
Abstract
Astrocytes are fundamentally important to the maintenance and proper functioning of the central nervous system. During the period of development when myelination is occurring, white matter astrocytes are particularly sensitive to ischemic injury and their failure to regulate glutamate during ischemic conditions may be an important factor in excitotoxic injury. Here, we have identified key mechanisms of injury that operate on the processes of immature white matter astrocytes during oxygen-glucose deprivation (OGD) using GFAP-GFP mice. Oxygen-glucose deprivation produced a parallel loss of astrocyte processes and somata, assessed by both the retention of GFP fluorescence within these structures and by quantitative electron microscopy. Oxygen-glucose deprivation-induced process loss was Ca2+ independent and had two distinct mechanisms. Substituting either extracellular Na+ or Cl−, or perfusion with the Na—K–Cl co-transport blocker bumetanide, provided protection up to 40 mins of OGD but not beyond that point. HCO−3 substitution or perfusion with 4,4′-diisothiocyanostilbene-2,2′-disulphonic acid provided complete protection of the processes up to 60 mins of OGD. Zero-Na+/zero-K+ conditions provided complete protection from OGD-induced injury of processes and somata at all time points. We conclude that acute ischemic-type injury of immature astrocytes follows a cytotoxic ion influx mediated in part by Na—K–Cl co-transport and in part by Na+- and K+-dependent HCO−3 transport, a mechanism that is common to both cell processes and somata. This work provides a basis on which preventative strategies may be developed to protect white matter astrocytes from ischemic injury in susceptible individuals.
Introduction
Astrocytes are multifunctional cells responsible for the homeostatic regulation of the extracellular space of the central nervous system (CNS). They have a complex morphology with numerous, often highly ramified, cell processes radiating from the soma to locations such as the node of Ranvier, the perisynaptic space, peri-vascular space, and the glial limitans (Butt and Ransom, 1993; Grosche et al, 1999; Skoff, 1990; Vaughn, 1969). Individual astrocytes are required to perform different functions at these various locations, and consequently astrocyte processes have a polar distribution of membrane proteins. For example, potassium channels are differentially expressed on astrocyte processes, allowing spatial regulation of extracellular [K+] (Newman, 1986), whereas high-affinity glutamate transporters are differentially expressed in perisynaptic processes allowing for effective neurotransmitter re-uptake at these sites (Gundersen et al, 1996). It is possible that the differential expression of membrane proteins in astrocyte processes, coupled with their large membrane to surface area ratio, may give these structures a different sensitivity to ischemic injury from that found in their somata. Loss of processes from astrocytes under ischemic conditions has been reported numerous times (Davies et al, 1998; Hulse et al, 2001; Kraig and Chesler, 1990; Thomas et al, 2004), although it is currently not known if injury mechanisms in the processes are distinct from those in the somata or indeed whether injury occurs more rapidly in processes relative to the somata.
Ischemia can evoke two basic responses in astrocytes. Astrocytes that experience severe ischemia can quite rapidly suffer irreversible injury leading to necrotic cell death (Fern, 1998, 2001; Thomas et al, 2004; Bondarenko and Chesler, 2001), or apoptosis (Giffard and Swanson, 2005). Alternatively, astrocytes that survive a period of ischemia can instigate reactive changes involving an increase in somata size and a thickening of proximal processes (Davies et al, 1998; Ridet et al, 1997), while the absence of fine processes in reactive astrocytes indicates their selective loss from the cell. We have recently reported that astrocytes in immature white matter are significantly more sensitive to acute ischemic injury than gray matter astrocytes (Shannon et al, 2007). This may be relevant to developmental disorders such as cerebral palsy that typically involve selective injury to white matter regions (Back and Rivkees, 2004; Volpe, 1995). Considering the importance of astrocyte processes for CNS function and the clinical relevance of CNS ischemia, we have now examined the time course and mechanism of ischemic-type injury to both the processes and somata of immature white matter astrocytes. Live confocal imaging of transgenic mouse optic nerve with green fluorescent protein (GFP) expression under the control of the astrocyte-specific promoter glial fibrillary acidic protein (GFAP) allowed real-time assessment of changes in astrocyte morphology. Surprisingly, the results show that the mechanism and time course of acute injury are the same in both cell compartments and involve a cytotoxic ionic influx mediated by Na—K–Cl co-transport (NKCC) and HCO−3 transporters, including a putative K—HCO−3 co-transporter.
Materials and methods
Transgenic mice, strain FVB/N-Tg(GFAPGFP)14Mes/J (jax stock number 003257), carrying GFP (hGFP-S65T) under the control of the human GFAP promoter were used for the confocal imaging studies. Heterozygous males were mated with wild-type females and transgenic littermates identified at post-partum day 4 (P4) using a blue light source and appropriate filters (the transgenic animals exhibited a characteristic green glow around their eyes). All animals were maintained in accordance with United Kingdom Home Office regulations. For all experiments, mouse optic nerves were dissected out at P7-13 (called ‘P10′ throughout) and maintained before mounting at 18°C in artificial cerebrospinal fluid (aCSF) aerated with hydrated 95% O2/5% CO2 to a maximum of 4 h. Table 1 shows the composition of the experimental solutions used. Unless otherwise stated all chemicals were purchased from Sigma (Gillingham, UK).
Solutions
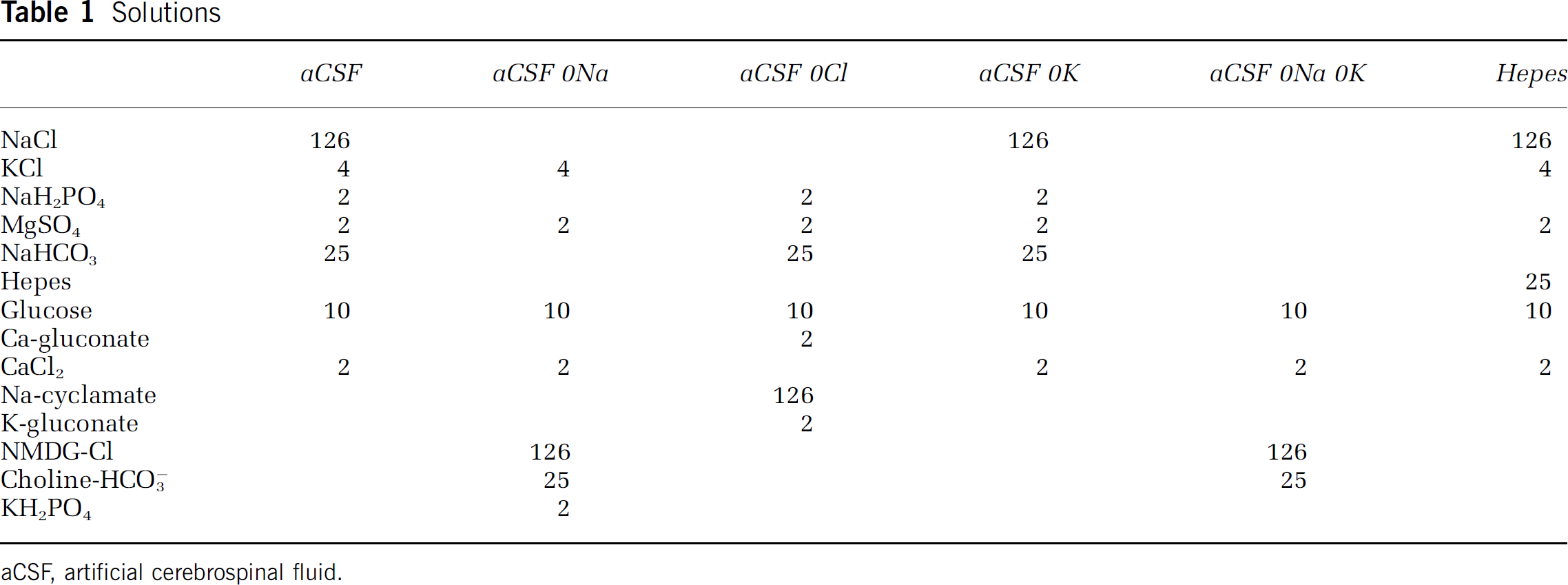
aCSF, artificial cerebrospinal fluid.
Immunohistochemistry
To examine the identity of GFAP-GFP(+) cells in P10 optic nerves, nerves from transgenic mice were dissected in 0.1 mol/L phosphate-buffered saline and fixed in 4% paraformaldehyde for 30 mins. The optic nerves were subsequently incubated in 0.1 mol/L phosphate-buffered saline plus 20% sucrose w/v for 5 mins before freeze-sectioning and subsequent blocking for 60 mins in 0.1 mol/L phosphate-buffered saline 10% fetal goat serum plus 0.5% Triton-X 100. They were incubated in this solution plus primary antibody at 4°C overnight. Immunohistochemistry was performed using antibodies raised against CD44 (1:100, AbD Serotec, Oxford, UK), O4 (1:100, conjugated to phycoerythrin, R&D Systems, Minneapolis, MN, USA), neurofilament-200 (1:100, Chemicon, Temecula, CA, USA), GFAP (conjugated to Cy3: 1:100, Sigma), NG-2 (Zymed Laboratories, Paisley, UK; 1:100), HuD (1:100, Chemicon), Thy-1 (1:3, supernatant from cell line T11D7e2, European Collection of Cell Cultures, Porton Down, UK), and GLT-1 (1:100, Alpha Diagnostics, San Antonio, TX, USA). After washing, the appropriate Alexa-conjugated secondary antibody (Cambridge Bioscience, Cambridge, UK, only for non-conjugated antibodies) was applied for 120 mins and single-plane fluorescent sections were imaged at × 20 on an Olympus scanning confocal microscope. GFP(+) cells were found to be largely CD44 (+) (Figure 1A), GFAP(+) (Figure 1B), GLT-1(+) (data not shown)/HuD(–) (Figure 1C, but note the presence of GFP(–)/HuD(+) cells, presumably oligodendroglia, see Kostyk et al, 1996), Thy-1(–) (data not shown), neurofilament-200(–) (data not shown)/NG-2(–) (Figure 1D), O4(–) (data not shown).
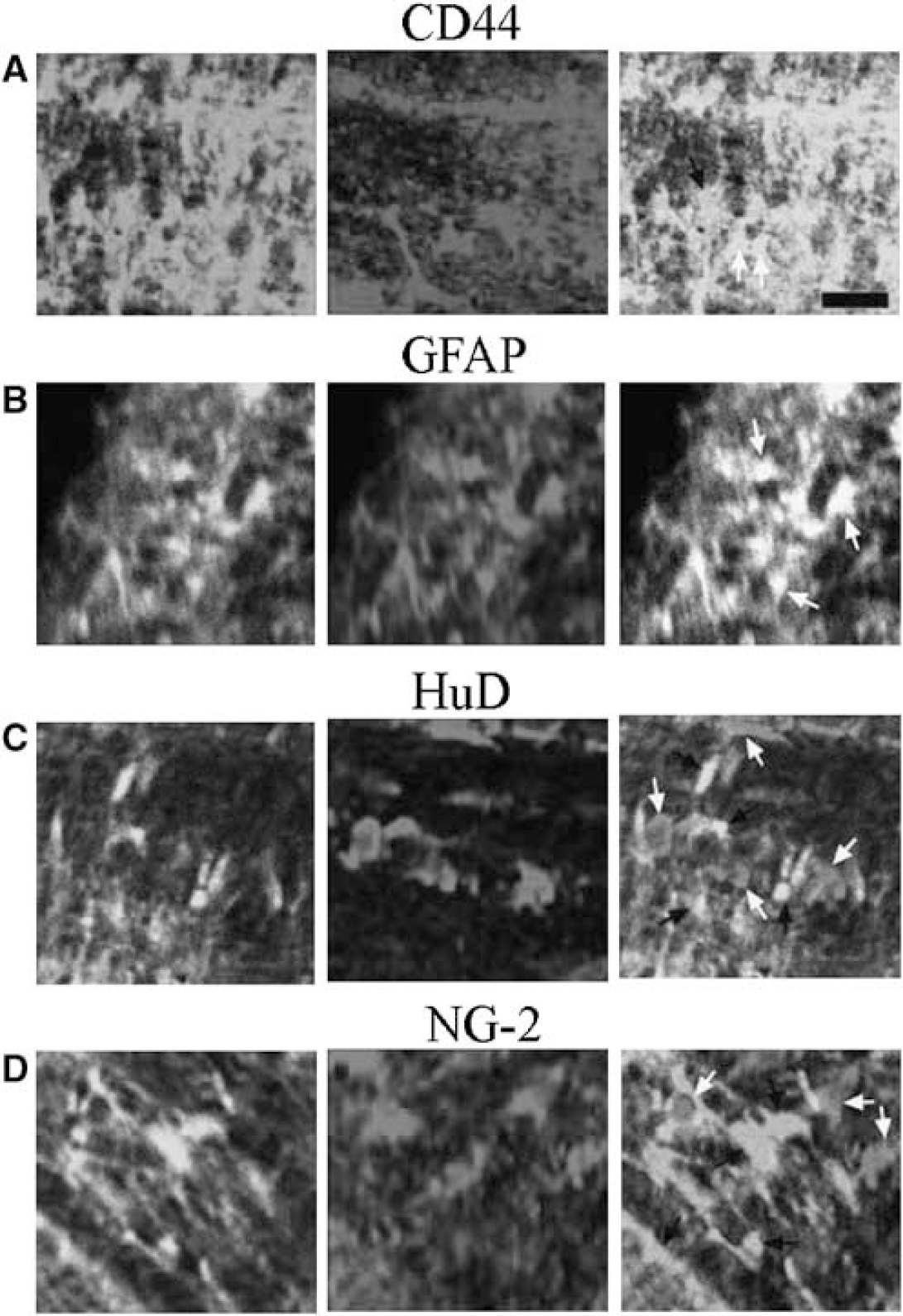
Immunostaining of GFP-GFAP(+) cells in the P10 mouse optic nerve. (
Confocal Microscopy
Optic nerves were attached to a 22 × 40 mm no. 1 microscope coverslip using a small amount of cyanocrylate glue at each end. The nerve was then sealed in a plexiglass perfusion chamber (Atmosphere chamber; Warner Instruments, Hamden, CT, USA) using silicone grease (see Fern, 1998). The nerve was perfused with aCSF or the appropriate experimental solution using a peristaltic pump (Gilson, Villiers, France) at a rate of 1.6 mL/min and aerated with either 95% O2/5% CO2 or 95% N2/5% CO2 at 120 mL/min. Solutions were heated just before entry into the chamber using an in-line heater (Warner Instruments) and the chamber temperature maintained using an objective heater (Bioptechs, Butler, PA, USA), using settings that would maintain chamber temperature at 37°C±0.5°C. The chamber was mounted on an Olympus IX70 invert microscope with a × 60 oil emersion objective. Samples were illuminated with an argon laser and images were captured using an Olympus FV300 Fluoview confocal image system. Image analysis was completed using MetaMorph software (Universal Imaging, Sunnyvale, CA, USA).
Scoring of Cell Death and Process Loss
To assess cell death, flattened stacks were opened in MetaMorph and a manual count was performed. Cell numbers were then expressed as a percentage of the number present at 0 min for each nerve and an overall percentage obtained by averaging the data for all nerves within a treatment group for each time point. To assess process loss, mean pixel intensity was measured within a selected region of the flattened image stack (200 × 200 × 16 μm thick; see Salter and Fern, 2005). Depending on image quality, up to five representative soma-free regions were selected per nerve. Mean pixel intensity was measured using MetaMorph and collated in Excel (Microsoft, Seattle, WA, USA). Mean pixel intensity was then normalized to the value recorded at 0 min for that region, with a mean percentage taken for all regions measured within that treatment group at each time point. Statistical significance was determined using analysis of variance or t-test, as appropriate (Prism, Graphpad, San Diego, CA, USA). Data are presented as mean±s.e.m.
Electron Microscopy
Optic nerves from P10 GFP-GFAP mice were perfused as above before post-fixing in 3% gluteraldehyde in Sorenson's solution. Nerves were then post-fixed with 2% osmium tetroxide and dehydrated before infiltration in epoxy. Sections were counterstained with uranyl acetate and lead citrate and examined with a Jeol 100CX electron microscope (see Thomas et al, 2004 for details). The identity of astrocytes was determined using established criteria, which included the presence of wide-bore endoplasmic reticulum, glycogen particles, and a characteristic irregular nucleus (Butt and Ransom, 1993; Grosche et al, 1999; Skoff, 1990; Vaughn, 1969). Astrocyte process length was measured using Photoshop (Adobe Software, San Jose, CA, USA), with two nerves examined blind for each condition. Process length was measured between the somata and the farthest point on the process that was continuous with the somata. No attempt was made to account for the often complex morphology of the processes and only the point that was most distant from the somata was measured. The number of processes that extended from each somata was also counted, and the data analyzed in turns of mean number of processes/somata and the mean length of the processes, under the various conditions.
Results
Imaging Of GFAP-GFP Astrocytes in the Mouse Optic Nerve
Optic nerves from mice transgenic for GFP under the control of the human GFAP promoter were live imaged ex vivo with a confocal microscope to characterize the morphology of astrocyte somata and processes under control conditions (Zhuo et al, 1997). Optic nerves were mounted in a perfusion chamber and imaged at 20 mins intervals in 1 μm increments from the glial limitans to a depth of 15 μm, a total of 16 images in each stack. The stacks were then flattened and analyzed for process loss and cell death. Astrocyte somata, the major processes radiating from the somata, and the fine distal processes were clearly visible in the image stacks (Figure 2A). Under control conditions where nerves were perfused for 60 mins with aCSF, both astrocyte somata and processes remained stable with fine processes intact and little cell death evident (Figure 2A). Cell process integrity was measured by assessing pixel intensity in defined regions that excluded cell soma and overall a ~15% decrease was measured after 60 mins (to 85.9±2.0% of the initial mean intensity, n = 7 nerves; Figure 2C). We have used this method previously to measure process injury in oligodendrocytes and find it to correlate closely with blinded, scored visual inspection of process loss (Salter and Fern, 2005).
Ischemic Injury to Astrocytes in the Optic Nerve
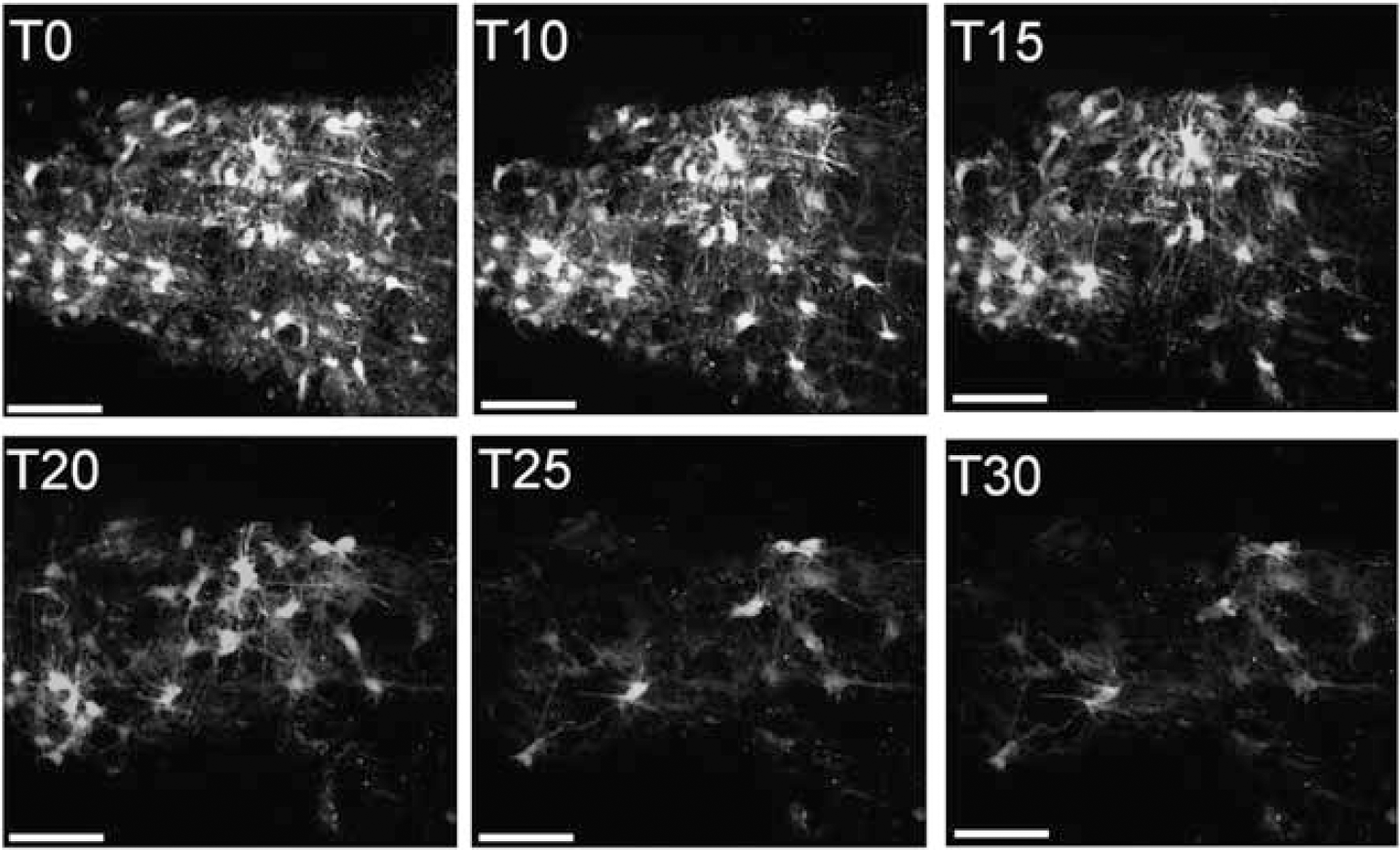
In contrast to control perfusion, switching to oxygen-glucose deprivation (OGD) resulted in severe progressive loss of cell processes (Figures 2B and 2C), which reached statistical significance after 40 mins (48.4±3.2% loss of pixel intensity, n = 14 nerves, compared with 12.1±2.5% in control; P < 0.001). Process loss was associated with a parallel loss of somata from the nerve (Figure 2B, arrow), and cell loss reached significance after 40 mins of OGD (Figure 2D; 66±4.4% cell loss compared with 4.8±2.5% in control; P < 0.001). Beading of processes with the formation of GFP-filled puncta was apparent on occasions (Figure 2B), a phenomenon that was always associated with the subsequent rapid loss of the somata. It was often the case that weakly fluorescent cells were apparent in the nerves, in particular at the end of the period of OGD. These cells presumably corresponded to the population of dull cells reported previously (Nolte et al, 2001), and appeared to have a high resistance to OGD and were not included in this study.
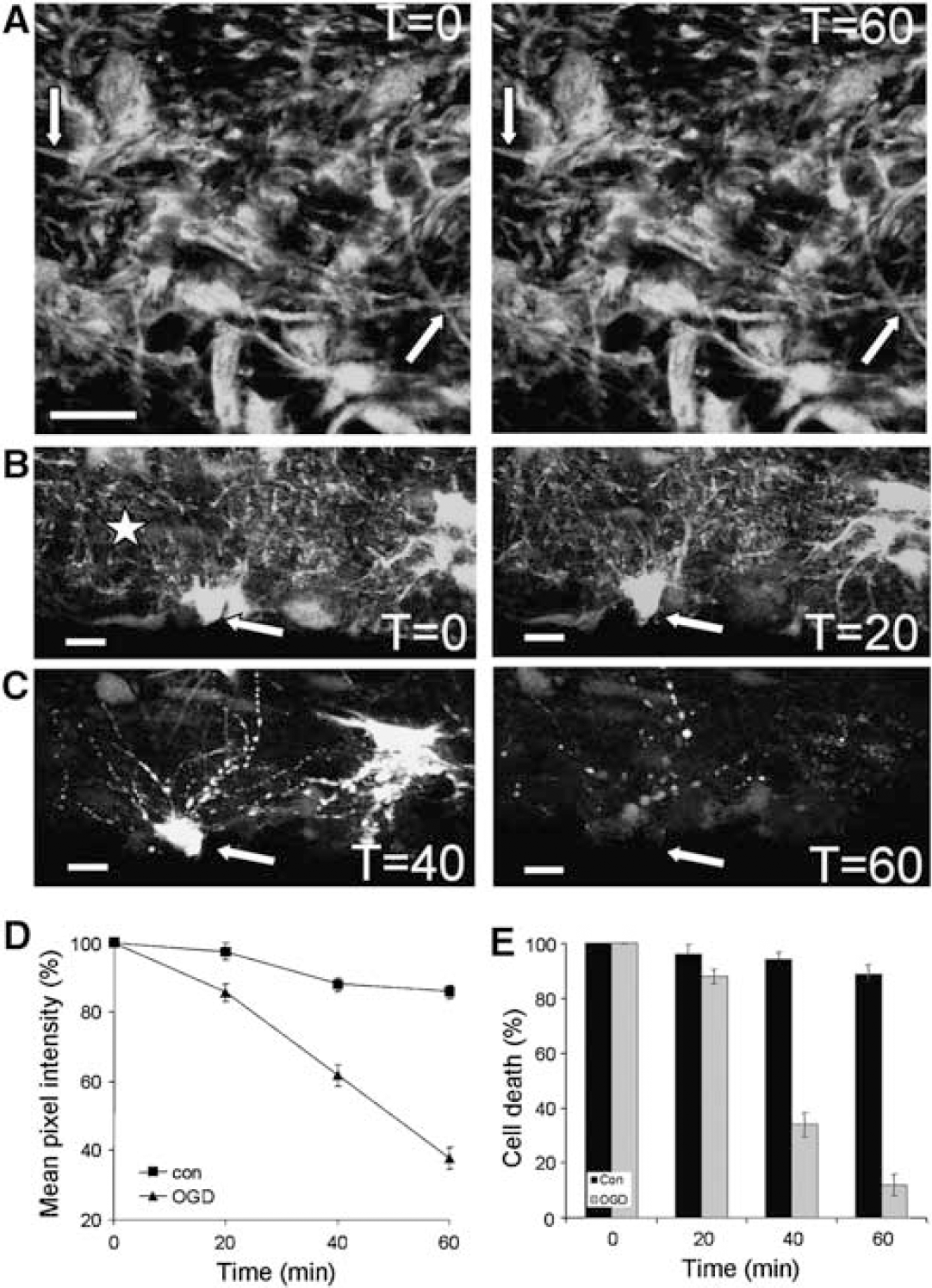
Live cell imaging of P10 optic nerve astrocytes: the effect of OGD. (
The ultrastructural features of OGD-induced injury were examined in P10 GFP-GFAP mice. At this point in development the mouse optic nerve was at a similar developmental point to the P10 rat optic nerves we have previously examined (Thomas et al, 2004), with the majority of axons pre-myelinated (Figures 3A and 3B). In most cases, glial cells could be readily identified using established criteria (see Materials and methods and Thomas et al, 2004). In control nerves perfused for 60 mins in aCSF before fixation, astrocyte processes radiated from the somata in the form of either thick primary processes or finer processes that originated directly from the somata or derived from branching of the primary processes. The fine processes, in particular, traveled circuitously around axon profiles (Figure 3B). Significant pathology was noted in nerves fixed after 60 mins of OGD (Figures 3C to 3H). In particular, examples were found where the thick proximal processes had detached from the soma (Figures 3C, 3D, and 3H arrows) and thick processes had parted along their length, most frequently in regions containing large cytoplasmic vacuoles (Figures 3E and 3F, arrow head). Examples of disintegrating processes were also found (Figure 3G, arrows). Separation of astrocyte processes from the somata was common even in micrographs from regions where axonal injury was minimal. For example, the astrocyte shown in Figures 4A to 4C is surrounded by numerous isolated sections of cell processes that appear to be disconnected from the somata and each other (e.g., arrows). Examples where small processes are parting are indicated by arrowheads. An example of a thick process that is separating into two sections is shown in Figures 4D to 4F. The section that is distal to the somata appears to be membrane delineated (small arrowheads, and note the nearby vacuole), while the proximal section appears open to the extracellular space (arrow).
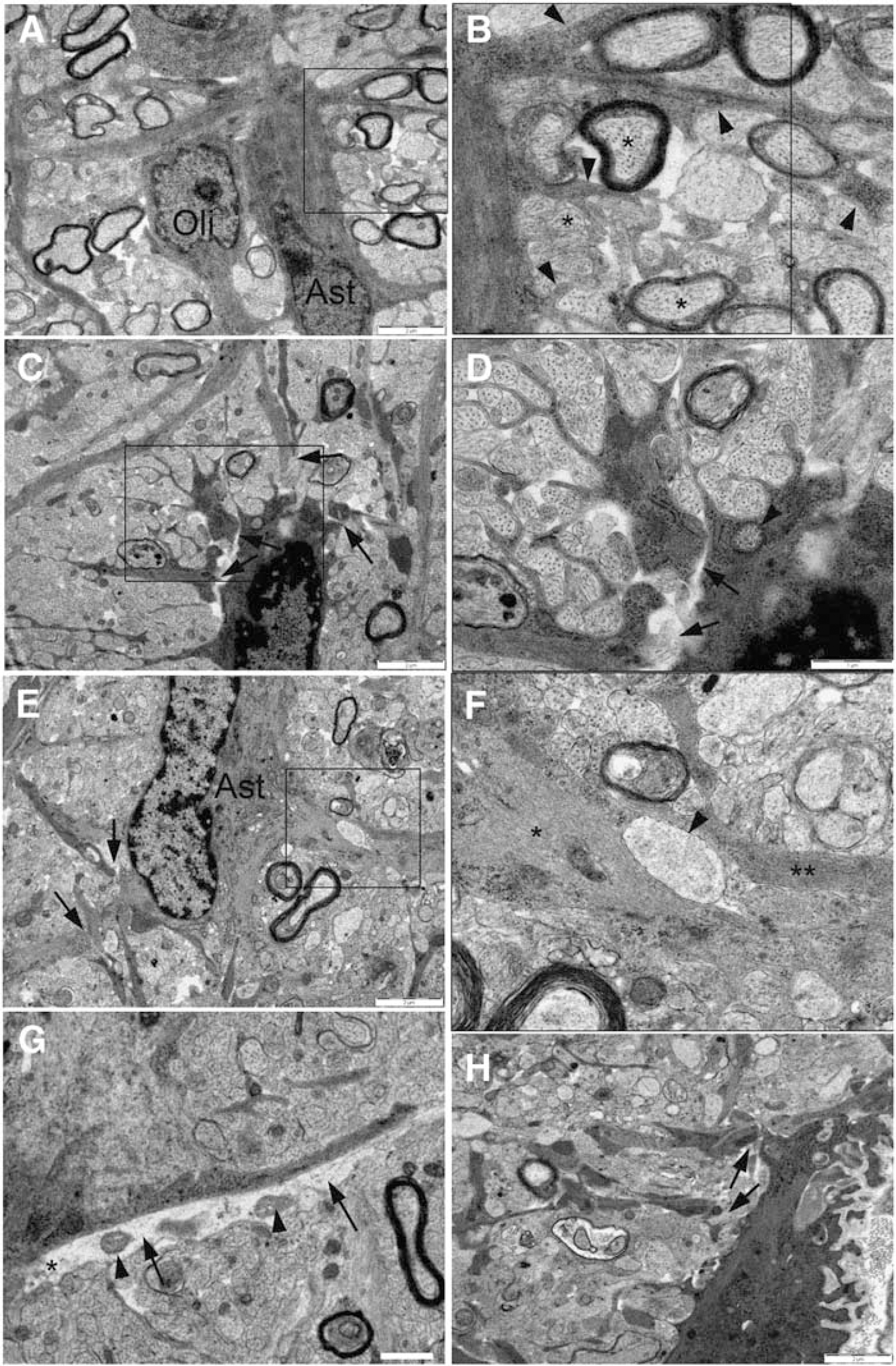
Ultrastructural features of OGD-induced changes in the P10 mouse optic nerve. (
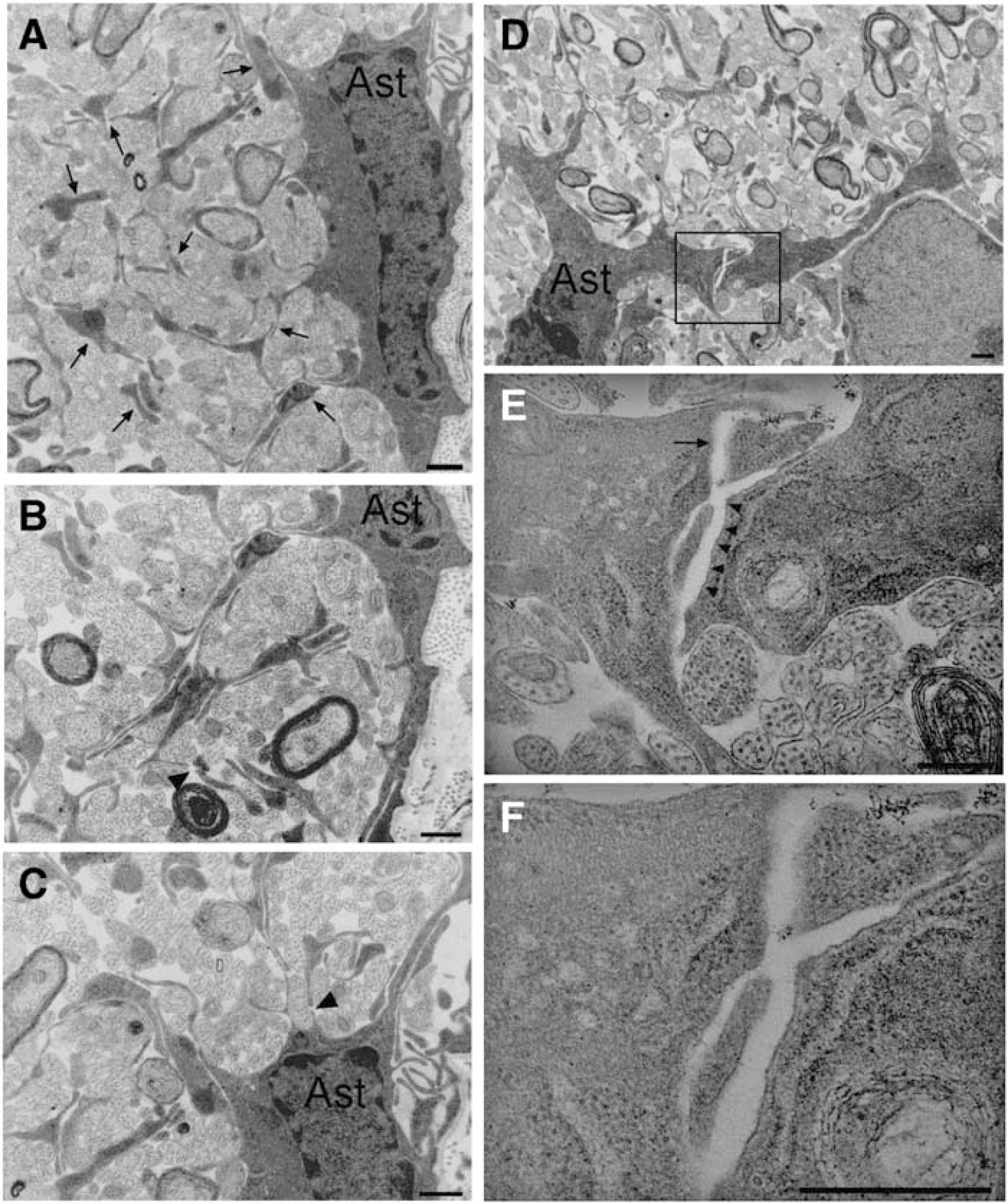
Ultrastructural features of OGD-induced changes in astrocyte processes in regions of mild axonal injury. (
Validation of The Use Of Green Fluorescent Protein Imaging to Assess Process Loss
To confirm that the changes in fluorescence seen in Figure 2 are due to loss of process integrity rather than to fluorescence quenching, a quantitative ultrastructural analysis was performed. The number and length of astrocyte processes in control nerves perfused for 60 mins with aCSF, nerves exposed to OGD for 60 mins, and nerves subjected to 60 mins of OGD in the absence of extracellular Na+ was determined (Figures 5A to 5E). Oxygen-glucose deprivation induced a ~33% reduction in the mean number of processes per astrocyte compared with control (Figure 5A), and a ~44% decrease in the average process length. This compares to an equivalent decrease of ~60% in the GFP fluorescence detected in astrocyte processes, which will presumably arise from a combination of these two factors (Figure 2C). The frequency distribution of process length showed the loss of longer processes after OGD (Figures 5C and 5D). A similar decrease in the mean number of processes per astrocyte was observed after OGD in the absence of extracellular sodium, but a significantly smaller reduction in process length was found (Figures 5A, 5B, and 5E). This is consistent with the intermediate level of protection observed under zero sodium conditions using the GFP-imaging approach as documented in Figure 9.
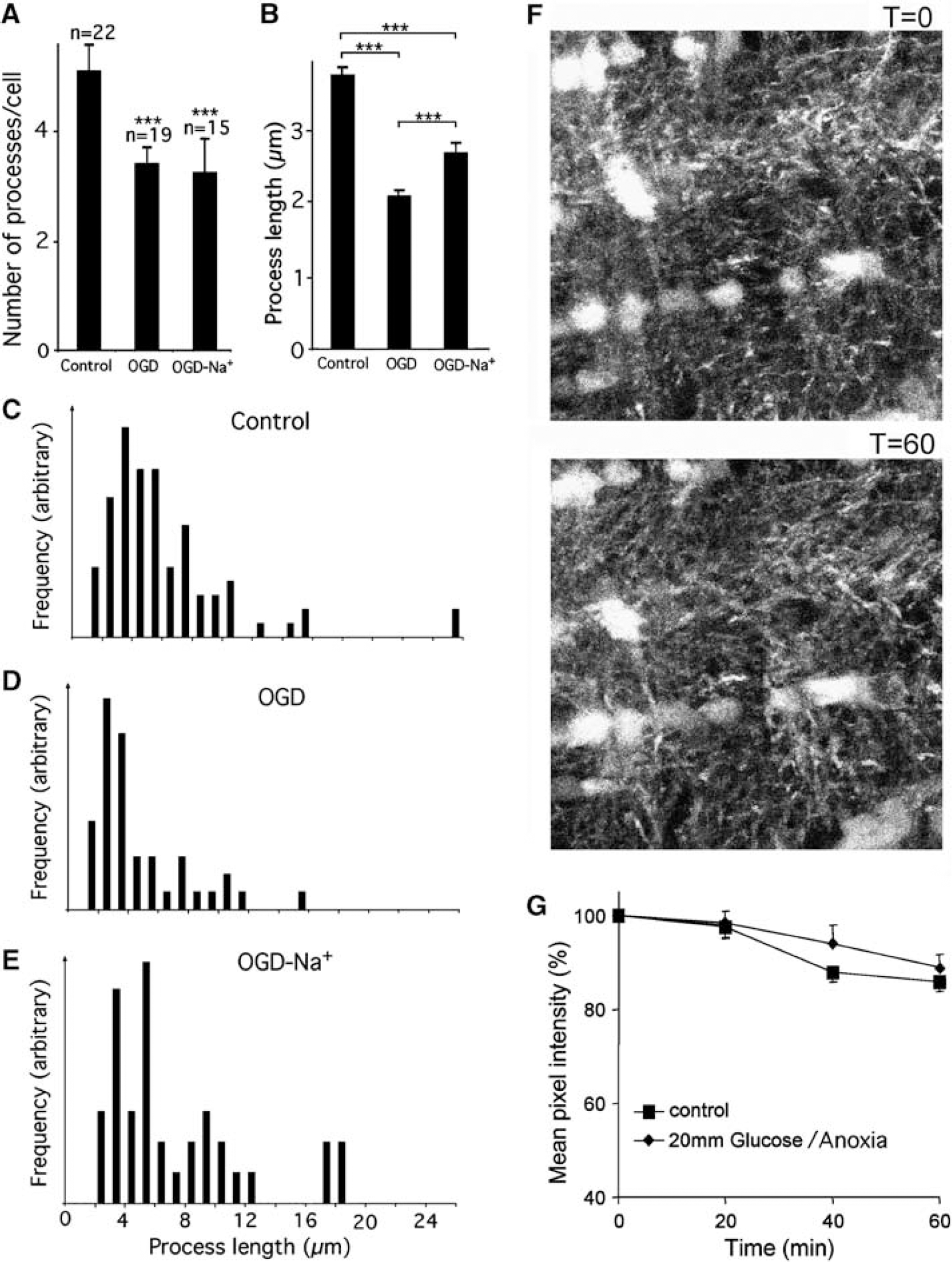
Validation of GFP-fluorescence imaging during OGD. (
Effects of metabolic arrest on GFP(+) astrocytes in P10 mouse optic nerve. A time series is shown illustrating the rapid loss of processes and somata on exposure to NaCN and iodacetic acid. Bar = 50 μm.
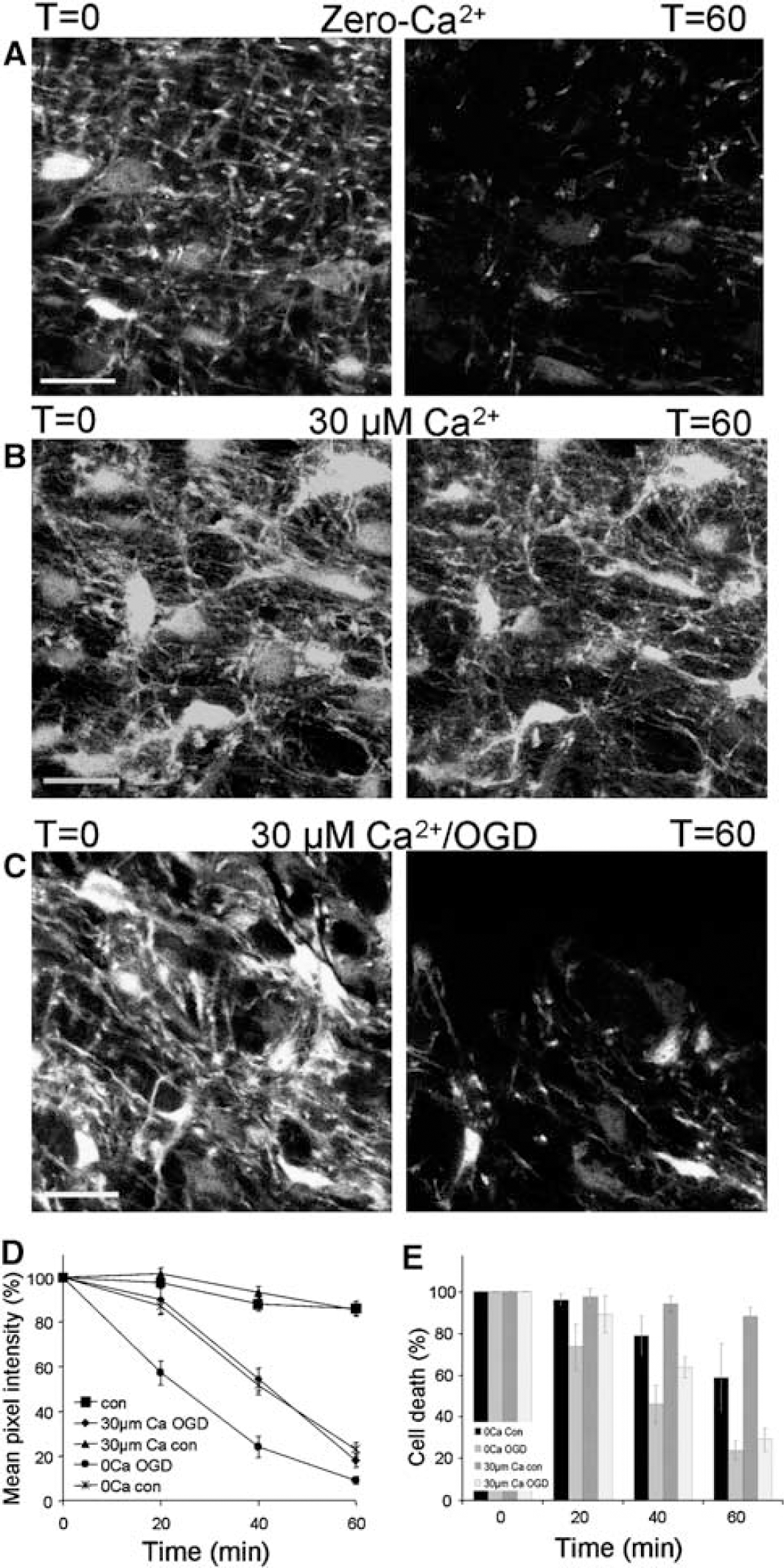
Ca2+ astrocyte injury. (
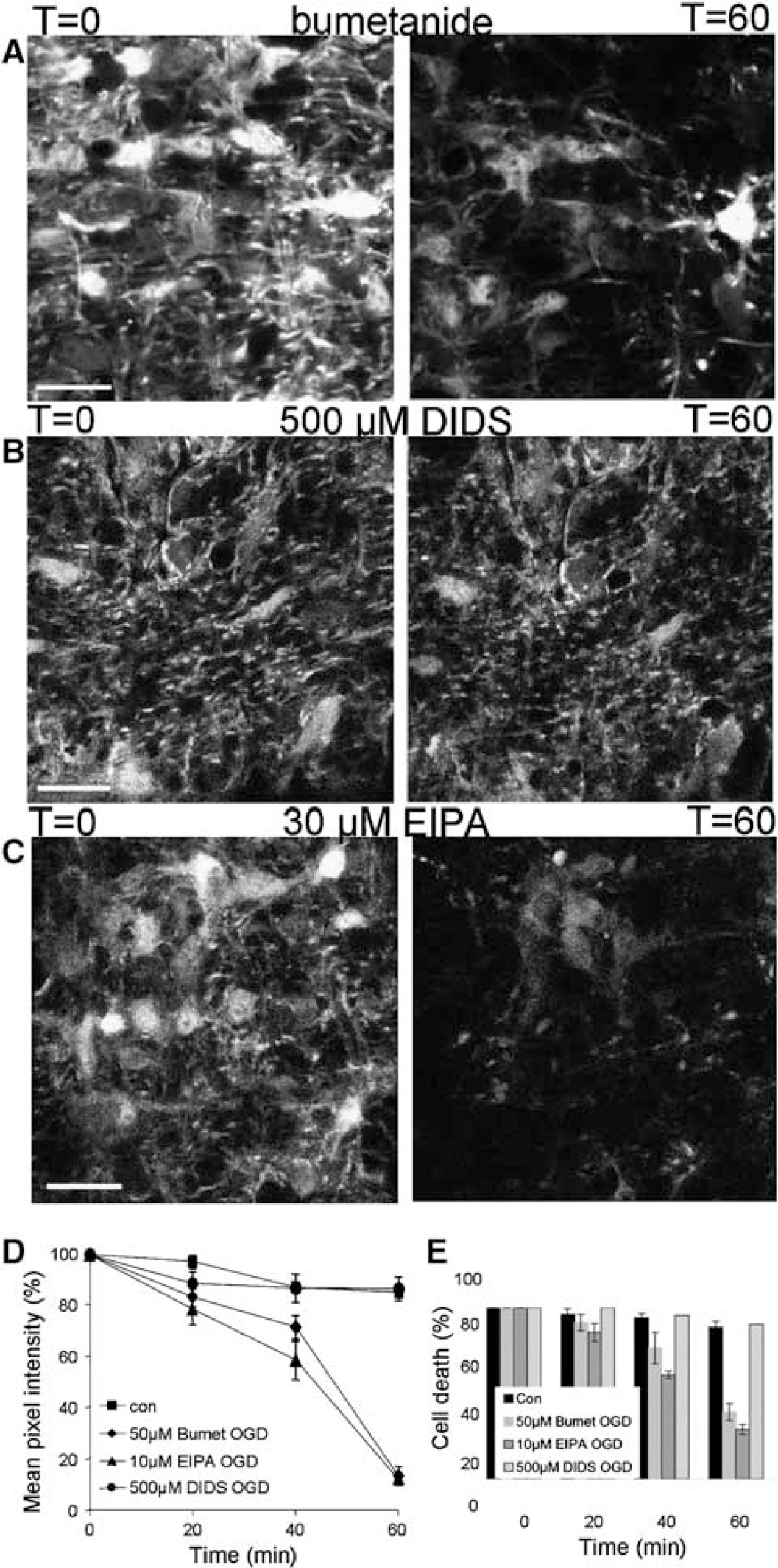
The effects of ion transport blockers. (
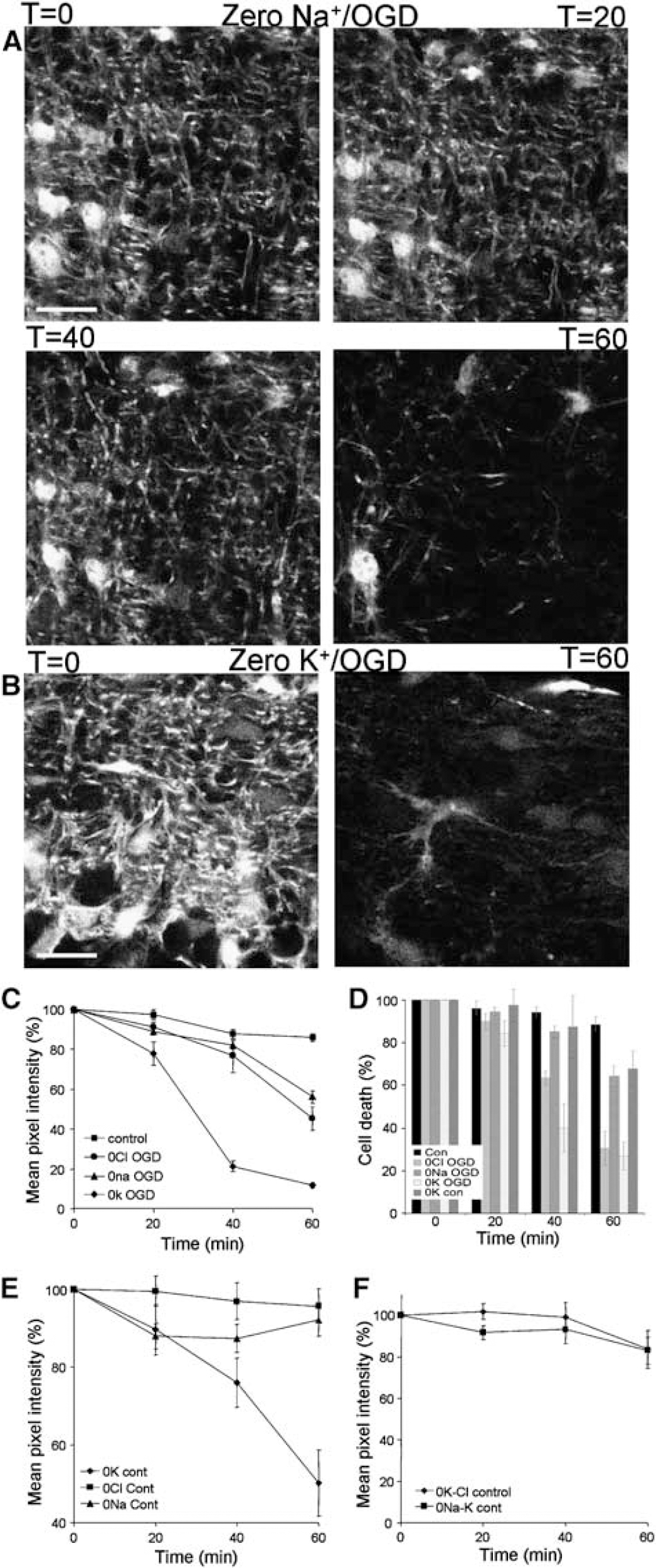
The effect of removing selected ions. (
It has previously been shown that elevating the extracellular glucose concentration potentiates acid changes produced by anoxia in the optic nerve (Ransom et al, 1992). The effect of 60 mins perfusion with 20 mmol/L glucose/anoxia are shown in Figures 5F to 5G, which show that this condition has no significant effect on the integrity of processes assessed using the fluorescence technique. Perfusion with sodium propionate (20 mmol/L), which will evoke an intracellular acid shift similar to that produced by OGD (Ritucci et al, 1998), was also without significant effect on process fluorescence (data not shown).
Effect of Metabolic Poisons
The effect of complete metabolic arrest was examined, using a combination of cyanide (2 mmol/L) and iodacetic acid (1 mmol/L) to block both glycolysis and oxidative phosphorlyation. The effects of metabolic inhibition were apparent more rapidly than were those of OGD (Figure 6). Complete loss of GFP(+) astrocytes was found after 40 mins of metabolic inhibition (n = 4 nerves), and almost complete loss of both somata and processes occurred within the first 30 mins (Figure 6).
The Role of Ca2+ in Astrocyte Injury
While ischemic cell death of pre-myelinating white matter astrocytes is Ca2+-dependent (Fern, 1998), we have recently shown that Ca2+ does not play a role in the death of astrocytes in white matter at a similar developmental stage to those studied here, at least during relatively short periods of ischemia (Thomas et al, 2004). In the current study, initial experiments showed that removing Ca2+ from the extracellular space by perfusing with aCSF to which no Ca2+ had been added and which contained 70 μmol/L EGTA (‘zero-Ca2+’), resulted in significant damage to processes at 40 mins (48.3±4.1% decrease in pixel intensity; n = 5 nerves; P < 0.001) and 60 mins (76.9±3.1%; P < 0.001; Figures 7A and 7D). Not surprisingly, therefore, zero-Ca2+ conditions failed to protect against OGD-induced process loss (Figure 7D). To prevent toxicity from Ca2+ removal, we employed a low Ca2+ solution (containing a calculated free [Ca2+] of 30 μmol/L). This solution was not toxic to astrocyte processes or somata (Figures 7B, 7D, and 7E) and had no protective effect against either process loss or soma loss during OGD (Figures 7C to 7E). Nerves showed significant loss of pixel intensity in 30 μmol/L Ca2+ after 40 mins of OGD (45.8±5% reduction, n = 5 nerves), a rate of process loss that continued to 60 mins. There was no significant difference between results for low Ca2+ ischemia and aCSF ischemia, indicating that injury to processes is not Ca2+-dependent and confirming that cell soma death is also not Ca2+-dependent.
The Effect of Transport Blockers
There is evidence that the NKCC is involved in injury to astrocyte somata via a loss of cell volume regulation (Su et al, 2002a, b ; Thomas et al, 2004), with the NKCC blocker bumetanide giving protection against soma death during a 40 mins period of OGD. The effect of bumetanide (50 μmol/L) on OGD-induced astrocyte process loss was tested, revealing significant protection at 40 mins of OGD (n = 6 nerves; P < 0.001 versus OGD) but not at 60 mins (P > 0.05; Figures 8A and 8D). Bumetanide had a similar effect on astrocyte somata with protection from injury at 40 mins (P < 0.001 versus OGD) with less significant protection at 60 mins (Figure 8E; P > 0.05). However, this protective effect of bumetanide was much reduced after 60 mins of OGD, although some benefit over ischemia in the absence of bumetanide remained (P < 0.01).
4,4′-Diisothiocyanostilbene-2,2′-disulfonic acid is a potent blocker of various Cl− transporters and channels. In a previous study, we found 100 μmol/L 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (DIDS) to be ineffective against OGD-induced astrocyte injury in P10 rat optic nerves (Thomas et al, 2004). In the current study, 500 μmol/L DIDS produced significant levels of protection from injury to both cell somata and processes throughout a 60 mins period of OGD (n = 5 nerves; Figures 8B, 8D, and 8E). There was no significant difference evident between the control nerves and nerves exposed to 60 mins OGD in the presence of this drug. In contrast, the Na—H exchanger blocker ethyl isopropyl amiloride (10 μm) failed to protect either astrocyte somata or processes from injury during OGD (n = 5 nerves; Figures 8C to 8E). We also used the Na—Cl transport blocker furosemide but found it to fluoresce under our imaging conditions, making measurement of pixel intensity difficult. We did however observe a loss of process integrity at 60 mins after visual inspection of images taken of nerves subjected to 60 mins OGD in the presence of furosemide.
Ionic Dependence of Process Loss in Astrocytes
Replacing either Na+ or Cl− was found to reduce process loss after 40 mins of OGD, a protective effect that was lost by 60 mins OGD (Figures 9A and 9C). When compared with control, there was no significant injury to astrocyte processes at 40 mins in either case (n = 11 and five nerves, respectively; P > 0.05). However, by 60 mins there was an acute deterioration of processes with a 43.8±2.9% loss of pixel intensity in the absence of Na+ (P < 0.001 versus, control), and a 54.8±5.7% loss of pixel intensity in the absence of Cl− (P < 0.001 versus, control). Consistent with previous results, either Na+ or Cl− substitution was highly protective of the astrocyte somata (compare Figure 9D with Figure 2D). We also looked at the effect of removing K+ from the media during OGD. We saw an immediate impact under these conditions with a 22.3±5.8% decrease in pixel intensity after 20 mins of OGD (Figures 9B and 9C; n = 5 nerves, P < 0.01 versus, control), which along with exposure to ischemic conditions in zero-Ca2+ was one of only two occasions in which we observed a significant effect at this time point. By 40 mins there was a catastrophic loss of processes compared with control with a 78.7±2.8% (P < 0.001) decrease in pixel intensity and the astrocytes continued to deteriorate through to 60 mins (Figure 9C). Cell somata also suffered under these conditions with significant losses compared with control at 40 mins (P < 0.001) and 60 mins (P < 0.001; Figure 9D).
We found that under control conditions removal of extracellular K+ was toxic to cell processes, with significant process loss compared with control after 60 mins (Figure 8E; n = 5 nerves; P < 0.001). In contrast, there was no significant loss of cell somata (Figure 9D). Neither Na+ nor Cl− substitution had any significant effect on astrocyte processes under control conditions (Figure 9E), and the toxic effect of K+ substitution was blocked by the co-substitution of either of these ions (Figure 9F). K+ substitution therefore evokes process injury that is dependent on both Na+ Cl−, presumably involving some form of cytotoxic volume change (e.g., Goldberg and Choi, 1993).
The data show complete protection of astrocyte processes by the Cl− transport and channel blocker DIDS, but only partial protection by Cl− substitution. We therefore examined the effect of HCO−3 removal by applying a HEPES-buffered aCSF HCO−3-free conditions were found to be completely protective against OGD-induced process and somata loss for more than 60 mins (n = 5 nerves; Figures 10A, 10C, and 10D). Since this experiment was performed in otherwise normal aCSF with a Cl− concentration of 131 mmol/L, this protection cannot be due to the loss of a HCO−3 component of a Cl− conductance. We therefore performed one further experiment to test the protective effect of the only major ion remaining that might contribute to injury, K+ (in the absence of Na+ to prevent K+ substitution toxicity). This condition was also completely protective against OGD-induced astrocyte process (n = 5 nerves; Figures 10B and 10C) and somata (Figure 10D) loss.
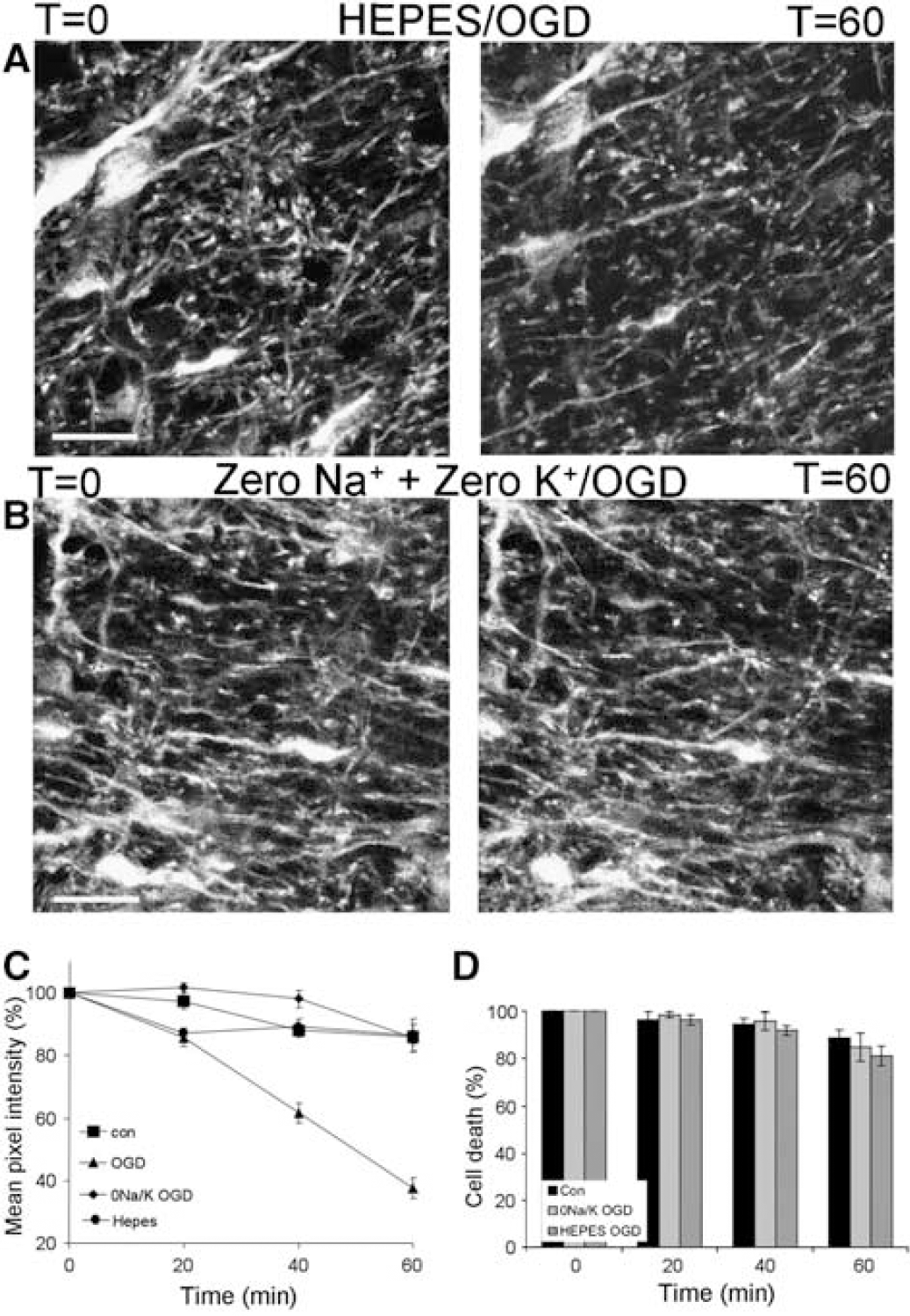
Some conditions that provide protection from injury. (
Discussion
Identity of GFAP-GFP(+) Cells in the Neonatal Optic Nerve
The optic nerve contains cells with neurogenic potential (Palmer et al., 1999), and several recent studies have described populations of GFAP(+) neurogenic progenitors. For example, GFAP-GFP(+) cells of the neonatal subventricular zone have been shown to have the potential to differentiate into neural stem cells (Raponi et al, 2007), while cultures of GFAP(+) cells from P0 to P2 brains contain a population of cells with neurogenic potential (Imura et al, 2006). However, it must be born in mind that GFAP-GFP(+) of the neonatal subventricular zone have many of the properties of mature astrocytes and behave functionally as astrocytes (Liu et al, 2006), while it is known that GFAP(+) cells of the neonatal rodent optic nerve are postmitotic in situ (Skoff, 1990). Furthermore, GFAP-GFP expression in somewhat more mature optic nerve is restricted to astrocytes identified on ultrastructural, electrophysiologic, morphologic, and immunolabeling criteria (Nolte et al, 2001). Consistent with this, Steiner et al, 2004 have reported that neurogenic hippocampal astrocytes become GFAP-GFP(–) before expression of the earliest neuronal markers (Steiner et al, 2004). The GFAP-GFP(+) cells examined in the current study therefore form a population of mature astrocytes, although possibly containing a small population of cells that have retained neurogenic potential.
Consistent with this, GFAP-GFP(+) cells of P10 optic nerve were positive for astrocyte markers such as CD44, GFAP, and GLT-1, were negative for oligodendroglial markers such as NG-2 and O4, and were negative for neuronal markers such as neurofilament, Thy-1 and HuD. We found some GFAP-GFP(+) cells that were CD44(–), which is consistent with prior reports of staining of a subpopulation of optic nerve astrocytes (Jones et al, 2000). Interestingly, while all GFAP-GFP(+) astrocytes were HuD(–), we observed a large population of HuD(+) cells that appear to be oligodendroglia. This is consistent with prior reports of Hu(+) glia in the rodent optic nerve (Kostyk et al, 1996).
The Effects of Ischemic Conditions on Astrocytes
Using GFAP-GFP mice, we report rapid changes in astrocyte processes in the whole-mount optic nerve during ischemia modeled by OGD. Injury to the processes was significant after 40 mins of OGD and pathologic changes in their morphology were apparent after 20 mins. No protection against injury was afforded to either the processes or somata of the astrocytes by reducing extracellular Ca2+ to 0 or 30 μmol/L, which confirms an earlier report that acute astrocyte injury is Ca2+ independent (Thomas et al, 2004), and extends this paradoxical finding to the processes of the cells. This is in contrast to the situation in pre-myelinating white matter astrocytes (Fern, 1998), neurons (Goldberg and Choi, 1993), and oligodendrocytes (Fern and Moller, 2000; Wilke et al, 2004; Micu et al, 2006; Salter and Fern, 2005), where Ca2+ influx is central to acute cell injury during ischemic conditions.
In the somata of P10 rat optic nerve astrocytes, acute cell death during 40 mins of OGD involves cell swelling associated with NKCC activation, which the cells counter by active volume regulation. The subsequent depletion of cellular energy reserves results in cell death, apparently by cytotoxic cell swelling (Thomas et al, 2004). The current findings show that a number of conditions protect astrocyte processes during the first 40 mins of OGD. These include replacing either extracellular Na+ or Cl− or perfusion with the selective NKCC blocker bumetanide, data that are consistent with early activation of NKCC, and injury to the processes via the same pathway that operates in the somata. However, the conditions that block the NKCC only delay the onset of process injury, which becomes significant after 60 mins of OGD in zero-Na+, zero-Cl−, or bumetanide. In contrast, block of HCO−3 transport by substitution of extracellular HCO−3, or perfusion with the broad-spectrum HCO−3-transport blocker DIDS (500 μmol/L), prevented both process and somata death over a 60 mins period of OGD. Presumably both DIDS and HCO−3-substitution are protective during the first 40 mins of OGD through block of HCO−3 transporters, indicating that DIDS-sensitive HCO−3 transport acts in parallel with NKCC, with their combined action producing early cytotoxic cell swelling.
The situation is different after 60 mins of OGD, where zero-Na+, zero-Cl−, or bumetanide are no longer protective and NKCC is presumably no longer a significant cause of cytotoxicity. Conversely, the prolonged protection provided by HCO−3-transport block implicates continued pathologic activation of an HCO−3 transporter in the cells during OGD. The identity of the HCO−3 transporter involved in this phenomenon may be unusual, because neither Na+ nor Cl− substitution was protective at 60 mins of OGD. One possibility is that a DIDS-sensitive, K-HCO−3 co-transporter is present in developing white matter astrocytes, which would be consistent with the protective effect at 60 mins of removing extracellular K+ (simultaneous with Na+ removal). K-HCO3 co-transport has been identified in squid giant axons (Hogan et al, 1995a, b ), but this is the first indication of its presence in the CNS. As with the NKCC and the other HCO−3-transporters, stimulation of K-HCO3 co-transport will produce cell swelling and potential cytotoxic injury, and the protective effect of combined Na+ K+ substitution will follow the block of NKCC and Na+- and K+-dependent HCO−3 transporters, explaining all of the data collected in the current investigation.
Clasmatodendrosis
In the current experiments, we found no condition under which the injury produced by OGD in astrocyte processes occurred more rapidly than the parallel loss of astrocyte somata, indicating that ‘clasmatodendrosis', as defined as a loss of distal processes before a loss of somata, was not seen during OGD in the current preparation. In this regard, it is worth noting that the only other modern study of clasmatodendrosis also observed somata death at short periods after a loss of distal processes when examined using cytoplasmic GFP fluorescence (Hulse et al, 2001). As in this previous study, we did observe beading of GFP-filled astrocyte processes during OGD, indicating the temporary formation of membrane-bound fragments of disintegrating processes. It must be born in mind that we have recently shown that immature white matter astrocytes are much more sensitive to ischemic conditions than are immature gray matter astrocytes (both in whole-mount preparations; Shannon et al, 2007). This may indicate that the astrocytes studied in the current investigation suffer a more severe form of injury than may occur in gray matter parts of the developing brain, and may therefore be less likely to exhibit a selective loss of cell processes. At the ultrastructural level we also found clear examples where processes were detaching from the soma, often associated with the presence of cytoplasmic vacuoles that may indicate local cell swelling. It would appear, therefore, that shedding of processes is a feature of acute ischemic injury of astrocytes, but that the underlying injury mechanism is not selective for processes and proceeds in parallel with injury to the somata.
We did observe a selective loss of astrocyte processes in the injuries produced by zero-Ca2+ or zero-K+ conditions. The effect of K+-removal was Na+- and Cl−-dependent, and we can speculate that the mechanism underlying this process loss may involve a localized form of cell swelling of unknown origin. Because volume regulation in astrocytes is partially dependent on Ca2+ (O'Connor and Kimelberg, 1993), a similar phenomenon may also explain the effect of Ca2+ substitution.
Validation of the Green Fluorescent Protein Imaging to Assess Cell Morphology Changes During Oxygen-Glucose Deprivation
A variety of approaches can be used to assess cell morphology changes in the brain during ischemic conditions. The most commonly used is immunohistochemistry at the light level. However, we have recently shown that astrocyte markers such as GFAP cannot be successfully applied to white matter preparations using this technique due to the low level of staining of the somata (Shannon et al, 2007). A more accurate approach is to use quantitative electron microscopy, although this can be laborious and does not allow the continuous assessment of cell shape with time. Confocal GFP-fluorescence imaging, in contrast, allows a time series of morphologic changes to be collected in an objective manner. This approach has been used recently to assess changes in oligodendrocyte processes (Salter and Fern, 2005). However, the application of GFP-fluorescent imaging under ischemic conditions may require additional validation because ischemia evokes changes in the ionic composition and pH of cells that may affect GFP-fluorescence.
In the current study, quantitative electron microscopy was used to measure the number of astrocyte processes per cell and the length of the processes under several conditions. The quantitative electron microscopy and the GFP-fluorescent imaging data correlated well, showing a similar degree of process loss after 60 mins of OGD compared with control perfusion, and an intermediate effect of OGD in the absence of extracellular Na+. Furthermore, conditions that evoke larger pH changes in the optic nerve than OGD, such as anoxia in the presence of 20 mmol/L glucose, failed to affect GFP fluorescence of astrocyte processes. Mature gray matter astrocytes show a small intracellular pH change during short (20 mins) periods of global ischemia in situ (Lascola and Kraig, 1997), while whole-mount P0–2 rodent optic nerve astrocytes show only a ~0.2 acid pH change during 60 mins of OGD (Garrido-Comas and Fern, 2004). Because the fluorescence of GFP is generally stable between pH 6 and 10, with the S65T mutant used here being only slightly more pH sensitive over this range (Patterson et al, 1997), the evidence is strong that GFP fluorescence of GFP-GFAP astrocytes in the current preparation is not significantly affected by pH changes during ischemic conditions.
Conclusions
The OGD-induced damage suffered by immature white matter astrocytes reported here may be relevant to the injuries most commonly associated with cerebral palsy. In many cases, these selective white matter lesions of the developing brain appear to be hypoxic—ischemic in origin (Back and Rivkees, 2004; Volpe, 1995). While astrocyte damage has not been well-studied in histopathologic studies, the sensitivity of these cells to ischemic-type injury together with the apparent loss of glutamate from them during ischemia (Back et al, 2006; Thomas et al, 2004), and the significance of extracellular glutamate to the injury process (Fern and Moller, 2000; Follett et al, 2000, 2004; Salter and Fern, 2005; Wilke et al, 2004), all suggest that the early astrocyte changes reported here will have clinically important consequences. The mechanisms of astrocyte injury involve overactivation of NKCC and DIDS-sensitive HCO−3 transport, leading to ion influx and loss of cell integrity as the cells swell. Complete interruption in the cellular energy supply resulted in accelerated cell death, indicating that astrocytes can use their glycogen energy reserve for a period to maintain volume regulation (Ransom and Fern, 1997). The current study indicates that at least three transport proteins are involved in ischemia-induced cytotoxic ionic influx into developing white matter astrocytes: NKCC, Na+-dependent HCO3 transport, and K+-dependent HCO−3 transport. This significantly increases the number of targets for potential therapeutic intervention in a highly prevalent and currently untreatable disease.