
Abstract
Oligodendrocytes, myelin-forming glial cells of the central nervous system, are vulnerable to damage in a variety of neurologic diseases. Much is known of primary myelin injury, which occurs in settings of genetic dysmyelination or demyelinating disease. There is growing awareness that oligodendrocytes are also targets of injury in acute ischemia. Recognition of oligodendrocyte damage in animal models of ischemia requires attention to their distinct histologic features or use of specific immunocytochemical markers. Like neurons, oligodendrocytes are highly sensitive to injury by oxidative stress, excitatory amino acids, trophic factor deprivation, and activation of apoptotic pathways. Understanding mechanisms of oligodendrocyte death may suggest new therapeutic strategies to preserve or restore white matter function and structure after ischemic insults.
The pathophysiology of neuronal death in cerebral ischemia has been intensively studied. In contrast, much less attention has focused on ischemic damage to glial cells. Oligodendrocytes are abundant in both gray and white matter of the brain and spinal cord. Because they are the only cells in the CNS capable of forming myelin, however, oligodendrocyte damage has most profound consequences for function in the myelinated CNS tracts, the white matter.
White matter is vulnerable in many clinical hypoxic–ischemic insults. In humans, almost all instances of focal ischemia (ischemic stroke) involve white matter. Some clinical strokes result from occlusion of small penetrating arteries that selectively perfuse subcortical white matter areas or fiber tracts. More commonly, even ischemic strokes caused by occlusion of major cerebral arteries cause infarction in equal portions of white and gray matter—because almost half of the human cortical volume is white matter (Miller et al., 1980). Some injured fibers belong to major ascending or descending cortical–spinal pathways, so lesions affect sensation or motor control. The great majority of white matter axons serve to connect local or contralateral brain areas. Accordingly, there is growing recognition of the potential neurocognitive consequences of white matter lesions (Filley, 2001).
In the perinatal period, damage to subcortical fiber tracts and oligodendrocyte progenitor cells results in periventricular leukomalacia and long-term demyelination, which is a major etiology of cerebral palsy (Volpe, 2001). White matter is also affected by many other acute conditions, including global ischemia (cardiac arrest), brain and spinal cord trauma, central venous occlusive infarcts, vascular dementia, cyanide intoxication, and hypoglycemia (Gilles and Murphy, 1969; Leviton and Gilles, 1971, 1984; Gledhill et al., 1973; Brierley et al., 1977; Caplan and Schoene, 1978; Griffiths and McCulloch, 1983; Pantoni and Garcia, 1995; Dambska et al., 1989; Volpe, 2001). In each of these situations, the pathology may selectively involve oligodendrocytes but more commonly affects other cellular elements including axons.
Central myelin loss is a critical element of pathology in other clinically important conditions, such as demyelinating disease, which seem to be etiologically and pathologically distinct from stroke. It is possible that even these very different diseases could share mechanistic features. Although this review focuses on ischemic injury in the mature brain, there is evidence that common pathways of injury occur in other conditions (Merrill and Scolding, 1999; Casaccia-Bonnefil, 2000), including perinatal injury (Volpe, 2001), trauma (Beattie et al., 2000), and multiple sclerosis (Pitt et al., 2000; Smith et al., 2000; Matute et al., 2001).
BIOLOGY OF OLIGODENDROCYTES
Histology
Oligodendrocytes were initially distinguished from other glia by Del Rio Hortega, who identified numerous small cells with few processes in metal-impregnated tissues and recognized that these cells were responsible for production of the axon myelin sheath (Szuchet, 1995). There is considerable morphologic heterogeneity of oligodendrocytes, and this structural variation is thought to be related to differences in the respective functions of different cell subtypes (Baumann and Pham-Dinh, 2001). In white matter, most oligodendrocytes have a characteristic intrafascicular pattern, arranged in horizontal rows of several adjacent cells interposed between axonal fibers (Fig. 1). The principal function of oligodendrocytes is to myelinate axons. Oligodendrocytes give off processes that enwrap the internodal segments of axons to form the myelin sheath that facilitates fast conduction of nerve impulses. One oligodendrocyte can myelinate the internodes of multiple axons (in contrast to the peripheral nervous system, in which each Schwann cell myelinates a single axon), and therefore damage to a single oligodendrocyte has the potential to alter function in many different axons.
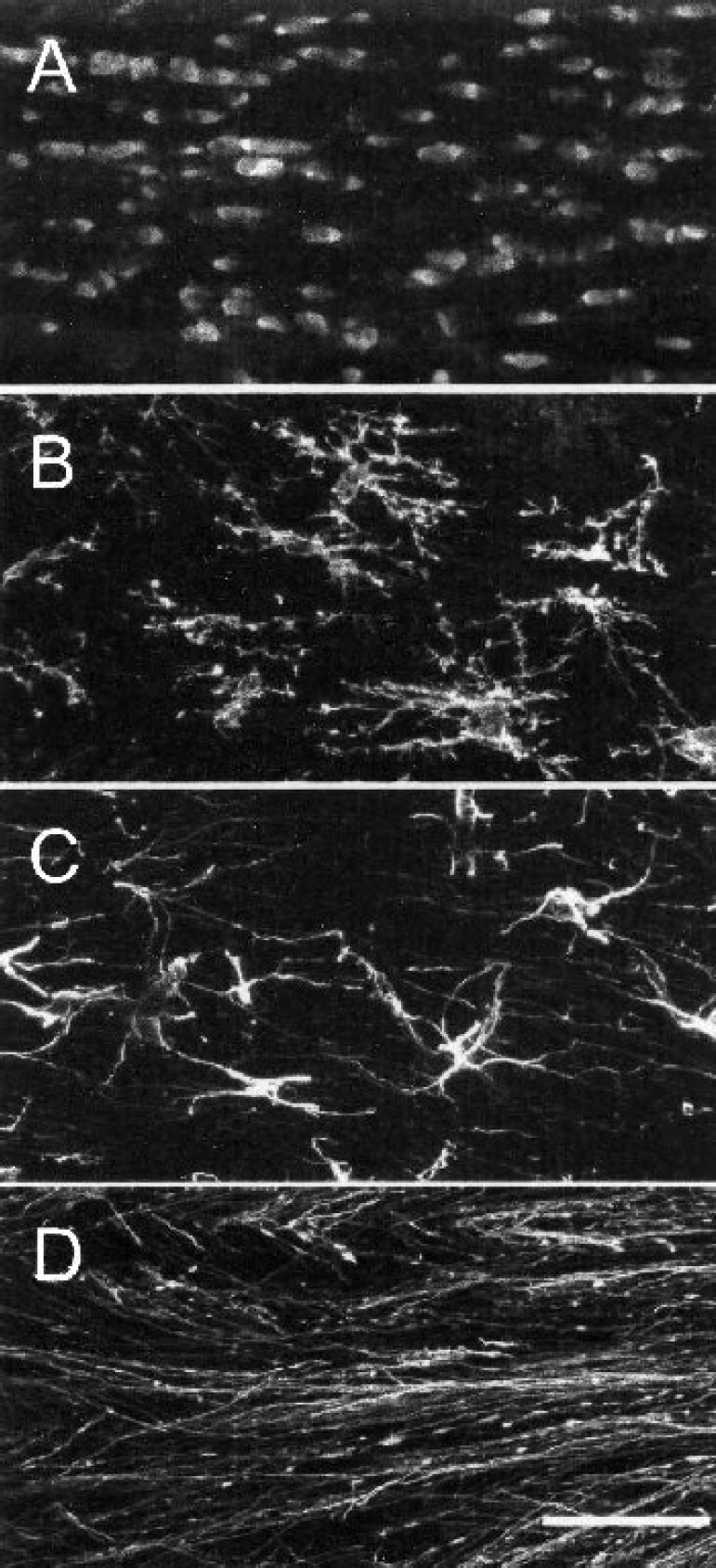
Cellular elements of white matter. Confocal fluorescence micrographs of the mature mouse corpus callosum show the arrangement of oligodendrocytes in white cerebral matter (
In addition to their intrafascicular disposition in white matter tracts, oligodendrocytes are distributed in gray matter regions of the brain. Although brain tissue is conventionally classified as either gray or white matter, it is worth noting that gray matter contains an abundance of axons that are visualized at the microscopic level using myelin stains. Presumably, the myelin of these axons is supplied by oligodendrocytes in gray matter. Oligodendrocytes in gray matter are observed juxtaposed to neuronal perikarya. These “satellite” oligodendrocytes may provide support for their associated neurons, perhaps by participating in regulation of the cellular microenvironment. Under pathologic circumstances, however, these satellite cells have been proposed to possess myelinating potential (Ludwin, 1984).
Oligodendrocyte development
The human brain contains little myelin at birth. During the early postnatal period, oligodendrocyte precursor cells migrate into axon tracts and gradually initiate the process of myelination necessary for acquisition of skilled motor behavior during development. Myelination occurs at different times in each brain region. In general, myelin formation is maximal in the first 3 weeks of the rodent and in the first year of human life (Rivkin et al., 1995; Back et al., 2001).
Cerebral oligodendrocytes arise from progenitors that originate in the subventricular zone and migrate into gray and white matter to differentiate into mature oligodendrocytes (Goldman, 1995; Miller, 1996). Cultured macroglia can be divided according to a morphologic and antigenic classification first proposed for the rat optic nerve (Raff, 1989). Oligodendrocytes may develop from common precursors known as O2A progenitor cells (Raff et al., 1983). O2A progenitor cells are characterized by a simple bipolar morphology, immunoreactivity for a surface antigen recognized by the A2B5 monoclonal antibody, and absence of specific oligodendrocyte or astrocyte markers. In culture, these cells can also differentiate into type 2 astrocytes, which express glial fibrillary acid protein (GFAP) and are readily distinguished from flat type 1 astrocytes by a distinct star-shaped morphology. The relevance of O2A differentiation in vivo is unclear. In vivo, oligodendrocyte progenitors can be identified by expression of the chondroitin sulfate proteoglycan NG2 (Levine and Card, 1987). Under conditions that favor oligodendrocyte differentiation, oligodendrocyte lineage cells develop a progressively more complex branching morphology (Behar, 2001) (Fig. 2). In addition, they express new surface markers, including antigens recognized by the O4 antisulfatide antibody, followed by expression of the glycolipid galactocerebroside (O1 antibody). Differentiated oligodendrocytes express myelin components, such as myelin basic protein (MBP) and proteolipid protein (PLP), and can myelinate axons in vitro (Lubetzki et al., 1993) as well as in vivo.
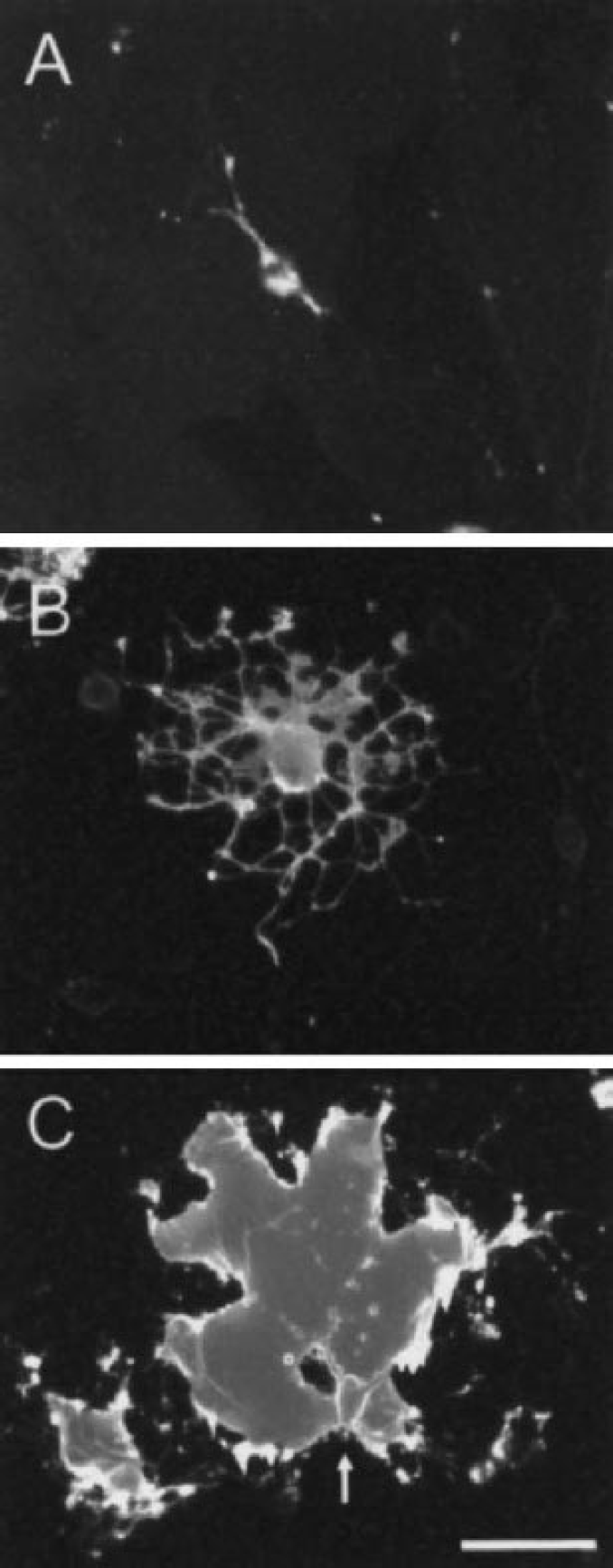
Oligodendrocytes in culture exhibit a defined morphology as they mature. Immature oligodendrocytes
The mature nervous system includes a large population of cells expressing NG2 characteristic of oligodendrocyte precursor cells. These unique glial cells, termed adult progenitor cells, are highly branched and mitotically active. They likely represent sources for repopulation of myelin-forming oligodendrocytes (Levine and Nishiyama, 1996; Levine et al., 2001). Ischemic damage of both oligodendrocytes and their progenitors could result in long-term loss of myelin and consequent white matter dysfunction.
Oligodendrocyte markers
White matter damage may be recognized by loss of myelin staining using standard histochemical procedures. Such loss, however, may be insensitive to partial injury and may take several days to develop. Histologic identification of intrafascicular oligodendrocytes in white matter tracts using light microscopy is relatively straightforward owing to their characteristic morphology, ultrastructure, and “string of pearls” organization. For the less-than-expert microscopist, however, it is much more challenging to identify oligodendrocytes in gray matter. Moreover, the difficulty of distinguishing damaged or shrunken oligodendrocytes from other glial cells after ischemia means that conventional histologic assessment has limited utility. Therefore, recognition of oligodendrocyte damage in animal models and clinical material requires use of more specific tools.
Several antibodies to oligodendrocyte cellular components have been used in animal ischemia models. These include antibodies to myelin proteins, such as MBP, PLP, and carbonic anhydrase (Roussel et al., 1979), and oligodendrocyte cell-surface sphingolipids, such as galactocerebroside (Raff et al., 1978). Glutathione-S-transferase, isoenzyme pi has been found in myelinating oligodendrocytes (Tansey and Crammer, 1991). The antigenic targets for other oligodendrocyte-specific antibodies remain to be identified; these include Rip (Friedman et al., 1989), and CC-1 (Fig. 1A), an antibody to the adenomatous polyposis coli tumor suppressor protein (Bhat et al., 1996). Cells can also be identified by in situ hybridization histochemistry for oligodendrocyte-specific messenger RNA (mRNA) such as PLP (Mandai et al., 1997). No single oligodendrocyte marker, however, has found universal acceptance for experimental studies. Some label only the cell bodies, whereas others label oligodendrocyte processes and myelin. Furthermore, many cellular markers are lost during the injury process, complicating attempts to identify dying oligodendrocytes.
Oligodendrocytes injured by ischemia in rat and human brain become immunoreactive for the cytoskeletal protein, tau (Dewar and Dawson, 1995; Irving et al., 1996b, 2001; Uchihara et al., 2000) (Fig. 3). Therefore, quantitation of tau immunoreactive glial cells can be used to assess potentially therapeutic interventions on ischemic oligodendrocyte pathology (Imai et al., 2001; Irving et al., 1997; McCracken et al., 2002; Valeriani et al., 2000). This can be achieved either by counting the number of tau-positive oligodendrocytes or by plotting out their distribution and measuring the volume of tissue in which they are contained (Fig. 4).
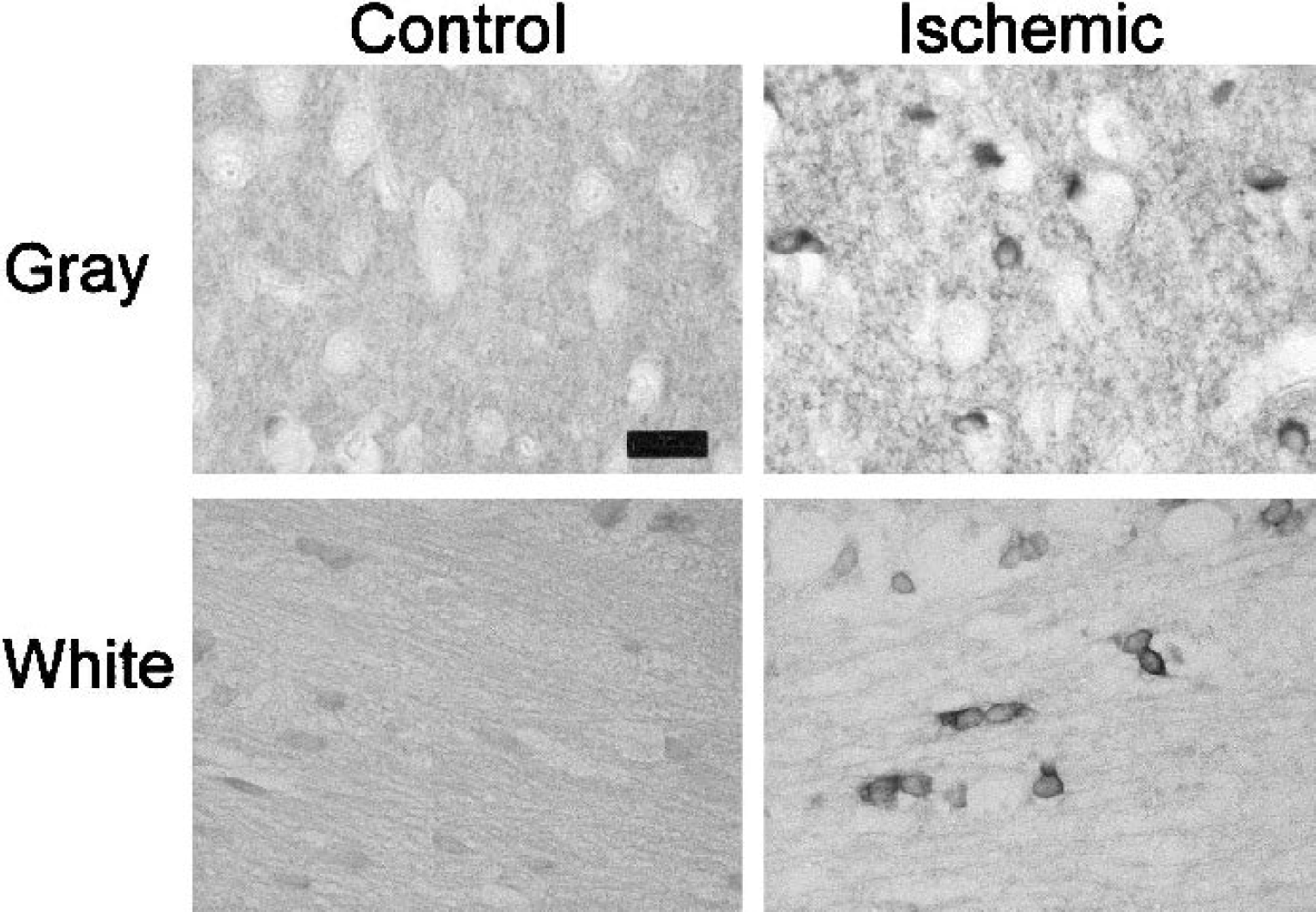
Immunostaining of tau protein reveals ischemic pathology in oligodendrocytes. Glial cells in gray and white matter show minimal reactivity to tau antibodies in control tissue. In ischemic gray and white matter, increased tau immunoreactivity is present in cells with the characteristic morphology of oligodendrocytes—a small, oval soma with a thin rim of cytoplasm located in white matter in intrafascicular rows or in gray matter adjacent to neuronal perikarya. Scale bar = 50 μm.
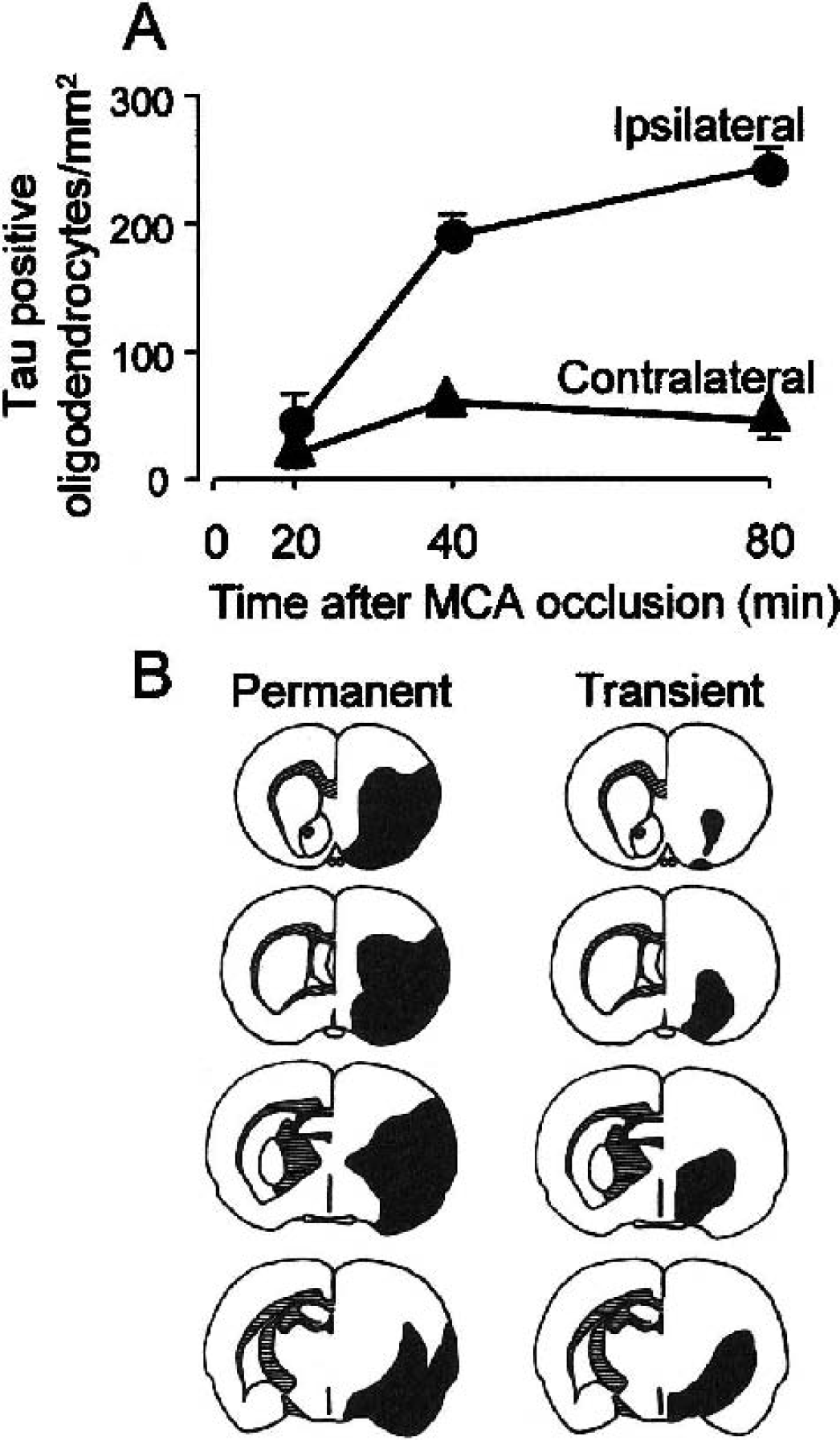
Quantitation of tau-positive oligodendrocytes in ischemic tissue.
It is clear that loss of oligodendrocytes in experimental ischemic models, and their protection by potential therapeutic drugs, can be recognized only if specific procedures are used to identify these cells. It is recommended that preclinical studies of drugs with therapeutic potential for acute stroke include assessment of white matter, including oligodendrocyte survival.
OLIGODENDROCYTES ARE VULNERABLE TO ISCHEMIA
Vulnerability of oligodendrocytes in culture
Oligodendroglia in primary culture are readily damaged by energy deprivation, oxidative stress, or exposure to endogenous or exogenous toxins. In vitro systems allow detailed molecular, cellular, and pharmacological characterization of cell death pathways. Experimental drugs can be examined without confounding effects on the systemic circulation, cerebral blood flow, or other cell types. The limitations of culture models, however, are widely recognized (Underhill et al., 2002b). In contrast to culture models, oligodendrocytes in vivo are never found in isolation; rather, they are in close proximity to neighboring astrocytes, neuronal cell bodies or axons, and endothelial cells in a three-dimensional architecture that is not reproduced in vitro. Furthermore, glial cultures are typically prepared from embryonic or early postnatal animals (McCarthy and de Vellis, 1980) and therefore may not reflect properties of the mature nervous system. Because many published studies report data on early oligodendroglial lineage cells, these results may be more immediately relevant to injury in the developing rather than the mature nervous system.
Combined deprivation of oxygen and glucose kills mature oligodendrocytes (McDonald et al., 1998; Lyons and Kettenmann, 1998; Yoshioka et al., 2000). Compared with other cell types tested under similar hypoxic conditions, oligodendrocytes are less vulnerable than neurons (Goldberg and Choi, 1993) but are much more quickly injured than astrocytes, microglia, or endothelial cells (Goldberg and Choi, 1993; Lyons and Kettenman, 1998; Xu et al., 2000). Within the oligodendroglial lineage, immature cells are much more vulnerable than mature oligodendrocytes to hypoxic–ischemic insults (Husain and Juurlink, 1995; Fern and Moller, 2000; Ness et al., 2001; Back et al., 2002).
Vulnerability of oligodendrocytes in vivo
By definition, oligodendrocytes are lost along with all other cell types when there is pan-necrosis (infarction), and the traditional view was that neurons are the cell type most susceptible to an ischemic insult (Garcia et al., 1977). More recent data, however, indicate that gray and white matter oligodendrocytes are as vulnerable as neurons to ischemia in vivo. Rapid morphologic changes in subcortical white matter oligodendrocytes occurred in response to permanent middle cerebral artery occlusion (MCAO) in the rat (Pantoni et al., 1996). In a few cells expansion of the cytoplasm and swelling of the nuclear compartment was observed as early as 30 minutes. After 3 h of ischemia most oligodendrocytes had a swollen appearance, when the features of pyknosis began to appear. Abnormalities of oligodendrocyte perikaryal structure after induction of ischemia were accompanied by loss of myelin staining. Vacuolation of myelin sheaths was present at 3 h, whereas at 6 h vacuoles were associated with detachment of the sheath from the axolemma. The temporal evolution of these morphologic changes over the first few hours of permanent ischemia before tissue infarction is coincident with that exhibited by neuronal perikarya. Infarction is not a consequence of a global or forebrain ischemic insult. Oligodendrocytes, however, are also vulnerable to these types of insult. Oligodendrocytes exhibited greater vulnerability than neurons in the cerebral cortex and thalamus after transient global ischemia in the rat. Oligodendrocytes showing evidence of apoptosis were present in these two brain regions that sustain minimal neuronal damage, suggesting that they may be selectively vulnerable (Petito et al., 1998). By contrast, after transient forebrain ischemia, the susceptibility of oligodendrocytes to transient forebrain ischemia seemed to parallel the severity of neuronal damage in the striatum relative to the cortex (Petito, 1986).
A variety of structural abnormalities were induced in oligodendrocytes after transient forebrain ischemia, including alterations in the appearance of the Golgi apparatus and endoplasmic reticulum and increases in perikaryal size. Another notable feature noted was abnormalities of microtubules (Petito, 1986). Oligodendrocytes are rich in microtubules, a function of which is to transport essential myelin proteins to their processes (Richter-Landsberg, 2000), and oligodendrocytes contain the microtubule-associated protein, tau (LoPresti et al., 1995; Migheli et al., 1988; Müller et al., 1997; Shin et al., 1991). Rapid alterations in the immunoreactivity of the microtubule-associated protein, tau, provide further evidence that gray and white matter oligodendrocytes are acutely sensitive to ischemia (Dewar and Dawson, 1995; Irving et al., 1997). Consistent with the time course of changes in morphology reported by Pantoni et al. (1996), increased tau immunoreactivity in oligodendrocytes was detectable within 40 minutes of ischemia (Fig. 4A). Overall, the evidence supports the view that oligodendrocytes are acutely vulnerable to ischemia and are not more resistant to it than neurons.
MECHANISMS OF OLIGODENDROCYTE INJURY
Development of therapeutic approaches to ischemic white matter injury requires improved understanding of how oligodendrocytes are irreversibly damaged. Pathogenic mechanisms identified in neurons may not apply to oligodendrocytes. As noted previously, both in vitro and in vivo systems can contribute to understanding of cellular mechanisms of oligodendrocyte injury in ischemia. Several mechanisms have been shown to initiate oligodendrocyte injury under hypoxic–ischemic conditions (Fig. 5). Oxidative stress and excitotoxic injury, discussed in detail further on, seem particularly likely to contribute to rapid oligodendrocyte death in acute ischemic stroke. Oligodendrocyte injury can also be initiated by specific pathways likely to be activated during development or immune-mediated injury. In the developing brain, oligodendrocyte survival requires sequential activation of trophic factors and sustained contact with normal targets such as axons or astrocytes (Barres and Raff, 1993, 1994; Noble and Murray, 1984). In immune-mediated disorders, oligodendrocytes are vulnerable to cytokines and ligand activation of specific cell-surface signaling mechanisms such as the tumor necrosis factor receptor, Fas receptor, or low-affinity nerve growth factor receptor (Casaccia-Bonnefil, 2000). These mechanisms represent well-characterized pathways leading to oligodendrocyte death, but their relevance in the setting of cerebral ischemia remains to be established.
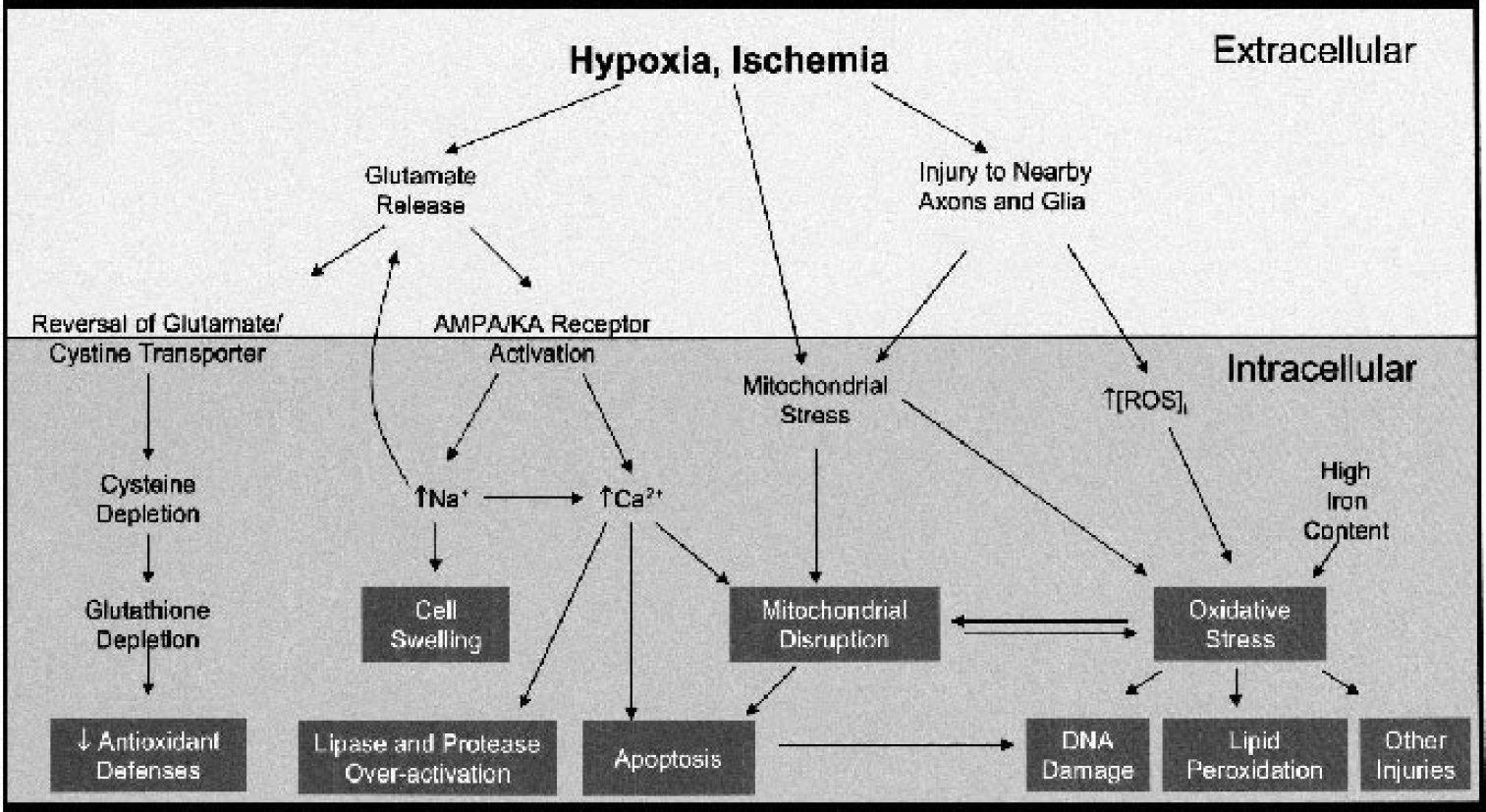
Potential mechanisms of oligodendrocyte injury after hypoxia or ischemia. Schematic shows cellular pathways contributing to oligodendroglial death. Pathways on left are triggered by accumulation of glutamate in the extracellular space. Glutamate may be toxic through inhibition of glutamate–cystine exchange, leading to depletion of glutathione and loss of antioxidant defenses. Glutamate is also toxic by direct activation of ionotropic AMPA and kainate glutamate receptors, leading to increased intracellular sodium and calcium. Sodium overload may cause injury by cell swelling, enhancement of further glutamate release, activation of voltagegated calcium channels or Na/Ca exchange reversal. Calcium overload has many potential consequences, including activation of calcium-dependent proteases, lipases, apoptosis, mitochondrial damage, and production of reactive oxygen species (ROS). Pathways on the right side are initiated by excessive formation of ROS, which may be produced by oligodendrocytes themselves or by neighboring cellular elements (such as axons, astrocytes, microglia, endothelial cells). There is considerable overlap among these pathways; for example, glutamate receptor–mediated excitotoxicity likely activates calcium-dependent injury, ROS formation, and caspase-dependent apoptosis pathways. AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid.
Oxidative stress
Neural cells require antioxidant defenses to deal with the continuous supply of reactive oxygen species (ROS) generated in the brain during aerobic metabolism; oxidative stress is greatly increased during anaerobic respiration occurring during ischemia and reperfusion. In culture, oligodendrocytes seem to be especially sensitive to oxidative stress. Some of the reasons proposed for the high vulnerability of oligodendrocytes, compared with other cells, include high lipid content, high iron content, decreased antioxidant enzymes, or limited substrates (Juurlink et al., 1998). A decreased ability to cope with an increased generation of ROS may be a key reason for oligodendrocyte loss in the ischemic brain. Reactive oxygen species may cause peroxidation of lipids, DNA damage, and oxidative injury of several key molecules essential for cell survival.
Oligodendrocytes have been noted to have low supplies of the cellular antioxidant, glutathione (Thorburne and Juurlink, 1996). Depletion of glutathione stores (e.g., by depletion of its precursor, cysteine) is toxic to oligodendrocytes or enhances other forms of injury, whereas supplementation of glutathione supply often conveys resistance to injury. Early oligodendrocyte lineage cells in vitro have been shown to be highly vulnerable to oxidative stress (Kim and Kim, 1991; Oka et al., 1993; Husain et al., 1995), whereas more mature oligodendrocytes in culture are resistant (Back et al., 1998). Oka et al. (1993) have described a model of oligodendrocyte death induced by extracellular glutamate, in which glutamate toxicity is mediated by interference with cysteine uptake and subsequent depletion of intracellular glutathione. In this model, glutamate toxicity is therefore a situation of primary oxidative stress (Back et al., 1998).
Excitotoxicity
Oligodendroglial lineage cells express most of the genes encoding α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) and kainate glutamate receptor subunits (Patneau et al., 1994; Yoshioka et al., 1995). Although mRNA encoding the N-methyl-d-aspartate (NMDA) type of glutamate receptor subunits can be also detected in oligodendroglia (Yoshioka et al., 1995), most studies suggest that these cells lack expression of functional NMDA receptors (Wyllie et al., 1991; Borges et al., 1994; Pende et al., 1994; Gallo et al., 1995; Liu and Almazan, 1995). AMPA and kainate receptor expression is especially prominent in oligodendrocyte progenitor cells (Barres et al., 1990; Wyllie et al., 1991; Kastritsis and McCarthy, 1993; Holzwarth et al., 1994), and there are fewer descriptions of functional glutamate receptors in differentiated oligodendrocytes. Glutamate receptor overactivation (excitotoxicity) seems to be an important mechanism of oligodendrocyte injury. Several observers have found that application of exogenous AMPA/kainate receptor agonists can be toxic to oligodendrocytes in vitro (Yoshioka et al., 1995; Matute et al., 1997; McDonald et al., 1998; Kelland and Toms, 2001; Liu et al., 2002; Ness and Wood, 2002). Importantly, AMPA/kainate receptor blockade protects cultured oligodendrocytes from hypoxic injury (McDonald et al., 1998; Fern and Moller, 2000; Yoshioka et al., 2000). In the mature white matter, most oligodendrocytes are myelinating cells; therefore, a major concern related to cell culture studies is whether the results could be relevant to the situation in vivo. Oligodendrocytes in adult rodent brain express AMPA/kainate receptor subunits and can be irreversibly damaged by microstereotaxic AMPA administration (McDonald et al., 1998). Similarly, Matute et al. (1997) found injury to oligodendrocytes in optic nerve exposed to kainate. In acute slice preparations of intact brain or spinal cord white matter, AMPA/kainate receptor blockade prevented hypoxia-induced oligodendrocyte death (Tekkök and Goldberg, 2001) and preserved electrical conduction after hypoxia or trauma (Agrawal and Fehlings, 1997; Li et al., 1999, 2000; Tekkök and Goldberg, 2001, Tekkök et al., 2002). These results support the hypothesis that release of endogenous excitatory amino acids and glutamate receptor overactivation may play a major role in hypoxic–ischemic oligodendrocyte death.
Other putative mechanisms
Ischemia-induced activation of the mitogen-activated kinase and extracellular signal-related kinase-signaling pathway (MAPK/ERK) in oligodendrocytes has been reported (Irving et al., 2000). Phospho-ERK1/2 immunoreactivity was increased in white matter oligodendrocytes 6 and 24 h after transient focal ischemia, indicating activation of this pathway. The transcription factor cyclic adenosine monophosphate response element–binding protein (CREB) can be activated through the MAPK pathway, and increased phosphorylation of CREB is essential for its activation. Oligodendrocytes exhibited increased phospho-CREB immunoreactivity after transient focal ischemia in the rat (Irving et al., 2000; Tanaka et al., 2001a). The number of phosphorylated CREB–positive oligodendrocytes was increased within 4 h of tissue reperfusion, but thereafter the number of positive cells within the ischemic area declined (Tanaka et al., 2001a). Barone et al. (1999) point out that treatment with MAPK/ERK inhibitors can reduce the extent of neuronal damage in rat models of focal ischemia and raise the possibility that oligodendrocytes may also be protected in this way. MAPKs, however, have been reported to lose their ability to phosphorylate CREB in mature oligodendrocytes (Sato-Bigbee and DeVreis, 1996; Wegner, 2000), and, at present, it is not clear if MAPK/ERK activation leads to oligodendrocyte death.
Degradation of MBP has been detected 24 h after ischemia, although the levels of other major myelin proteins, PLP, and DM20 were preserved. Myelin proteins are substrates for a variety of proteases, and degradation of MBP after ischemia was reduced in mice lacking matrix metalloproteinase-9 (Asahi et al., 2001). Because the only time point after ischemia examined in this study was 24 h, however, it is not clear whether matrix metalloproteinase attack on myelin is a primary mediator of oligodendrocyte death. Pathologic changes in the microtubule-associated protein, tau, may reflect abnormalities of microtubule structure. Toxic concentrations of glutamate, as well as ischemia, induce increased staining of dephosphorylated tau in vivo (Irving et al., 1996a, Irving 1997). Dephosphorylated tau binds more avidly to microtubules and membranes and may act to stabilize their structure (Arraste et al., 2000; Drewes et al., 1998). Dephosphorylation of tau can also be induced by exposure of cultured oligodendrocytes to oxidative stress, and this precedes fragmentation of oligodendrocytes processes indicative of cytoskeletal breakdown (LoPresti and Konat, 2001). At present, it is not clear whether alterations in tau phosphorylation are directly involved in, or are epiphenomena to, pathogenic processes.
Necrosis and apoptosis
Like other cells, oligodendrocytes can die by way of either necrotic or apoptotic pathways. In cell culture and brain slices, oligodendrocyte excitotoxicity depends on extracellular calcium (Yoshioka et al., 1996; Sanchez-Gomez and Matute, 1999; Tekkök and Goldberg, 2001). Elevated cytosolic calcium may lead to rapid oligodendroglial death by activation of a variety of calcium-dependent processes such as kinases, proteases, and phospholipases. Excitotoxic oligodendrocyte death may also involve generation of intracellular free radicals (e.g., Liu et al., 2002).
Other oligodendrocyte insults trigger injury by relatively well-characterized caspase-dependent apoptotic pathways (Casaccia-Bonnefil, 2000). Trophic factor addition and caspase inhibition did not reduce excitotoxic or hypoxic death of cultured cerebral oligodendrocytes (McDonald et al., 1998) or oligodendroglia lineage cells (Yoshioka et al., 2000), but have been shown to protect immature oligodendrocytes in other models in vitro (Kavanaugh et al., 2000; Ness and Wood, 2002; Liu et al., 2002) and in vivo (Shibata et al., 2000; Han et al., 2000). Numerous intracellular pathways can mediate oligodendrocyte death, and the relative importance of apoptotic and necrotic mechanisms likely depends on cell maturation, injury type, and insult intensity.
Caspase-dependent injury may contribute to oligodendrocyte apoptosis in ischemia in vivo. In models of permanent focal ischemia, oligodendrocyte injury was attenuated in transgenic mice with deletion of caspase-11 or with oligodendrocyte-specific overexpression of the broad-spectrum caspase inhibitor, p35 (Shibata et al., 2000). A role for caspase activation in ischemia-induced oligodendrocyte death is further suggested by the increased expression of Bim, a member of the proapoptotic BH3-only proteins, in oligodendrocytes at 3 to 6 h after permanent focal ischemia in mice (Shibata et al., 2002).
PROTECTION OF OLIGODENDROCYTES FROM ISCHEMIA IN VIVO
To determine if putative antiischemic agents can reduce the extent of oligodendrocyte damage in animal models of stroke, it is necessary to use methods to quantify the extent of the pathology. As noted previously, oligodendrocyte injury may be assessed quantitatively by determination of tau immunoreactivity after ischemia (Fig. 4). For example, in the study of Valeriani et al. (2000), the volume of tissue containing tau-positive oligodendrocytes, in animals that had experienced early reperfusion of the tissue after 2 h of intraluminal-thread MCAO, was reduced compared with that in animals subjected to permanent artery occlusion. Early tissue reperfusion significantly reduced oligodendrocyte pathology in the cerebral cortex but reduced it to a much lesser degree in subcortical areas. The greater severity of ischemia in deep brain areas may account for the lack of a protective effect of reperfusion and indicates that, as with neuronal damage, there are areas within the ischemic territory where it may not be possible to protect oligodendrocytes.
Glutamate receptor antagonists
Despite the compelling evidence from in vitro studies that excitotoxicity causes oligodendrocyte damage, to date, there are only two studies that have investigated this issue after ischemia in vivo in mature animals. Pretreatment of rats subjected to 40 minutes of focal ischemia with the NMDA antagonist, MK-801, or the AMPA antagonist, NBQX, both at doses previously shown to reduce neuronal damage, did not reduce the number of tau-positive oligodendrocytes in subcortical white matter (Irving et al., 1997). The failure of MK-801 to attenuate oligodendrocyte pathology is consistent with their lack of NMDA receptors (Gallo and Russell, 1995; Patneau et al., 1994). The ineffectiveness of NBQX, however, contrasts with the substantial in vitro evidence for AMPA-mediated toxicity in oligodendrocytes (see section on Excitotoxicity). A more recent study of oligodendrocyte pathology after ischemia, however, used the novel AMPA antagonist SPD 502. This agent does not have the adverse properties of poor solubility and nephrotoxicity that have compromised previous in vivo use of antagonists such as NBQX (Nielsen et al., 1999). Administration of SPD 502 to rats subjected to 3-h MCAO significantly reduced the extent of ischemic oligodendrocyte pathology determined 21 h later (McCracken et al., 2002). The extent of oligodendrocyte pathology, however, was significantly reduced in the cerebral cortex but not in subcortical areas. It was suggested that this is related to the severity of ischemia, which after MCAO is far greater in subcortical compared with cortical regions. Thus, when the reduction in blood flow is profound it may not be possible to protect oligodendrocytes with AMPA antagonists because the severity of ischemia is too great and sufficient delivery of protective agent is not possible. Interestingly, however, SPD 502 reduced the extent of neuronal damage in both cortical and subcortical regions, and it also reduced the amount of axonal pathology located predominantly in subcortical regions. These data suggest that AMPA receptor antagonists have the ability to protect multiple cellular elements in both white and gray matter from the effects of ischemia, and further studies of these compounds are warranted to determine whether they are able to improve functional recovery.
In addition to cerebral ischemia, AMPA receptor antagonists have proved to be efficacious in improving outcome in other models of CNS injury that include oligodendrocyte and/or white matter pathology. The severity of white matter damage, assessed by MBP immunostaining, in a rat model of perinatal hypoxia–ischemia was reduced by systemic administration of NBQX (Follett et al., 2000). This agent also attenuated the extent of axonal damage, assessed by neurofilament immunostaining, and improved locomotor function in a rat model of spinal cord ischemia (Kanellopoulos et al., 2000). After traumatic injury to the spinal cord of rats, NBQX not only improved the survival of oligodendrocytes but also reduced axonal injury and attenuated impairment of locomotor function (Rosenberg et al., 1999; Wrathall et al., 1994, 1996, 1997). Finally, in the chronic condition induced by experimental autoimmune encephalomyelitis in rats, a model of multiple sclerosis, NBQX ameliorated neurologic symptoms and also reduced oligodendrocyte and axonal loss in the spinal cord (Pitt et al., 2000; Smith et al., 2000). Thus, in a variety of conditions with differing etiologies, including ischemia, AMPA receptor antagonists may prove to be beneficial.
Antioxidants
The susceptibility of cultured oligodendrocytes to oxidative damage has been discussed previously, and, therefore, antioxidant treatments may also hold promise for protection of gray and white matter in vivo. In support of this, pretreatment with the spin-trap agent α-phenyl-tert-butyl-nitrone significantly reduced the amount of oligodendrocyte pathology induced by focal cerebral ischemia, although the effect was only examined at 40 minutes after the insult (Irving et al., 1997). Using a posttreatment strategy initiated at the time of tissue reperfusion after transient focal ischemia, Imai et al. (2001) showed the protective effects of ebselen, a potent antioxidant. Ebselen infusion reduced the hemispheric extent of oligodendrocyte pathology by 60% after transient MCAO, with protection afforded in both cortex and striatum. The amounts of both neuronal and axonal damage were also reduced by ebselen treatment. The ability of ebselen treatment to reduce ischemic damage in oligodendrocytes, axons, and neurons was reflected in its ability to improve neurologic function in the animals.
OLIGODENDROCYTES AND RECOVERY AFTER ISCHEMIA
What are the functional consequences of oligodendrocyte loss in cerebral ischemia? Selective loss of oligodendrocytes and their myelin will impede axonal saltatory conduction, leading to reduction in transmission speed and possible conduction failure. There may be additional functional sequelae. Loss of oligodendrocytes may deprive axons of trophic or metabolic support, and myelin damage may reveal internodal ion channels or immune targets. Recent evidence suggests that excitotoxic injury of oligodendrocytes causes secondary damage to axons under hypoxic conditions in brain slices (Underhill et al., 2002a).
Alterations in the structure and function of surviving tissue are generally thought to be involved in processes mediating recovery after stroke. Oligodendrocyte survival and maintenance of myelin in the periinfarct zone has been linked to the activation of CREB (Tanaka et al., 2001a). CREB may indirectly regulate the expression of myelin protein genes and is an important mediator of oligodendrocyte differentiation and process outgrowth (Sato-Bigbee and DeVreis, 1996; Wegner, 2000), so it has been suggested that this gene response may mediate oligodendrocyte survival in periinfarct tissue (Tanaka et al., 2001a).
The presence of oligodendrocyte precursors in the mature brain may provide the opportunity for significant renewal of oligodendroglial numbers after injury. Proliferation of oligodendrocytes at the margin of an infarct, as detected by PLP or MBP mRNA, has been reported to occur between 1 and 7 d after ischemia (Gregersen et al., 2001; Mandai et al., 1997; Mabuchi et al., 2000). Increased numbers of cells were detected both in gray matter around the infarct and in the corpus callosum adjacent to the lesion. Proliferation of oligodendrocytes has been suggested to be a response to a short, sublethal ischemic injury that causes myelin damage (Liu et al., 2001). Consistent with postischemic proliferation is the finding of increased immunolabeling of cell cycle proteins in oligodendrocytes after focal ischemia (Li et al., 1997). Proliferation of oligodendrocyte progenitor cells that express the chondroitin sulphate proteoglycan, NG2, also occurred at the margin of an infarct 2 weeks after ischemia (Tanaka et al., 2001b). The authors of this study raise the possibility that proliferation of progenitors may constitute an attempt to remyelinate periinfarct tissue. Even though progenitor proliferation occurs in the periinfarct region, however, it remains purely speculative at present that these cells have the capacity to myelinate any newly formed axons. Indeed, the presence of myelinating oligodendrocytes may hinder rather than promote tissue reorganization essential for recovery of function, since regeneration and structural plasticity in the adult brain are inhibited by myelin-associated neurite growth inhibitors (Bandtlow and Schwab, 2000). The effect of oligodendrocyte proliferation on periinfarct recovery remains unknown.
CONCLUSIONS
The recognition that, in addition to neuronal perikarya, oligodendrocytes (and axons) are vulnerable to ischemia means that only targeting pathophysiologic mechanisms of neuronal death does not represent an optimal therapeutic strategy. Although there are multiple reasons why clinical trials of NMDA receptor antagonists failed to improve outcome in stroke patients, a fundamental biologic issue may relate to their inability to prevent death of oligodendrocytes and damage to axons—neither of which express NMDA receptors. A major challenge in the development of new pharmacological treatments for cerebral ischemia is to identify agents that will protect the cellular elements of gray and white matter, both of which are affected in almost all cases of stroke. Potential targets include oxidative stress and AMPA receptor–mediated excitotoxicity, blockade of which has been shown to attenuate neuronal, axonal, and oligodendrocyte damage in rodent models of stroke. These mechanisms have also been implicated in other clinical conditions such as multiple sclerosis and spinal cord injury. Thus, although distinct primary pathogenic events are causative, excitotoxicity and oxidative stress may represent common mechanisms that lead to oligodendrocyte damage in a variety of neurologic conditions.