
Abstract
Multiple sclerosis (MS) is a disease of the central nervous system characterized by patchy areas of demyelination, inflammation, axonal loss and gliosis, and a diffuse axonal degeneration throughout the so-called normal-appearing white matter (NAWM). A number of recent studies using perfusion magnetic resonance imaging in both relapsing and progressive forms of MS have shown a decreased perfusion of the NAWM, which does not appear to be secondary to axonal loss. The reduced perfusion of the NAWM in MS might be caused by a widespread astrocyte dysfunction, possibly related to a deficiency in astrocytic β2-adrenergic receptors and a reduced formation of cAMP, resulting in a reduced uptake of K+ at the nodes of Ranvier and a reduced release of K+ in the perivascular spaces. Pathologic and imaging studies suggest that ischemic changes might be involved in the development of a subtype of focal demyelinating lesions (type III lesions), and there appears to exist a relationship between decreased white matter perfusion and cognitive dysfunction in patients with MS.
Keywords
Introduction
The pathology of multiple sclerosis (MS) is characterized by inflammation, demyelination, axonal loss, and gliosis. Most people with MS have a relapsing—remitting course, often followed by a more gradual decline in neurologic function, termed secondary progressive MS. Some patients have gradual worsening from the beginning, termed primary progressive MS. Relapses are caused by focal demyelinating lesions (plaques) in the brain or spinal cord. The progressive neurologic disability of secondary and primary MS is mainly based on a slowly progressive widespread axonal degeneration (Frohman et al, 2005; Keegan and Noseworthy, 2002).
The classic teaching is that MS is a T-cell-mediated autoimmune disorder of the central nervous system. However, a number of pathophysiological observations cannot be simply explained on the basis of autoimmune mechanisms. First, the progressive (neurodegenerative) component of the disease continues despite intense immunosuppressive interventions that effectively stop inflammatory disease activity (Coles et al, 1999; Metz et al, 2007; Roccatagliata et al, 2007; Samijn et al, 2006). Second, pathologic studies have shown that some demyelinating lesions develop without a preceding inflammatory reaction (Barnett and Prineas, 2004; Gay, 2007, 2006; Guseo and Jellinger, 1975; Lucchinetti et al, 2000). Third, another intriguing finding difficult to explain by autoimmune phenomena is the finding of a diffuse cerebral white matter hypoperfusion, which is the subject of this review.
White matter hypoperfusion
A number of studies using single photon emission computed tomography or positron emission tomography found reduced cerebral blood flow (CBF) in the gray and white matter of patients with MS, but these findings received little attention in terms of their possible pathophysiological significance (Brooks et al, 1984; Lycke et al, 1993; Sun et al, 1998; Swank et al, 1983).
Renewed interest in this matter arose from perfusion studies with magnetic resonance imaging, a technique allowing differentiation between white matter plaques and normal-appearing white matter (NAWM). Dynamic susceptibility contrast-enhanced magnetic resonance imaging is based on the acquisition of a series of images during the transit of a bolus of contrast material through the vasculature of brain parenchyma. It is a very fast imaging technique typically using echo planar with a temporal resolution of 1 or 2 secs. The method provides parameters of brain perfusion, such as CBF, cerebral blood volume (CBV), and vascular mean transit time. By using this technique, Law et al (2004) found a significantly decreased CBF by about 50%, and a more than twofold prolonged mean transit time, throughout the NAWM in patients with relapsing—remitting MS compared with controls. Adhya et al (2006) studied the regional pattern of tissue perfusion in the NAWM of patients with primary progressive MS, relapsing—remitting MS, and healthy controls. The CBF and CBV were significantly decreased in all NAWM regions in both forms of MS compared with controls. This shows that a globally decreased white matter perfusion is a consistent phenomenon in MS, whatever the disease subtype of MS.
The majority of nonenhancing (chronic) focal lesions in MS white matter show a more decreased perfusion when compared with contralateral NAWM (Law et al, 2004). Conversely, gadolinium-enhancing (active) lesions show increased CBF and CBV, indicative of inflammation-mediated vasodilation (Ge et al, 2005; Haselhorst et al, 2000).
Hypoperfusion of gray matter in MS (Brooks et al, 1984; Inglese et al, 2007; Lycke et al, 1993; Sun et al, 1998; Swank et al, 1983) may be caused by the same mechanism underlying the reduced CBF in NAWM, or may be the consequence of a disconnection between the cerebral cortex and the subcortical structures because of the white matter damage. A similar reduced perfusion of the cerebral cortex is also observed in ischemic white matter damage of the Binswanger type (Yao et al, 1990).
Primary or secondary phenomenon?
To assess whether hypoperfusion in NAWM in MS may be related to a primary vascular etiology or is secondary to decreased metabolic demand from axonal degeneration, Saindane et al (2007) studied the relation between perfusion and diffusion tensor imaging in normal-appearing corpus callosum in patients with relapsing—remitting MS and control subjects. Diffusion tensor imaging is a technique that quantifies the amount of nonrandom water diffusion within tissues, providing information about processes such as ischemia and axonal (Wallerian) degeneration that can affect water diffusion as a result of microstructural changes (Alexander et al, 2007). The mean diffusivity (MD) measures the magnitude of diffusion of the water molecules, whereas diffusion anisotropy indices, such as fractional anisotropy (FA), indicate the deviation from pure isotropic diffusion of water mobility in vivo.
In acute ischemia, MD is decreased because of cellular swelling, restricting the diffusion of water molecules (Munoz Maniega et al, 2004). After the acute phase, MD increases to normal values, and in the chronic phase, becomes elevated because of loss of membrane integrity and tissue necrosis, and FA progressively decreases, which is also consistent with loss of integrity of the tissue structure (Munoz Maniega et al, 2004). In MS patients, there was a highly significant correlation between CBF and MD (decreasing CBF was associated with decreasing MD), but not with FA. This finding argues against the possibility that the white matter hypoperfusion in MS is secondary to axonal degeneration, as this would be characterized by increased MD and decreased FA (Saindane et al, 2007). However, it does not exclude the possibility that a diffuse reduced axonal activity underlies or contributes to the reduced perfusion of the NAWM.
Vascular pathology in MS white matter?
Ge et al (2005) interpreted the hypoperfusion in NAWM as a vasculopathy in the context of the perivascular inflammations that occur in focal MS lesions. However, although inflammatory infiltrates in MS are typically located around smallor medium-sized veins (Adams, 1989) and in the perivascular spaces surrounding arterioles (Gay, 2006; Gay et al, 1997), microvessel thrombosis is only exceptionally being observed within these lesions (Aboul-Enein and Lassmann, 2005; Wakefield et al, 1994). A primary vascular pathology, as seen in vasculitis, would lead to regional cerebral perfusion defects rather than a generalized microvascular hemodynamic impairment as observed in MS. Furthermore, the finding that enhancing white matter lesions show increased CBF and CBV, likely caused by inflammation-mediated vasodilatation (Ge et al, 2005; Wuerfel et al, 2004), strongly argues against inflammation as the causal factor. The absence of structural (peri)vascular pathology (Aboul-Enein and Lassmann, 2005) indicates that hypoperfusion of the cerebral white matter in MS has a functional origin. The cells involved in regulating cerebral microcirculation in the central nervous system are the astrocytes.
Astrocytes and white matter perfusion
Astrocytic end-feet occupy a great proportion of the surface of intracerebral blood vessels, and convincing evidence has emerged for a direct role of astrocytes in influencing their vascular diameter and the CBF (Filosa et al, 2004; Metea and Newman, 2006; Zonta et al, 2003). Astrocytes in the white matter play a crucial role in the maintenance of ion balance at the nodes of Ranvier, where their perinodal processes form contacts with the axonal membrane (Butt et al, 1994; Raine, 1984). K+ released during action potential propagation at the nodes of Ranvier is soaked up by astrocytes through inward rectifying potassium channels (Kir), especially Kir4.1, and is ‘spatially buffered’ to restore the ionic balance in the axonodal region (Butt and Kalsi, 2006). A highly negative resting membrane potential of the astrocytes is considered critical for their K+ spatial buffering function (Bolton et al, 2006). White matter astrocytes, just as gray matter astrocytes, contain Ca2+-activated K+ channels in their perivascular end-feet (Price et al, 2002), through which the buffered K+ is released into the perivascular space to activate Kir channels, in particular Kir2.1, in the vascular smooth muscle cells. As a consequence, the vascular smooth muscle cells hyperpolarize, which closes voltage-dependent Ca2+ channels in the smooth muscle cells, promoting vasodilation (Figure 1) (Butt and Kalsi, 2006; Knot and Nelson, 1998). Thus, diminished axonal activity leading to reduced K+ efflux at the nodes of Ranvier might represent one mechanism by which less K+ is buffered and released into the perivascular space.
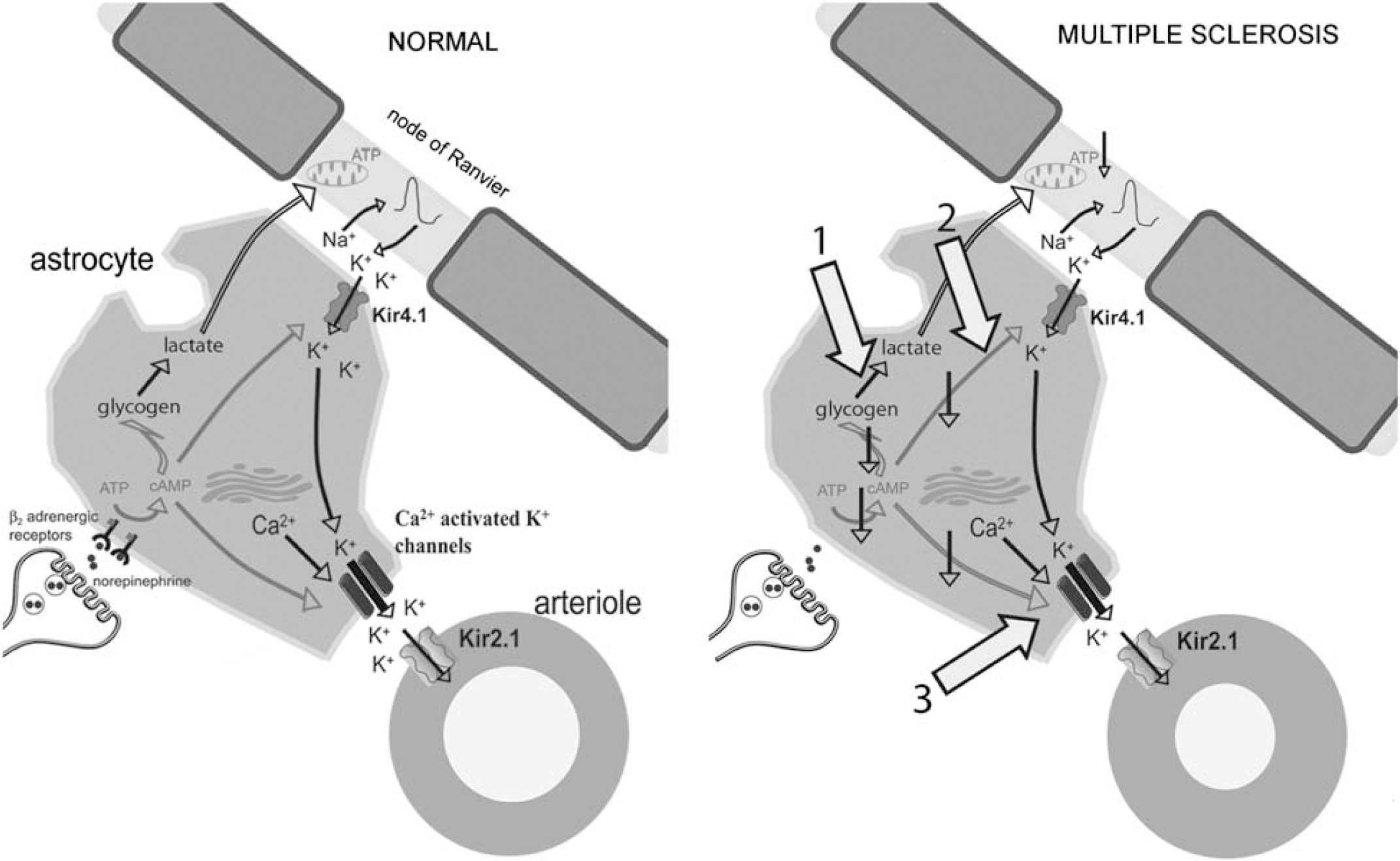
Hypothetical model of axon-vascular coupling in physiologic conditions and in MS. K+ released at the nodes of Ranvier during axonal activity is taken up by astrocytes through Kir4.1 channels and released at perivascular end-feet through Ca2+-activated K+ channels. K+ released into the perivascular space induces hyperpolarization of arteriole smooth muscle cells through activation of Kir2.1 channels, to promote vasodilation. Norepinephrine, via activation of β2-adrenergic receptors and cAMP elevation, provides energy to axons and enhances the uptake of K+ and its release in the perivascular space. In MS, a lack of β2-adrenergic receptors on astrocytes may lead to a reduced concentration of K+ in the perivascular space. Three mechanisms related to decreased intracellular cAMP signaling may be involved: (1) reduced K+ efflux at the nodes of Ranvier because of impaired axonal activity, (2) reduced uptake of K+ by the astrocytes, (3) reduced activity of Ca2+ -activated K+ channels.
Dysfunction of astrocytes in multiple sclerosis
Astrocytes possess a wide variety of neurotransmitter receptors (Porter and McCarthy, 1997). They express adrenergic receptors and receive noradrenergic innervation originating from the locus ceruleus (Cohen et al, 1997; Paspalas and Papadopoulos, 1996; Raichle et al, 1975; Stone and Ariano, 1989). Astrocytes in MS white matter lesions and NAWM were found to be deficient in β2-adrenergic receptors (De Keyser et al, 1999; Zeinstra et al, 2000a). In view of important homeostatic functions mediated by these receptors, it is tempting to speculate that this abnormality plays a pathogenetic role. Activation of astrocytic β2-adrenergic receptors by norepinephrine increases the formation of intracellular cAMP, which prevents astrocytes from transforming into facultative antigen-presenting cells (De Keyser et al, 2003; Zeinstra et al, 2003, 2000b), induces the release of trophic factors for oligodendrocytes (such as brain-derived neurotrophic factor, nerve growth factor, and neuregulin), and stimulates the conversion of glycogen to lactate, which is transported to axons as an energy source (De Keyser et al, 2004a, b ). We have hypothesized that alterations in these homeostatic mechanisms may explain the occurrence of both inflammatory and noninflammatory (primarily caused by oligodendrocyte apoptosis) focal demyelinating lesions, as well as the diffuse axonal degeneration that underlies the progressive phase of MS (De Keyser et al, 2004a, b ). Why and how β2-adrenergic receptors are lost on astrocytes in MS is unknown. Interestingly, a disappearance of astrocytic β2-adrenergic receptors has also been observed in canine distemper encephalitis, which can cause a chronic demyelinating disease that resembles MS (De Keyser et al, 2001).
The astrocytic β2-adrenergic receptor deficit in MS might also be responsible for a reduced CBF in the white matter. Impaired cAMP signaling in astrocytes may limit the proportion of K+ released from perivascular astrocytic end-feet, and thereby reduce the degree of vasodilation by three possible mechanisms (Figure 1). First, reduction in cAMP-mediated formation of lactate in astrocytes may lead to decreased axonal activity and a reduced K+ efflux at the nodes of Ranvier. Second, K+ buffering by astrocytes may be impaired because of a decreased cAMP-mediated hyperpolarization of astrocytes, which is a driving force for the uptake of K+ by the astrocytes (Bolton et al, 2006). Third, the proportion of K+ released in the perivascular space may be limited because of a decreased cAMP-mediated stimulation of Ca2+ -activated K+ channel activity (Bates et al, 1977; Schopf et al, 1999).
Systemic vascular dysregulation?
Another factor that might contribute to reduced perfusion of the white matter in MS is a more systemic vascular dysregulation associated with increased endothelin-1 plasma levels (Pache et al, 2003). Endothelin-1 is a potent vasoconstrictor peptide, and plasma levels are increased in many disease conditions (Shah, 2007). One study found that blood flow velocities in extraocular blood vessels were reduced in patients with MS with increased endothelin-1 plasma levels (Pache et al, 2003). However, a possible effect of circulating endothelin-1 on the CBF in MS and other diseases remains to be clarified.
Pathophysiological role of white matter hypoperfusion in multiple sclerosis
Four fundamentally different patterns of demyelination have been described in MS, defined on the basis of myelin protein loss, the geography and extension of plaques, patterns of oligodendrocyte destruction, and immunopathological evidence of complement activation (Lucchinetti et al, 2000). One type of actively demyelinating lesions in MS (named type III) shows similarity with those found in acute ischemic conditions in the white matter, with a preferential loss of myelin-associated glycoprotein (located in the most distal periaxonal oligodendrocyte processes) and apoptotic-like oligodendrocyte destruction (Aboul-Enein et al, 2003). In these lesions, other myelin proteins, such as myelin basic protein and proteolipid protein (both located in compact myelin) and myelin Oligodendroglia glycoprotein (located in the cytoplasm and the more proximal processes of Oligodendroglia) remain well preserved. The same pattern of demyelination is found in acute ischemic lesions of the white matter.
Active MS lesions and acute ischemic white matter lesions with myelin-associated glycoprotein loss and oligodendrocyte apoptosis reveal a prominent nuclear expression of hypoxia inducible factor-1α (HIF-1α) in glial cells (Aboul-Enein and Lassmann, 2005; Aboul-Enein et al, 2003). HIF-1α is expressed in conditions of hypoxia and forms heterodimers with HIF-1β, which is expressed constitutively in all cells and does not respond to changes in oxygen tension, but is essential for hypoxia-induced transcriptional changes. The HIF-1 heterodimers are translocated to the nucleus where they act as transcription factors, which induce gene expression of various other molecules involved in vasomotor control, angiogenesis, cell growth (growth factors), and energy metabolism (Sharp and Bernaudin, 2004). Altogether, these proteins are expressed to render the tissue more resistant to further hypoxia-induced injury. Micro-array data provide further support to the concept that a diffuse (hypoxic?) tissue-preconditioning in NAWM is a feature of MS pathology. In a study comparing gene expression in the NAWM of patients with secondary progressive MS with that in controls, one of the most consistent differences was found in the expression of HIF-1α and its downstream genes (Graumann et al, 2003). A similar upregulation of HIF-1α in cerebral white matter has been found in leukoaraiosis attributed to white matter hypoperfusion (Fernando et al, 2006).
Could white matter ischemia contribute to focal lesion formation in MS? A longitudinal study in relapsing—remitting MS patients found that a focal increased perfusion in active lesions preceded blood—brain barrier breakdown visualized by gadolinium leakage in the lesion. However, in lesions that developed ring enhancement after contrast agent injection, patterns of CBV and CBF changes comparable to non-ring-enhancing lesions were seen only in the ‘ring tissue‘, whereas inside the ring there was a decrease in CBF and CBV, suggestive of ischemia (Wuerfel et al, 2004). In addition, a few patients have been described who developed new focal lesions with diffusion-weighted magnetic resonance imaging characteristics (reduced apparent diffusion coefficient) suggestive of acute ischemic lesions (Rosso et al, 2006; Rovira et al, 2002). From a historical perspective it is worth mentioning that a number of scientists since the middle of the nineteenth century have already put forward the hypothesis that demyelinating lesions in MS may have an ischemic origin (Gottlieb et al, 1990; Murray, 2005; Putnam, 1933), but these ideas have not gained wide acceptance by the scientific community.
Subcortical ischemic brain damage leading to leukoaraiosis is associated with reduced CBF in white matter (Markus et al, 2000) and represents a common cause of cognitive impairment. The cognitive manifestations in leukoaraioisis are very similar to those observed in MS (Schmidt et al, 2006). In a preliminary study, Inglese et al (2008) found a relationship between perfusion changes in deep gray matter and NAWM and neuropsychological dysfunction in patients with both relapsing—remitting and primary progressive MS. Further studies are required to confirm this interesting finding and to investigate whether interventions that increase white matter perfusion in MS might have a beneficial effect on cognitive functions.
Conclusions
Accumulating evidence indicates that there is a decreased perfusion throughout the NAWM in patients with MS. It occurs in both relapsing—remitting and primary progressive MS, strongly suggesting that it represents an integral part of the disease process. Ischemic changes might be involved in the development of a subtype of focal demyelinating lesions (type III lesions). There appears to be a relationship between reduced white matter perfusion and cognitive dysfunction in patients with MS. We provide a hypothetical framework for the reduced perfusion, implicating a key role of astrocyte dysfunction, possibly related to a deficiency in β2-adrenergic receptors resulting in an impaired siphoning and release of K+ in the perivascular spaces. The underlying pathophysiological mechanisms need to be further elucidated as it could ultimately allow us to understand and treat this complex disease better.