effected by intravascular pressures associated with reperfusion (Halldorsson et al, 2000). However, the major focus of postconditioning mechanisms is the role of the effector protein kinases and the question of the similarity or difference of these effectors versus those thought to be involved in preconditioning (Hausenloy et al, 2005).
Here we show that postconditioning produces robust neuroprotection resulting from modulating vascular reperfusion after the occlusion of a cerebral vessel in a model of focal ischemic stroke. We corroborate these findings by studies in vitro conducted using oxygen and glucose deprivation (OGD) in neuronal cultures subjected to conditions mimicking postconditioning in vivo. To obtain insight into the molecular mechanisms which may govern postconditioning neuroprotection, we investigated the activation of protein kinases that have been associated with mediating ischemic protection in the heart and brain: extracellular signal-regulated kinase 1/2 (ERK1/2), P38 mitogen-activated protein kinase (MAPK), AKT, and Jun N-terminal kinase (JNK) (Meller et al, 2005a; Nozaki et al, 2001; Shamloo et al, 1999; Yano et al, 2001) and suggest a role for AKT. As such, this neuroprotective strategy, supported by other studies, may pave the road for a clinically applicable stroke treatment. We compare and contrast these results to those of preconditioning.
Materials and methods
Experimental Groups
Male Sprague—Dawley rats (Charles River) weighting 250 to 300 g were housed under diurnal lighting conditions (12 h darkness/light). Experiments were performed according to the international guidelines for animal research. All experiments were performed in accordance with the American animal protection legislation and approved by the Institutional Animal Care and Use Committee of Legacy Research.
Focal Ischemia
Transient focal ischemia was induced by suture occlusion of the middle cerebral artery (MCA) in male rats anesthetized using 1.5% isofluorane, 70% N2O, and 28.5% O2 (Xiong et al, 2004). Ischemia was induced by introducing a 3-O surgical monofilament nylon suture from the external carotid artery into the internal carotid artery and advancing it into the circle of Willis to the branching point of the MCA, thereby occluding the MCA (Longa et al, 1989). Achievement of ischemia was confirmed by monitoring regional cerebral blood flow in the area of the right MCA. Cerebral blood flow was monitored through a disposable microtip fiber optic probe (diameter 0.5 mm) connected through a Master Probe to a laser Doppler computerized main unit (PF5001; Perimed, Sweden) and analyzed using PSW Perisoft 2.5 (Kawano et al, 2006). Animals that did not show a cerebral blood flow reduction of at least 70% were excluded from the experimental group, as well as animals that died after ischemia induction. Rectal and temporalis muscle temperature was maintained at 37 ± 0.5°C with a thermostatically controlled heating pad and lamp. All surgical procedures were performed under an operating stereomicroscope.
Evaluation of the Infarct Volume
Animals were killed with isofluorane 24 h or 7 days after ischemia. Brains were quickly removed, sectioned coronally at 1 mm intervals, and stained by immersion in the vital dye (2%) 2,3,5-triphenyltetrazolium hydrochloride (TTC) (Bederson et al, 1986). The infarct volume was calculated by summing infarction areas of all sections and multiplying by slice thickness. The percentage of the infarct was calculated by dividing the infarct volume by the total ipsilateral hemispheric volume. This ratio was then multiplied by 100 (Pignataro et al, 2004; Swanson et al, 1990).
Experimental Protocol
To characterize the temporal profile of ischemia and reperfusion resulting in postconditioning, animals were assigned to six groups (see Figure 1); animals were killed 24 h after the final ischemic insult. The control group: MCA was occluded for 100 mins. The preconditioned group: a 30-min MCA occlusion (MCAO) was followed, 72 h later, by a MCAO of 100 mins.
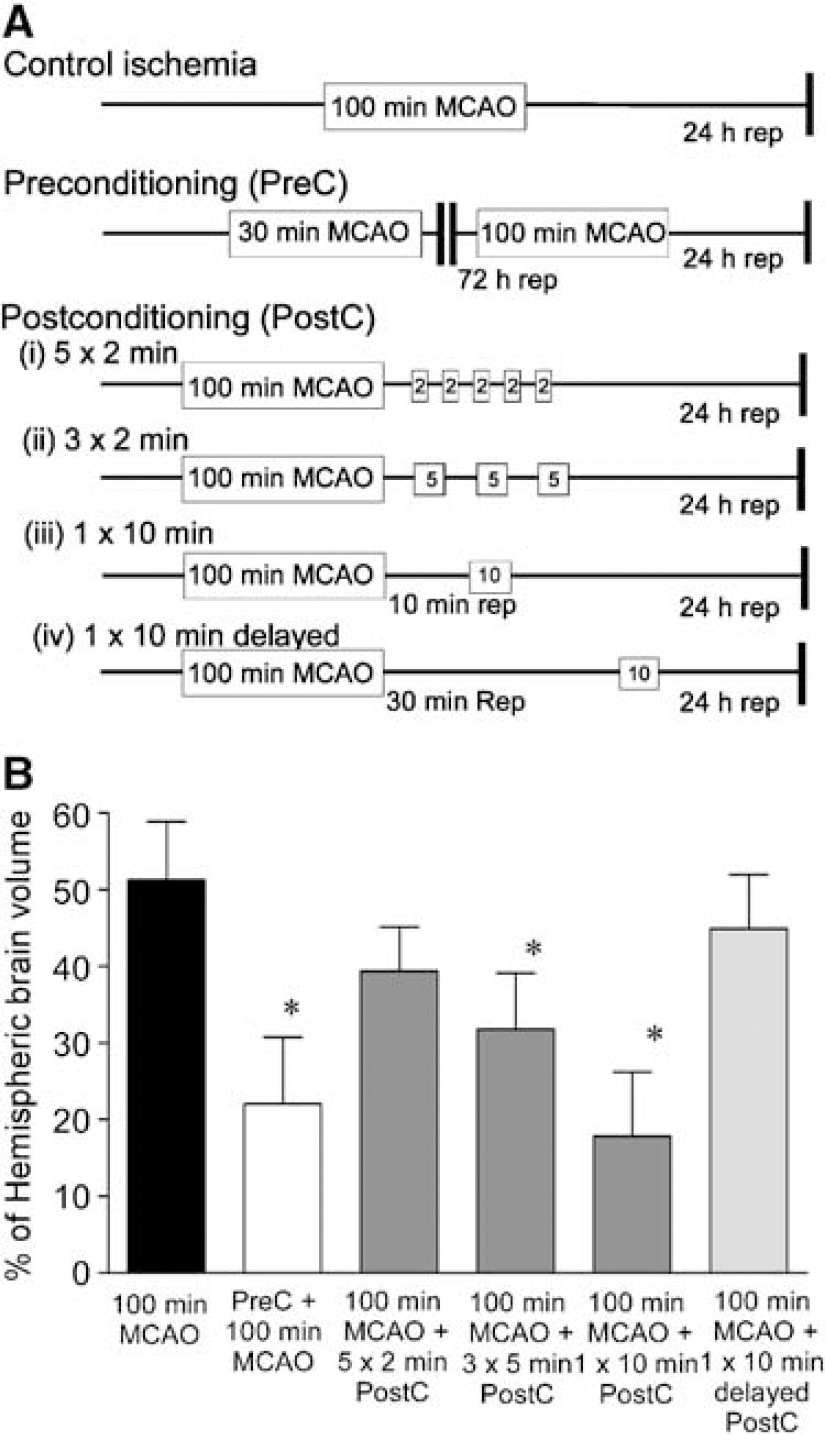
Defining the therapeutic window of postconditioning in a focal model of ischemia. (
The first postconditioned group: after 100 mins of MCAO, reperfusion was established for 2 mins after which the MCA was occluded again for 2 mins, followed with another four cycles of 2 mins of reperfusion and 2 mins of occlusion (total of five cycles). Animals were then recovered for 24 h.
The second postconditioned group: after 100 mins of MCAO, reperfusion was established for 5 mins after which the MCA was occluded again for 5 mins, followed with another two cycles of 5 mins of reperfusion and 5 mins of occlusion (total of three cycles). Animals were then recovered for 24 h.
The third postconditioned group: after 100 mins of MCAO, reperfusion was established for 10 mins after which the MCA was occluded for 10 mins. Animals were then recovered for 24 h.
The fourth postconditioned group: after 100 mins of MCAO, reperfusion was established for 30 mins afterwhich the MCA was occluded for 10 mins. Animals were then recovered for 24 h.
To determine whether neuroprotection results in extended reduction in infarct volume, some animals were subject to postconditioning and then recovered for 7 days.
To assess whether the neuroprotective effect exerted by preconditioning was additive to that exerted by postconditioning, rats (n = 5) were subjected to both pre- and postconditioning: animals were subject to 30 mins of MCAO, followed by 72 h of reperfusion (the time at which preconditioning would be maximally protective), then 100 mins of MCAO, followed by 10 mins of reperfusion and finally 10 mins of MCAO (the maximally protective postconditioning parameter). Animals were then recovered for 24 h.
Protein Kinase Inhibitor Study
To assess the effect of ERK, p38MAPK, and AKT on the neuroprotective effect exerted by postconditioning, four groups of rats (N = 5) were treated with either the Mek inhibitor (which activates ERK1/2) U0126 (5 μL, 100 μmol/L, in 3% dimethyl sulfoxide) (Favata et al, 1998); the P38MAPK inhibitor SB203580 (5 μL, 100 μmol/L, in 3% dimethyl sulfoxide) (Cuenda et al, 1995), the inhibitor of phosphatidylinositol-3 kinase (which activates AKT) LY294002 (5 μL, 10 mmol/L, in 3% dimethyl sulfoxide) or vehicle only (3% dimethyl sulfoxide). All drugs or vehicle, were intracerebroventricularly injected at the following coordinates from the bregma: anteroposterior, −0.8; laterolateral, −1.5; depth, −4.0 (Paxinos and Watson, 1997). Drugs or vehicle were administered either 15 mins before postconditioning, or 15 mins before preconditioning ischemia.
Cortical samples were harvested from ischemic brains of rats subjected to 100 mins of MCAO or from brains of postconditioned rats. In both experimental conditions, four groups of ipsilateral and contralateral cortex as well as striatum samples were obtained at different reperfusion times after the last occlusion: (a) 10 mins; (b) 30 mins; (c) 5 h; and (d) 24 h. Four further groups of samples were obtained from brains of sham-operated animals.
Rat brain samples were homogenized using an 18-gauge needle in a lysis buffer (50 mmol/L Tris—HCl, pH 7.5, 100 mmol/L NaCl, 1% Triton X–100) containing protease inhibitors (aprotinin, leupeptin, phenylmethylsulfonyl fluoride, and pepstatin), and phosphatase inhibitor (Sigma cocktail). After centrifugation at 12,000 g at 4°C for 5 mins, the supernatants were collected. Protein concentration was estimated using the Badford reagent (Sigma, St Louis, MO, USA). Then, 50 μg of protein was mixed with a Laemmli sample buffer and boiled at 95°C for 5 mins. The samples were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis, and transferred to polyvinylidene difluoride membranes. Blots were probed with antibodies to phospho-ERK (1:1,000; Cell Signaling, Beverly, MA, USA), phospho-p38 MAPK (1:500; Cell Signaling), phospho-JNK (1:1,000; Cell Signaling), phospho-AKT (1:1,000; Cell Signaling), or α-actin (1:2,000; Abcam, Cambridge, MA, USA) diluted in tris buffered saline (TBS-T) 1% bovine serum albumin overnight (4°C), and detected using horseradish peroxidase-conjugated secondary antibody (1:2,000; Cell Signaling; 60 mins at room temperature in 5% non-fat milk) and an enhanced luminescence kit (Amersham Pharmacia Biotech, Piscataway, NJ, USA).
In Vitro Studies
Sprague—Dawley rat pups were used to prepare cortical neuronal cultures, as described previously (Meller et al, 2005a). After dissection, cortices were enzymatically dissociated with papain (Worthington Biochemicals, Lakewood, NJ, USA) and triturated with a fire-polished pipette. Cells were plated out at a density of 400,000 cells per cover slip for cell viability assays. Cells were maintained in Neurobasal-A/B27 media (Invitrogen, Carlsbad, CA, USA) for 7 to 10 days.
Oxygen and glucose deprivation was performed by washing the cells with phosphate-buffered saline (NaCl 1.37, KCl 2.7, Na2HPO4 10, KH2PO4 1.7 (all in mmol/L) pH 7.4) supplemented with 0.5 mmol/L CaCl2, 1.0 mmol/L MgCl2, and placing culture dishes in an anaerobic chamber for various times (85% N2, 5% H2, 10% CO2; 35°C for 10, 30, or 120 mins; Forma Scientific, Marjetta, OH, USA). Anaerobic conditions in the chamber were monitored using Gaspack anaerobic indicator strips (Becton Dickinson, San Jose, CA, USA). Oxygen and glucose deprivation was terminated by removing cells from the anoxia chamber, replenishing with Neurobasal A media, and replacing them back into the normoxic incubator.
Cell Death Assay
Cell death was determined by propidium iodide exclusion assay as described previously (Meller et al, 2005a). Cortical cells were incubated with propidium iodide (1.5 μg/mL) for 2 mins, washed with phosphate-buffered saline, and fixed with 4% formaldehyde. Cells were permeabilized with 0.1% Triton X-100 and then mounted onto glass slides using Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (Vector Labs, Burlingame, CA, USA). Cell death was determined as the ratio of propidium iodide-stained cells, to the total number of 4′,6-diamidino-2-phenylindole-stained cells, visualized using a fluorescence microscope. Cell counts were made from three random sections of the coverslip and repeated on six to eight independent sets of cultures.
Statistical Analysis
Values are expressed as mean ± s.d. Statistical analysis was performed with analysis of variance (ANOVA) followed by Dunnet's or Bonferroni's post hoc test. Seven-day survival data were analyzed using Student's t-test. Statistical significance was accepted at the 95% confidence level (P < 0.05).
Results
Effect of Pre- and Postconditioning on Infarct Volume
Subjecting animals to a period of 100 mins of MCAO results in a significant infarct (51.3 ± 3.4% of hemispheric brain volume), as determined by TTC staining (Figure 1B). In agreement with our previous studies (Meller et al, 2005a; Stenzel-Poore et al, 2003), preconditioning the brain with 30 mins of MCAO, 72 h before harmful ischemia, reduced infarct volume (22 ± 3.8%, Figure 1B).
Subjecting the brain to 100 mins of MCAO and then manipulating reperfusion also resulted in reduced infarct volume compared to 100 mins of MCAO alone. Maximal protection was seen if the 100 mins of MCAO was followed by 10 mins of MCAO, with a reperfusion interval of 10 mins (Figure 1). Such postconditioning reduced infarct volume to 17.9 ± 3.7% of hemispheric brain volume (Figure 1B). Infarct volume was also reduced with a postconditioning protocol of three cycles of 5-min reperfusion followed by 5-mins MCAO, but not five cycles of a 2-min reperfusion, followed by 2-min occlusion (31.8 ± 3.3 and 39.4 ± 2.6% infarct volume, respectively; Figure 1B). When the duration of reperfusion after 100 mins of MCAO is extended to 30 mins before a 10-min postconditioning MCAO, protection was lost (44.9 ± 3.1 infarct volume; Figure 1B).
To determine whether ischemic postconditioning results in permanent protection, we subject some animals to 10 mins of postconditioning, 10 mins after harmful ischemia, and then allowed the animals to recover for 7 days. Profound neuroprotection was observed in the postconditioned group versus non-postconditioned group after 7 days of recovery (9.7 ± 4.2% of hemispheric volume versus 39.3 ± 9.9% hemispheric volume, respectively; n = 5, P < 0.01).
An In Vitro Model of Postconditioning
To determine whether postconditioning is a cellular event, or a vascular or endothelial event due to reperfusion, we examined postconditioning in an in vitro model. We have previously defined models of ischemic preconditioning whereby 30 mins of modeled ischemia (OGD) was used to precondition cortical cultures against a harmful 120-min OGD insult, in a long-term/delayed tolerance model (Meller et al, 2005a). Using this OGD dose as a starting point, we subjected cortical cells to 120-min OGD, and allowed the cells to recover for 60, 30, and 10 mins before subjecting the cells to a further 30-min OGD (Figure 2A). Pairing the harmful 120 mins of OGD with 30 mins of OGD after 10, 30, or 60 mins of OGD termination did not significantly reduce cell death compared to 120 mins of OGD alone (Figure 2B).
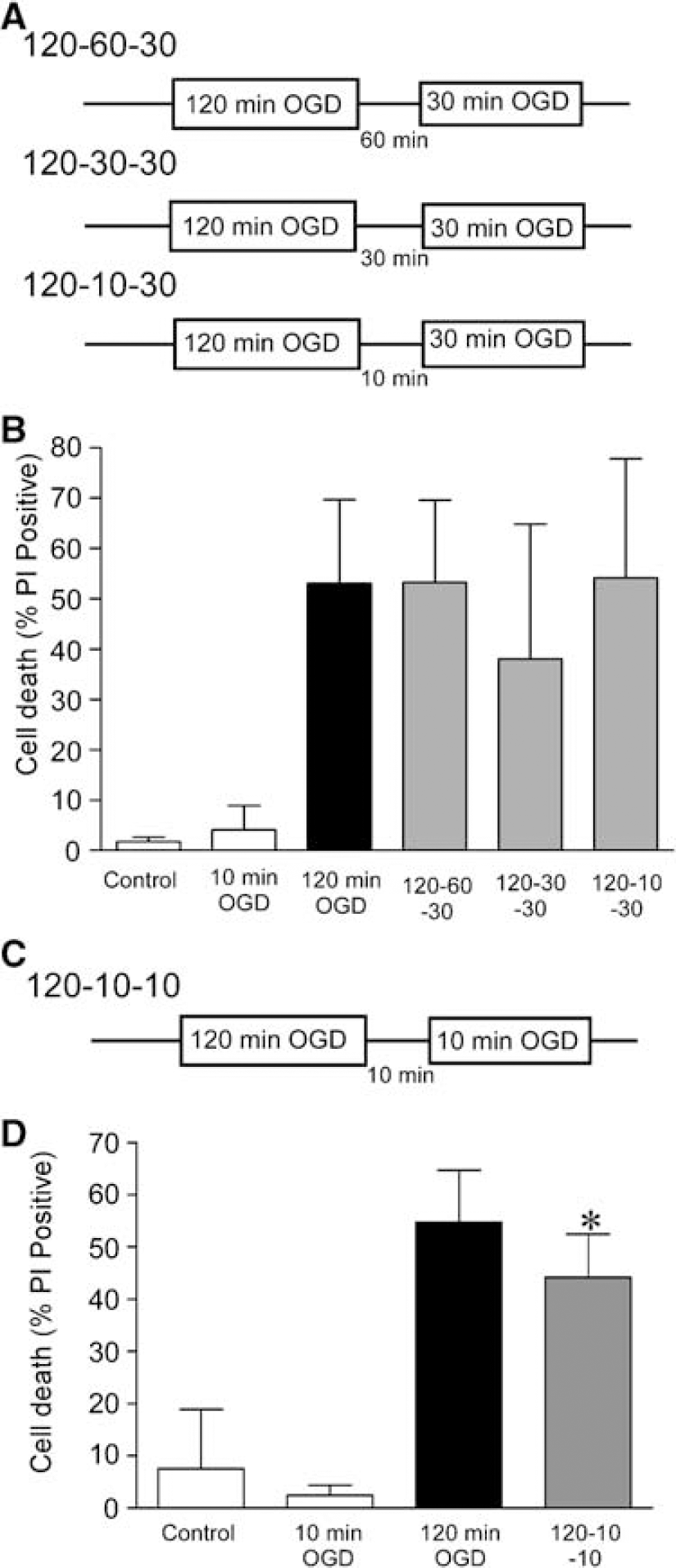
Postconditioning in an in vitro model of ischemia. (
However, when we subjected the cells to 120 mins of OGD, followed by 10 mins of recovery and then 10 mins of OGD (Figure 2C), we observed a significant reduction in cell death compared to 120 mins of OGD (Figure 2D). There was no change in the total number of cells counted in these experiments (data not shown). However, of note, the protection observed in our postconditioning model in vitro was less than the protection observed in our model of preconditioning in vitro (Meller et al, 2005a). As the postconditioning response was more robust in our in vivo model, we focused our experiments there to determine possible effector mechanisms for postconditioning.
Pre- and Postconditioning are Nonadditive Protective Mechanisms
To determine whether the protective effects of pre- and postconditioning could be added together to give a greater neuroprotection, we subjected rats to30 mins of MCAO preconditioning ischemia, followed by 100 mins of MCAO 72 h later, reperfused for 10 mins, and then subject the rat to an additional 10 mins of MCAO (see Figure 3A). There was no enhancement of neuroprotection when pre- and postconditioning were combined (19.1 ± 3.0; Figure 3B) compared to each alone (Figure 3B). This suggests that these two endogenous protective mechanisms are not additive.
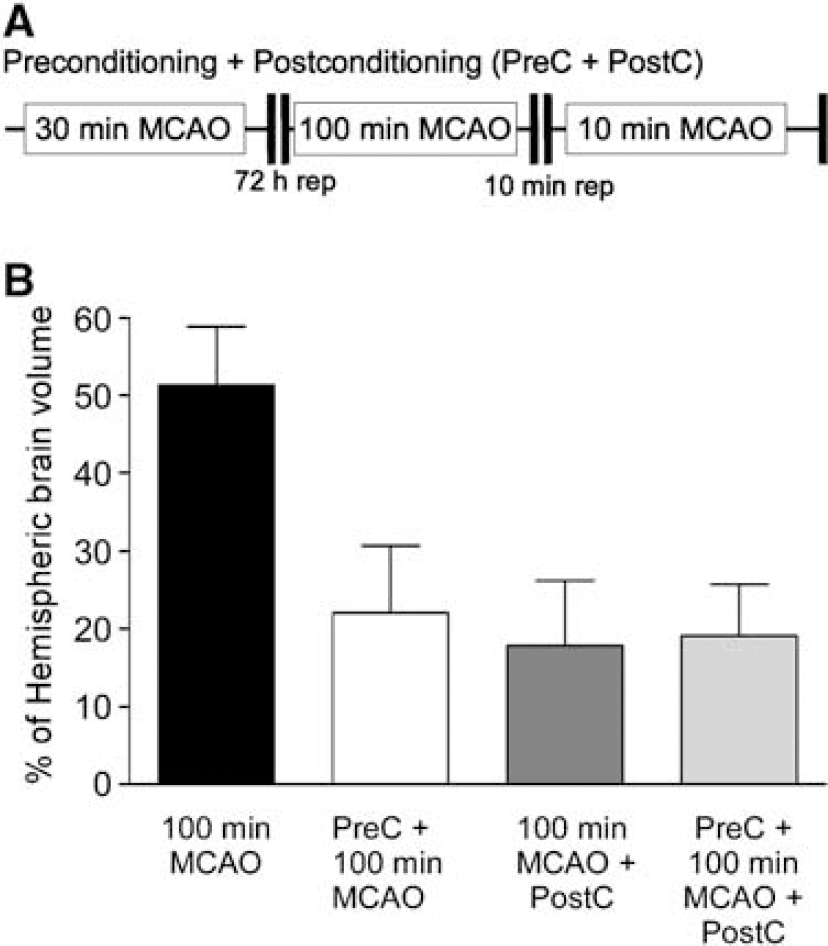
Nonadditive nature of pre- and postconditioning neuroprotection in the brain. (
Time course of ERK, JNK, p38 MAPK, and Akt Phosphorylation after Harmful Ischemia and Postconditioning
The MAPKs (ERK1/2, JNK, and P38 MAPK) have been shown to regulate cell survival and death after preconditioning ischemia in the heart and brain (Hausenloy et al, 2005; Meller et al, 2005a). Intimately implicated with MAPK signaling pathways, the prosurvival protein kinase AKT has also been shown to play a role in mediating neuroprotection after preconditioning ischemia (Hausenloy et al, 2005; Hillion et al, 2006; Yano et al, 2001). To assess the effect of harmful ischemia and of postconditioning on these protein kinases, the phosphorylation of ERK, JNK, p38 MAPK, and AKT was evaluated at different times after either 100 mins of MCAO (Figure 4A) or 100 mins of MCAO followed by postconditioning (Figure 4B): 10-min reperfusion and then a further 10 mins of MCAO. After harmful ischemia, there was a transient activation of Akt and ERK1/2 in the ipsilateral cortex at 30 and 10 mins, respectively (Figure 4A). In contrast, postconditioning ischemia induced a prolonged phosphorylation of these protein kinases up to 5 and 24 h duration for ERK and AKT, respectively (Figure 4B). JNK phosphorylation did not appear to change after either harmful MCAO or postconditioning. P38 MAPK phosphorylation was weaker compared to the other protein kinases investigated. P38 MAPK phosphorylation did not change after harmful ischemia (Figure 4A), but was moderately increased after postconditioning reaching significance at 10 mins and at 24 h (Figure 4B). In contrast to the ipsilateral (protected) cortex, we did not observe a significant change in ERK, JNK, P38 MAPK, or AKT phosphorylation in the contralateral (nonprotected) cortex after either harmful ischemia or postconditioning (Figures 4A and 4B, white bars). Furthermore, no significant changes were observed in the phosphorylation of ERK, JNK, P38 MAPK, and Akt in the striatum of harmful ischemia-treated or postconditioned animals (data not shown).
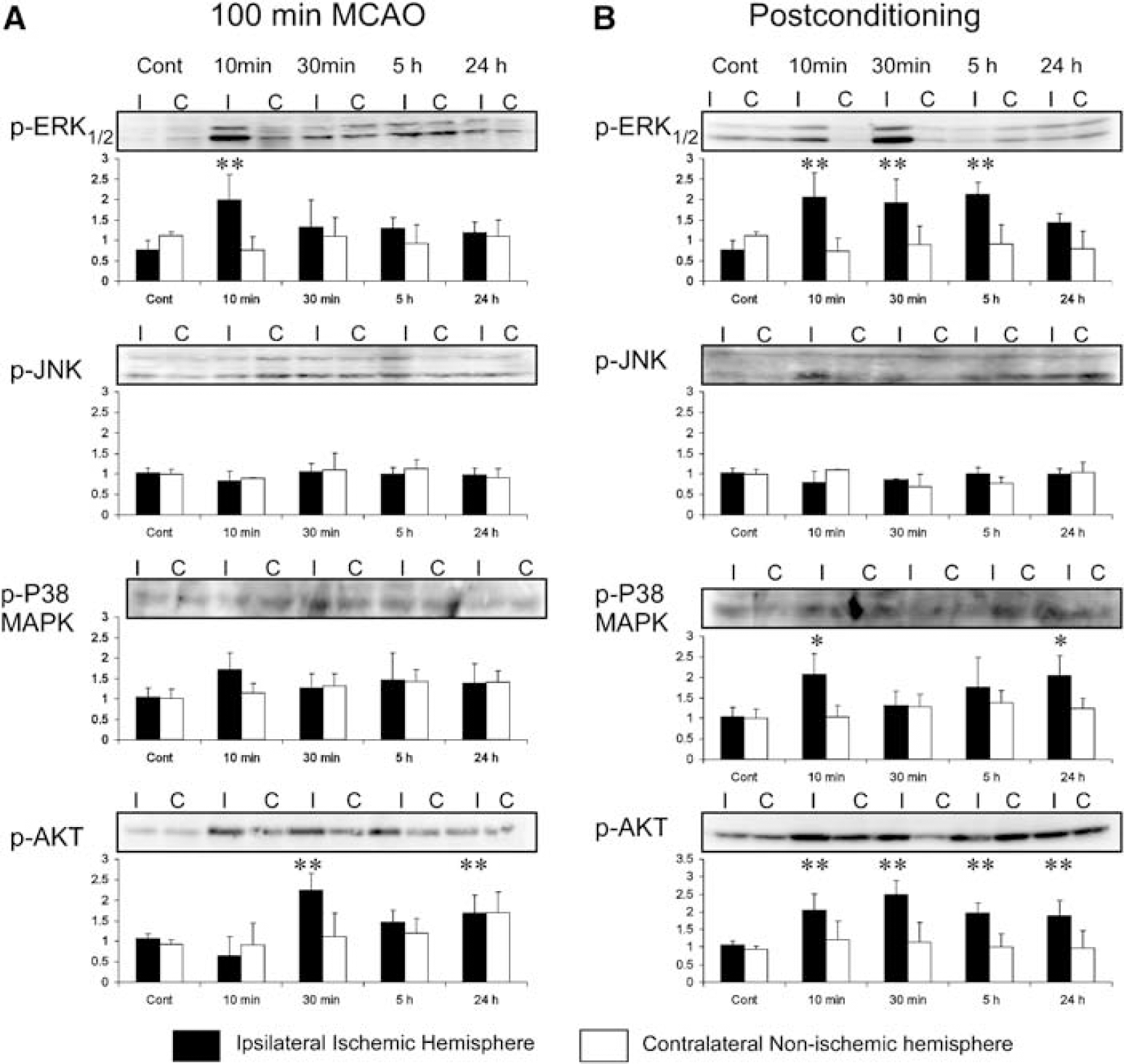
Postconditioning induces activation of stress-associated protein kinases. (
To determine if the neuroprotective effects of post-and preconditioning are mediated by the activation of the same pathways, the activation of ERK, JNK, P38 MAPK, and AKT was evaluated 72 h after a preconditioning dose of ischemia. There was an increase in Akt phosphorylation after preconditioning ischemia (Supplementary Data). In contrast, we only detected a slight increase in phosphorylation of ERK, JNK, and P38 MAPK in the ipsilateral cortex of preconditioned brains, 72 h after the application of the preconditioning stimulus, when we observe tolerance.
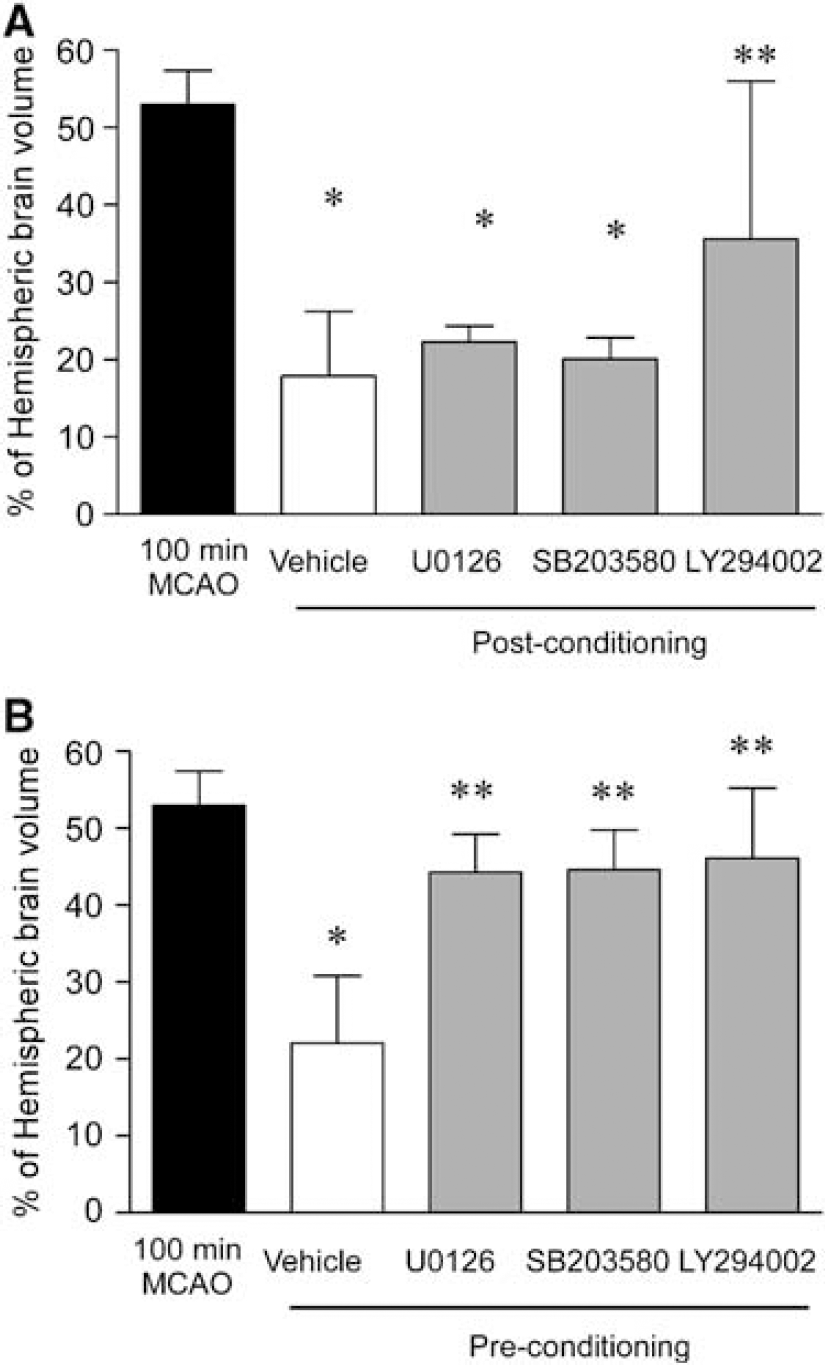
Effect of Protein Kinase Inhibitors on Neuroprotection Exerted by Postconditioning Treatment
To clarify the role of kinases activated by postconditioning (ERK, P38 MAPK, and AKT), potent and selective inhibitors of each kinase was administered to postconditioned rats. The neuroprotective effect triggered by postconditioning were reduced when AKT activation was inhibited by LY294002 (Figure 5). However neither the inhibition of ERK activation by U0126 nor the inhibition of P38MAPK by SB203580 was able to reverse the neuroprotective effect exerted by postconditioning (Figure 5A). Interestingly, each of the inhibitors tested—U0126, LY294002, and SB203580—inhibited the protection observed by preconditioning the animals with 30 mins of MCAO 72 h before 100 mins of MCAO (Figure 5B). None of those inhibitors were able to induce any significant change in the infarct volume of rats subjected to 100 mins of MCAO alone (Supplementary Data Figure 1B).
(
Discussion
The failure over the past decades of multiple trails of exogenously administered drugs as potential stroke neuroprotectants has enhanced the ongoing search to identify endogenously modulated mechanisms activated after cerebral ischemia, which might be harnessed as neuroprotectants in stroke (Dirnagl et al, 2003; Gidday, 2006; Kirino, 2002). Such molecular mechanisms might be those evolutionarily conserved to counteract the damage induced by a disruption to the cerebral blood supply (Gladstone et al, 2002). Our approach has been to investigate intrinsic mechanisms of neuroprotection that can be triggered by sublethal stresses, that is, ischemic- and lipopolysaccharide-induced preconditioning (Dahl and Balfour, 1964; Kitagawa et al, 1991; Meller et al, 2005a; Schurr et al, 1986; Stenzel-Poore et al, 2003, 2007; Tasaki et al, 1997). However, in addition to being preconditioning stimuli, both ischemia and lipopolysaccharide have been reported as postconditioners in the brain (Davis et al, 2005; Zhao et al, 2006). Although these preconditioning strategies confer robust neuroprotection in in vitro and in vivo models of cerebral ischemia, their translational relevance is limited by the fact that the preconditioning stimulus must be applied 24 or more hours before the onset of harmful ischemia. For this reason, the goal of ischemic tolerance experiments is to understand mechanisms that confer neuroprotection, which might enhance neuroprotective cell signaling after stroke. Postconditioning, however, has potential translational advantages: it is effective after stroke onset, and full knowledge of its mechanism of action is not necessary for clinical trial.
Most background studies of postconditioning have been performed in the heart (Yellon and Hausenloy, 2005); however, some recent studies have been performed in the brain. Using global ischemia as the postconditioning stimulus, Zhao et al (2006) showed that postconditioning neuroprotection could occur in a model of permanent focal ischemia in the brain. Postconditioning in focal ischemia can be described as a modified form of reperfusion (Zhao et al, 2003). It has been established that brief, intermittent, repetitive interruptions to reperfusion, at the onset of reperfusion after a prolonged period of ischemia, reduced myocardial injury to an extent comparable to ischemic cardiac preconditioning (Darling et al, 2005; Hausenloy et al, 2005; Kin et al, 2005; Yellon and Hausenloy, 2005). A similar phenomenon has been reported in the brain where graded reperfusion attenuates injury to hippocampal pyramidal neurons after global ischemia (Burda et al, 1991, 1995). The protection is substantial as 10, but not 15 min of global ischemic injury can be reversed with this technique (Burda et al, 2006).
Our study addresses focal ischemia with postconditioning in the same vascular bed induced by repeated periods of focal ischemia/reperfusion. We first addressed the temporal profile of efficacy of the postconditioning stimulus (Figure 1). Multiple brief, intermittent periods of interrupted reperfusion were studied, as cardiac literature shows that in the heart reperfusion in a ‘stuttering manner’ is protective and specifically three 10-sec cycles of reperfusion—re-occlusion in the first minute is optimal. Delaying postconditioning for even 1 min eliminates protection in the heart (Darling et al, 2005; Kin et al, 2005). Results in the brain are different. Optimal protection was obtained with a single 10-min period of re-occlusion administered 10 mins after the 100-min MCAO. Brief, repetitive re-occlusions were less effective (Figure 1). However, timing of the initiation of postconditioning is critical in the brain, as well as in the heart. If we delayed the onset of postconditioning to 30 mins, the neuroprotection was lost (Figure 1). Furthermore, the protection was robust as we observe less infarction in the brain 7 days after postconditioning, which suggests that postconditioning results in permanent neuroprotection.
In cardiomyocytes, protection by postconditioning can be shown in vitro with propidium iodide staining and intracellular calcium concentrations as end points (Sun et al, 2005). Accordingly, we further addressed neural postconditioning using primary neuronal cortical cultures in vitro. After 120 mins of OGD, an additional period of 10 or 30 mins of OGD was induced after 10, 30, or 60 mins of recovery (Figures 2A and 2C). The 10-min OGD duration and the 10-min interval resulted in protection from cell death (Figure 2C). Although other paradigms of in vitro postconditioning did not result in protection in these studies, the power of the experiments performed here is limited. Nevertheless, these data eliminate the possibility that the postconditioning neuroprotective effect is due solely to a modification of cerebral blood flow, which is relevant as graded reperfusion has been invoked as a mechanism of postconditioning in the brain (Burda et al, 1991, 1995).
Are Pre- and Postconditioning Additive?
In the heart, some but not all studies of preconditioning plus postconditioning have reported a cumulative protective effect (reviewed in Yellon and Hausenloy, 2005). We therefore explored this possibility by combining our maximally effective preconditioning parameter (30 mins of transient MCAO and 72 h of reperfusion; Stenzel-Poore et al, 2003) with the most effective postconditioning parameter from this study (10 mins of re-occlusion after the first 10 minutes of reperfusion; Figure 2). Although both pre- and postconditioning were protective, a cumulative effect was not shown (Figure 2). Hence, with the current parameters, pre- and postconditioning neuroprotection do not appear additive in the brain.
Postconditioning-Induced Neuroprotection is Mediated by Protein Kinases
In cardiac postconditioning, the signaling pathways involved include the activation of prosurvival protein kinases and these same kinases may be effectors of cardiac preconditioning as well (reviewed by Hausenloy et al, 2005). Our studies suggest postconditioning in the brain may also involve the activation of the survival associated with protein kinases. Akt and ERK have been shown to regulate ischemic preconditioning-induced neuroprotection both in the heart and brain (Gu et al, 2001; Meller et al, 2005a; Shamloo et al, 1999). That pre-and postconditioning were not cumulative in our study (Figure 3B) is compatible with the two mechanisms having common activation pathways. We show that after harmful ischemia, Akt is phosphorylated transiently at 30 mins after reperfusion, whereas after postconditioning, the activation of Akt starts 10 mins after the second occlusion and is still upregulated 24 h later. Erk and P38 MAPK also show similar activation profiles after postconditioning (Figure 4B).
We then addressed the relative roles of these survival kinases using specific kinase inhibition. Here postconditioning was only reduced by blockade of AKT activation with LY294002 (Figure 5A). This is in contrast to preconditioning, which was blocked by inhibition of ERK, p38MAPK, and AKT (Figure 5B). These data support distinct cell-signaling mechanisms of preconditioning versus postconditioning in the brain and suggest a role for AKT and its downstream signaling cascade in mediating postconditioning-induced neuroprotection.
In our studies, inhibition of AKT with LY294002 blocked ischemic preconditioning-induced neuroprotection. AKT has been reported to play a role in various neuroprotective strategies in the brain. The neuroprotective effects of estrodiol, osteopontin, erythropoietin, and preconditioning to reduce ischemia-induced brain injury are mediated by AKT (Kilic et al, 2005; Meller et al, 2005b; Won et al, 2006; Yin et al, 2005). The pharmacological postconditioning effect of the activation of AKT after harmful ischemia may account for the delayed activation of cell death pathways, as inhibition of AKT has been shown to speed cell death after ischemia (Noshita et al, 2001). Although the role of AKT in global ischemia-induced preconditioning is not as clear (Namura et al, 2000; Yin et al, 2005). AKT appears to play a neuroprotective role in focal ischemic tolerance (Nakajima et al, 2004; Yin et al, 2005). Although specific targets of AKT have not been shown in postconditioning, potential effectors include responsive element binding protein (CREB) and bcl-2 upregulation (Willaime-Morawek et al, 2005), both involved in preconditioning neuroprotection (Chen et al, 2000; Meller et al, 2005a), activation of the Akt/GSK3β (glycoden synthase kinase 3 beta) signaling pathway (protective in global ischemia (Endo et al, 2006) or prosurvival effect of Akt on Bad and caspase 9 (recently suggested by Chan, 2004).
Summary
Overall, the results of this study show a new endogenous neuroprotective strategy—that of postconditioning—is relevant to focal ischemia and reperfusion in the brain. The postconditioning stimulus appears to result in the prolonged activation of AKT, which has also been shown to mediate intrinsic protection to ischemia in models of ischemic preconditioning. Although the time window of effectiveness of postconditioning-induced europrotection shown here is narrow, proof of principle of postconditioning as a neuroprotective strategy in the brain is supported by these studies. As such, postconditioning may have translational relevance to reperfusion and thrombolitic treatments in acute brain ischemia.